
Facile N9-Alkylation of Xanthine Derivatives and Their Use as Precursors for N-Heterocyclic Carbene Complexes



Abstract
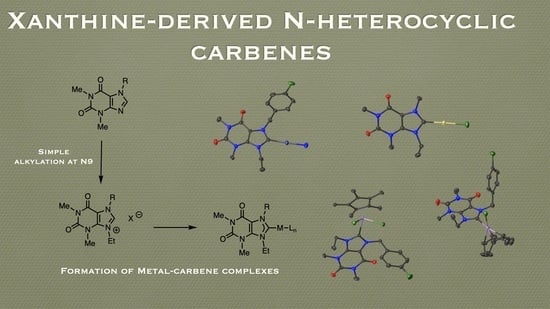
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Díez-González, S. (Ed.) N-Heterocylic Carbenes. From Laboratory Curiosities to Efficient Synthetic Tools; RSC Publishing: Cambridge, UK, 2011. [Google Scholar]
- Kühl, O. Functionalised N-heterocyclic Carbene Complexes; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Hindi, K.M.; Panzner, M.J.; Tessier, C.A.; Cannon, C.L.; Youngs, W.J. The Medicinal Applications of Imidazolium Carbene-Metal Complexes. Coord. Chem. Rev. 2009, 109, 3859–3884. [Google Scholar] [CrossRef] [Green Version]
- Diez-Gonzalez, S.; Marion, N.; Nolan, S.P. N-heterocyclic carbenes in late transition metal catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- de Frémont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Edn. Engl. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P. (Ed.) N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Crudden, C.M.; Allen, D.P. Stability and reactivity of N-heterocyclic carbene complexes. Coord. Chem. Rev. 2004, 248, 2247–2273. [Google Scholar] [CrossRef]
- Dröge, T.; Glorius, F. The Measure of All Rings. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2010, 49, 6940–6952. [Google Scholar] [CrossRef]
- Liu, W.; Gust, R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef]
- Liu, W.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Nelson, D.J. Accessible syntheses of late transition metal (pre)catalysts bearing N-heterocyclic carbene ligands. Eur. J. Inorg. Chem. 2015, 2015, 2012–2027. [Google Scholar] [CrossRef]
- Cisnetti, F.; Gautier, A. Metal N-heterocyclic carbene complexes: Opportunities for the development of anticancer metallodrugs. Angew. Chem. Int. Ed. 2013, 52, 11976–11978. [Google Scholar] [CrossRef]
- Teyssot, M.L.; Jarrousse, A.S.; Manin, M.; Chevry, A.; Roche, S.; Norre, F.; Beaudoin, C.; Morel, L.; Boyer, D.; Mahiou, R.; et al. Metal-NHC complexes: A survery of anti-cancer properties. Dalton Trans. 2009, 35, 6849–6902. [Google Scholar] [CrossRef] [PubMed]
- Beck, W.; Kottmair, N. Neue Übergangsmetallkomplexe mit Nuclein-Basen und Nucleosiden. Chem. Ber. 1976, 109, 970–993. [Google Scholar] [CrossRef]
- Kascatan-Nebioglu, A.; Panzner, M.J.; Garrison, J.C.; Tessier, C.A.; Youngs, W.J. Synthesis and Structural Characterization of N-Heterocyclic Carbene Complexes of Silver(I) and Rhodium(I) from Caffeine. Organometallics 2004, 23, 1928–1931. [Google Scholar] [CrossRef]
- Schütz, J.; Herrmann, W.A. Purine-based carbenes at rhodium and iridium. J. Organomet. Chem. 2004, 689, 2995–2999. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Schütz, J.; Frey, G.D.; Herdtweck, E. N-Heterocyclic carbenes: Synthesis, structures, and electronic properties. Organometallics 2006, 25, 2437–2448. [Google Scholar] [CrossRef]
- Kascatan-Nebioglu, A.; Melaiye, A.; Hindi, K.; Durmus, S.; Panzner, M.J.; Hogue, L.A.; Mallett, R.J.; Hovis, C.E.; Coughenour, M.; Crosby, S.D.; et al. Synthesis from Caffeine of a Mixed N-Heterocyclic Carbene–Silver Acetate Complex Active against Resistant Respiratory Pathogens. J. Med. Chem. 2006, 49, 6811–6818. [Google Scholar] [CrossRef] [PubMed]
- Hindi, K.M.; Siciliano, T.J.; Durmus, S.; Panzner, M.J.; Medvetz, D.A.; Reddy, D.V.; Hogue, L.A.; Hovis, C.E.; Hilliard, J.K.; Mallet, R.J.; et al. Synthesis, Stability, and Antimicrobial Studies of Electronically Tuned Silver Acetate N-Heterocyclic Carbenes. J. Med. Chem. 2008, 51, 1577–1583. [Google Scholar] [CrossRef]
- Panzner, M.J.; Deeraksa, A.; Smith, A.; Wright, B.D.; Hindi, K.M.; Kascatan-Nebioglu, A.; Torres, A.G.; Judy, B.M.; Hovis, C.E.; Hilliard, J.K.; et al. Synthesis and in vitro efficacy studies of silver carbene complexes on biosafetly level 3 bacteria. Eur. J. Inorg. Chem. 2009, 1739–1745. [Google Scholar] [CrossRef] [Green Version]
- Knapp, A.R.; Panzner, M.J.; Medvetz, D.A.; Wright, B.D.; Tessier, C.A.; Youngs, W.J. Synthesis and antimicrobial studies of silver N-heterocyclic carbene complexes bearing a methyl benzoate substituent. Inorg. Chim. Acta 2010, 364, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youngs, W.J.; Panzner, M.J.; Cannon, C.L. Metal Complexes of N-Heterocyclic Carbenes. U.S. Patent 8,648,205, 11 February 2014. [Google Scholar]
- Panzner, M.J.; Hindi, K.M.; Wright, B.D.; Taylor, J.B.; Han, D.S.; Youngs, W.J.; Cannon, C.L. A theobromine derived silver N-heterocyclic carbene: Synthesis, characterization, and antimicrobial efficacy studies on cystic fibrosis relevant pathogens. Dalton Trans. 2009, 35, 7308–7313. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.J.; Bai, S.Q.; Yeh, H.H.; Young, D.J.; Chi, Y.; Hor, T.S.A. N-heterocyclic carbene Pt(II) complexes from caffeine: Synthesis, structures and photoluminescent properties. Dalton Trans. 2011, 40, 4402–4406. [Google Scholar] [CrossRef]
- Zhang, J.J.; Che, C.M.; Ott, I. Caffeine derived platinum(II) N-heterocyclic carbene complexes with multiple anti-cancer activities. J. Organomet. Chem. 2015, 782, 37–41. [Google Scholar] [CrossRef]
- Skander, M.; Retailleau, P.; Bourrié, B.; Schio, L.; Mailliet, P.; Marinetti, A. N-Heterocyclic carbene-amine Pt(II) complexes a new chemical space for the develpment of platinum based anticancer drugs. J. Med. Chem. 2010, 53, 2146–2154. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.T.; Lo, H.K. Short synthesis of bis-NHC-Pd catalyst derived from caffeine and its applications to Suzuki, Heck, and Sonogashira reactions in aqueous solution. J. Organomet. Chem. 2011, 696, 1262–1265. [Google Scholar] [CrossRef]
- Lo, H.K.; Luo, F.T. Synthesis of PS-supported NHC-Pd catalyst derived from theobromine and its applications in Suzuki-Miyaura reaction. J. Chin. Chem. Soc. 2012, 59, 394–398. [Google Scholar] [CrossRef]
- Landaeta, V.R.; Rodríguez-Lugo, R.E.; Rodríguez-Arias, E.N.; Coll-Gómez, D.S.; González, T. Studies on the coordination chemistry of methylated xanthines and their imidazolium salts. Part 1: Benzyl derivatives. Transit. Met. Chem. 2010, 35, 165–175. [Google Scholar] [CrossRef]
- Scattolin, T.; Giust, S.; Bergamini, P.; Caligiuri, I.; Canovese, L.; Demitri, N.; Gambari, R.; Lampronti, I.; Rizzolio, F.; Visentin, F. Palladacyclopentadienyl complexes bearing purine-based N-heterocyclic carbenes: A new class of promising antiproliferative agents against human ovarian cancer. Appl. Organomet. Chem. 2019, 33, e4902. [Google Scholar] [CrossRef]
- Scattolin, T.; Caligiuri, I.; Canovese, L.; Demitri, N.; Gambari, R.; Lampronti, I.; Rizzolio, F.; Santo, C.; Visentin, F. Synthesis of new allyl palladium complexes bearing purine-based NHC ligands with antiproliferative and proapoptotic activities on human ovarian cancer cell lines. Dalton Trans. 2018, 47, 13616–13630. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; Stefan, L.; Pirrotta, M.; Monchaud, D.; Bodio, E.; Richard, P.; Le Gendre, P.; Warmerdam, E.; de Jager, M.H.; Groothuis, G.M.M.; et al. Caffeine-Based Gold(I) N-Heterocyclic Carbenes as Possible Anticancer Agents: Synthesis and Biological Properties. Inorg. Chem. 2015, 53, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Eslava-Gonzalez, I.; Valdés, H.; Ramírez-Apan, M.T.; Hernandez-Ortega, S.; Zermeño-Ortega, M.R.; Avila-Sorrosa, A.; Morales-Morales, D. Synthesis of theophylline-based iridium(I) N-heterocyclic carbene complexes including fluorinated-thiophenolate ligands. Preliminary evaluation of their in vitro anticancer activity. Inorg. Chim. Acta 2020, 507, 119588. [Google Scholar] [CrossRef]
- Mohamed, H.A.; Lake, B.R.M.; Laing, T.; Phillips, R.M.; Willans, C.E. Synthesis and anticancer activity of silver(I) N-heterocyclic carbene complexes derived from the natural xanthine products caffeine, theophylline and theobromine. Dalton Trans. 2015, 44, 7563–7569. [Google Scholar] [CrossRef]
- Holmes, J.; Kearsey, R.J.; Paske, K.A.; Singer, F.N.; Atallah, S.; Pask, C.M.; Phillips, R.M.; Willans, C.E. Tethered N-Heterocyclic Carbene-Carboranyl Silver Complexes for Cancer Therapy. Organometallics 2019, 38, 2530–2538. [Google Scholar] [CrossRef]
- Sitalu, K.; Babu, B.H.; Naveena Lavanya Latha, J.; Rao, A.L. Synthesis, characterization and antimicrobial activities of copper derivatives of NHC complexes. Pak. J. Biol. Sci. 2017, 20, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Valdés, H.; Canseco-González, D.; Germán-Acacio, J.M.; Morales-Morales, D. Xanthine based N-heterocyclic carbene (NHC) complexes. J. Organomet. Chem. 2018, 867, 51–54. [Google Scholar] [CrossRef]
- Pelz, S.; Mohr, F. “Oxide Route” for the Preparation of Mercury(II) N-Heterocyclic Carbene Complexes. Organometallics 2011, 30, 383–385. [Google Scholar] [CrossRef]
- Rubbiani, R.; Schuh, E.; Meyer, A.; Lemke, J.; Wimberg, J.; Metzler-Nolte, N.; Meyer, F.; Mohr, F.; Ott, I. TrxR inhibition and antiproliferative activities of structurally diverse gold N-heterocyclic carbene complexes. Medchemcomm 2013, 4, 942–948. [Google Scholar] [CrossRef]
- Citta, A.; Schuh, E.; Mohr, F.; Folda, A.; Massimino, M.L.; Bindoli, A.; Casini, A.; Rigobello, M.P. Fluorescent silver(I) and gold(I) N-heterocyclic carbene complexes with cytotoxic properties: Mechanistic insights. Metallomics 2013, 5, 1006–1015. [Google Scholar] [CrossRef]
- Schuh, E.; Pflüger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) carbene complexes causing Thioredoxin 1 and Thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef]
- Schuh, E.; Valiahdi, S.M.; Jakupec, M.A.; Keppler, B.K.; Chiba, P.; Mohr, F. Synthesis and biological studies of some gold(I) complexes containing functionalised alkynes. Dalton Trans. 2009, 48, 10841–10845. [Google Scholar] [CrossRef]
- Mell, B.; Rust, J.; Lehmann, C.W.; Berger, R.J.F.; Otte, D.; Ertl, M.; Monkowius, U.; Mohr, F. Arylamidoethyl-functionalized imidazolium salts: Precursors for dianionic [C,N,C]2– carbene ligands at a platinum center. Organometallics 2021, 40, 890–898. [Google Scholar] [CrossRef]
- Ivanov, E.I.; Kalayanov, G.D.; Yaroshchenko, I.M.; Stepanov, D.E. New synthesis of caffeine methiodide and its homolog. Chem. Heterocycl. Compd. 1989, 25, 1318. [Google Scholar] [CrossRef]
- Bredereck, H.; Christmann, O.; Koser, W.; Schellenberg, P.; Nast, R. Synthesen in der Purinreihe, XV. Synthesen von Xanthinium-und Guaninium-betainen. Chem. Ber. 1962, 95, 1812–1819. [Google Scholar] [CrossRef]
- Hori, M.; Kataoka, T.; Shimizu, H.; Imai, E.; Matsumoto, Y.; Kawachi, M.; Kuratani, K.; Yokomoto, M. Generation and Properties of N7-Xanthinium Ylides: Reactions of N7-Xanthinium Ylides with Diphenylcyclopropenone and Acetylenic Compounds. Chem. Pharm. Bull. 1986, 34, 1328–1332. [Google Scholar] [CrossRef] [Green Version]
- Hori, M.; Kataoka, T.; Shimizu, H.; Imai, E.; Matsumoto, Y. Reactions of 8, 9-Dihydroxanthines with Acetylenic Compounds. Formation of Heteropropellanes. Chem. Pharm. Bull. 1985, 33, 3681–3688. [Google Scholar] [CrossRef] [Green Version]
- Gibard, C.; Fauché, K.; Guillot, R.; Jouffret, L.; Traïkia, M.; Gautier, A.; Cisnetti, F. Access to silver-NHC complexes from soluble silver species in aqueous or ethanolic ammonia. J. Organomet. Chem. 2017, 840, 70–74. [Google Scholar] [CrossRef]
- Su, H.L.; Pérez, L.M.; Lee, S.J.; Reibenspies, J.H.; Bazzi, H.S.; Bergbreiter, D.E. Studies of ligand exchange in N-heterocyclic carbene silver(I) complexes. Organometallics 2012, 31, 4063–4071. [Google Scholar] [CrossRef]
- Newman, C.P.; Clarkson, G.J.; Rourke, J.P. Silver(I) N-heterocyclic carbene halide complexes: A new bonding motif. J. Organomet. Chem. 2007, 692, 4962–4968. [Google Scholar] [CrossRef]
- Tulloch, A.A.D.; Danopoulos, A.A.; Winston, S.; Kleinhenz, S.; Eastham, G. N-Functionalised heterocyclic carbene complexes of silver. J. Chem. Soc. Dalton Trans. 2000, 4499–4506. [Google Scholar] [CrossRef]
- Zhang, J.J.; Muenzner, J.K.; Abu el Maaty, M.A.; Karge, B.; Schobert, R.; Wölfl, S.; Ott, I. A multi-target caffeine derived rhodium(I) N-heterocyclic carbene complex: Evaluation of the mechanism of action. Dalton Trans. 2016, 45, 13161–13168. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.T.Y.; Hahn, F.E. Synthesis of Iridium(III) and Rhodium(III) Complexes Bearing C8-Metalated Theophylline Ligands by Directed C–H Activation. Organometallics 2019, 38, 2250–2258. [Google Scholar] [CrossRef]
- Bhushan, K.; Lister, J.H. Purine studies. XVIII. The C- and N-alkylation of theophylline and derivatives. Aust. J. Chem. 1976, 29, 891–897. [Google Scholar] [CrossRef]
- Daly, J.W.; Padgett, W.L.; Shamim, T. Analogues of Caffeine and Theophylline: Effect of Structural Alterations on Affinity at Adenosine Receptors. J. Med. Chem. 1986, 29, 1305–1308. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M. Tetrahydrothiophenegold(I) or gold(III) complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 2, 233–241. [Google Scholar] [CrossRef]
- El Amouri, H.; Gruselle, M.; Jaouén, G. Bis[dichloro(η5-pentamethylcyclopentadienyl)rhodium(III). Synth. React. Inorg. Met. Org. Chem. 1994, 24, 395–400. [Google Scholar] [CrossRef]
- Giordano, G.; Crabtree, R.H. Di-μ-chloro-bis(η4-1,5-cyclooctadiene)dirhodium(I). Inorg. Synth. 1990, 28, 88–90. [Google Scholar]
- Sheldrick, G.M. SADABS 2.03; Universiät Göttingen: Göttingen, Germany, 2002. [Google Scholar]
- CrysAlisPro 40.53, 40.53; Rigaku Oxford Diffraction Ltd.: Oxford, UK, 2019.
- Kabsch, W. XDS. Acta Cryst. 2010, D66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokfi, M.; Rust, J.; Lehmann, C.W.; Mohr, F. Facile N9-Alkylation of Xanthine Derivatives and Their Use as Precursors for N-Heterocyclic Carbene Complexes. Molecules 2021, 26, 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123705
Mokfi M, Rust J, Lehmann CW, Mohr F. Facile N9-Alkylation of Xanthine Derivatives and Their Use as Precursors for N-Heterocyclic Carbene Complexes. Molecules. 2021; 26(12):3705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123705
Chicago/Turabian StyleMokfi, Moloud, Jörg Rust, Christian W. Lehmann, and Fabian Mohr. 2021. "Facile N9-Alkylation of Xanthine Derivatives and Their Use as Precursors for N-Heterocyclic Carbene Complexes" Molecules 26, no. 12: 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26123705