
Benzothiadiazole vs. iso-Benzothiadiazole: Synthesis, Electrochemical and Optical Properties of D–A–D Conjugated Molecules Based on Them

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
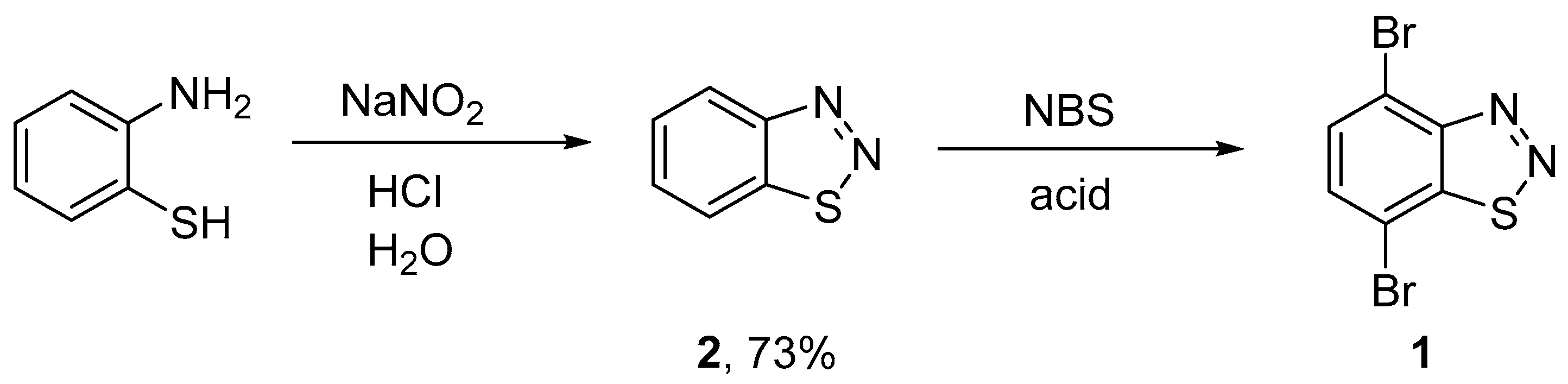
2.1. Synthesis of 4,7-Dibromobenzo[d][1,2,3]thiadiazole
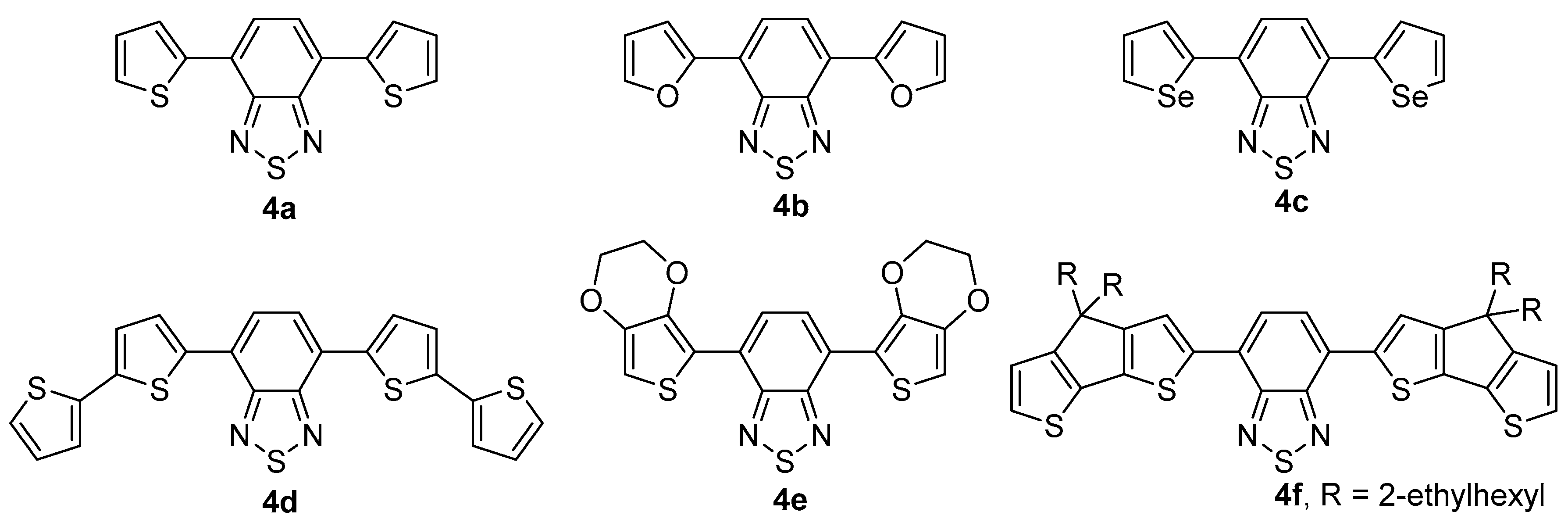
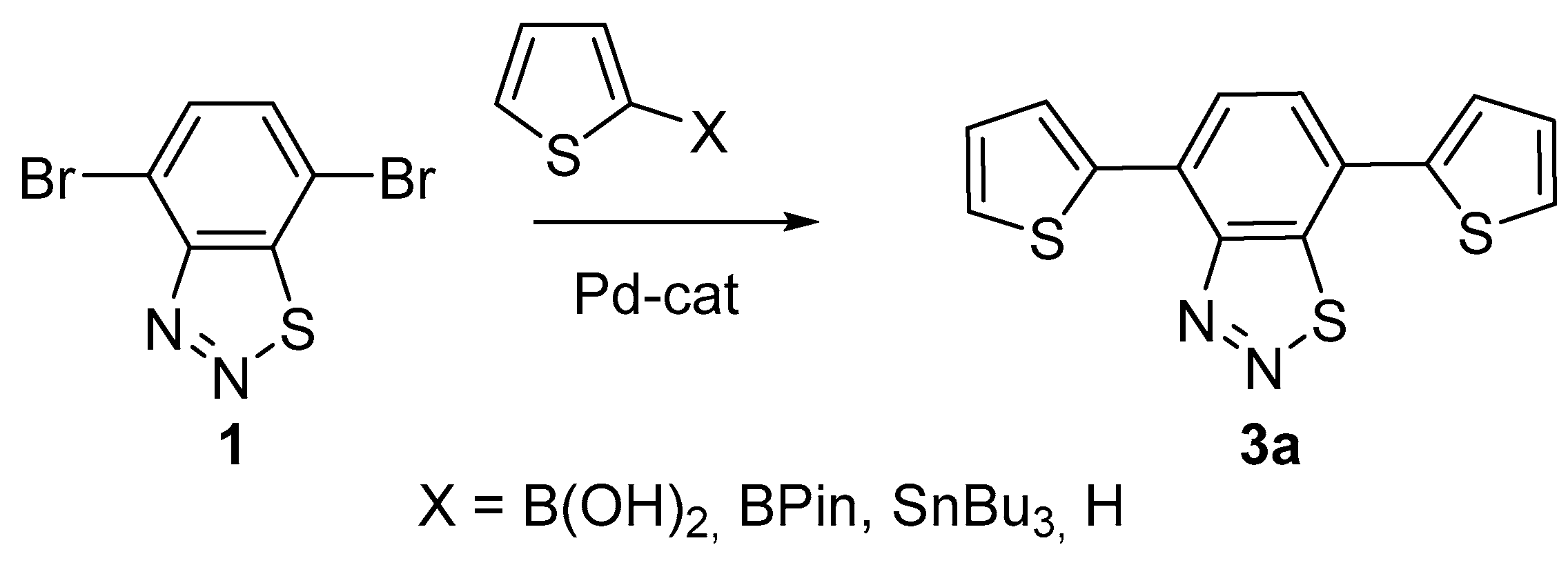
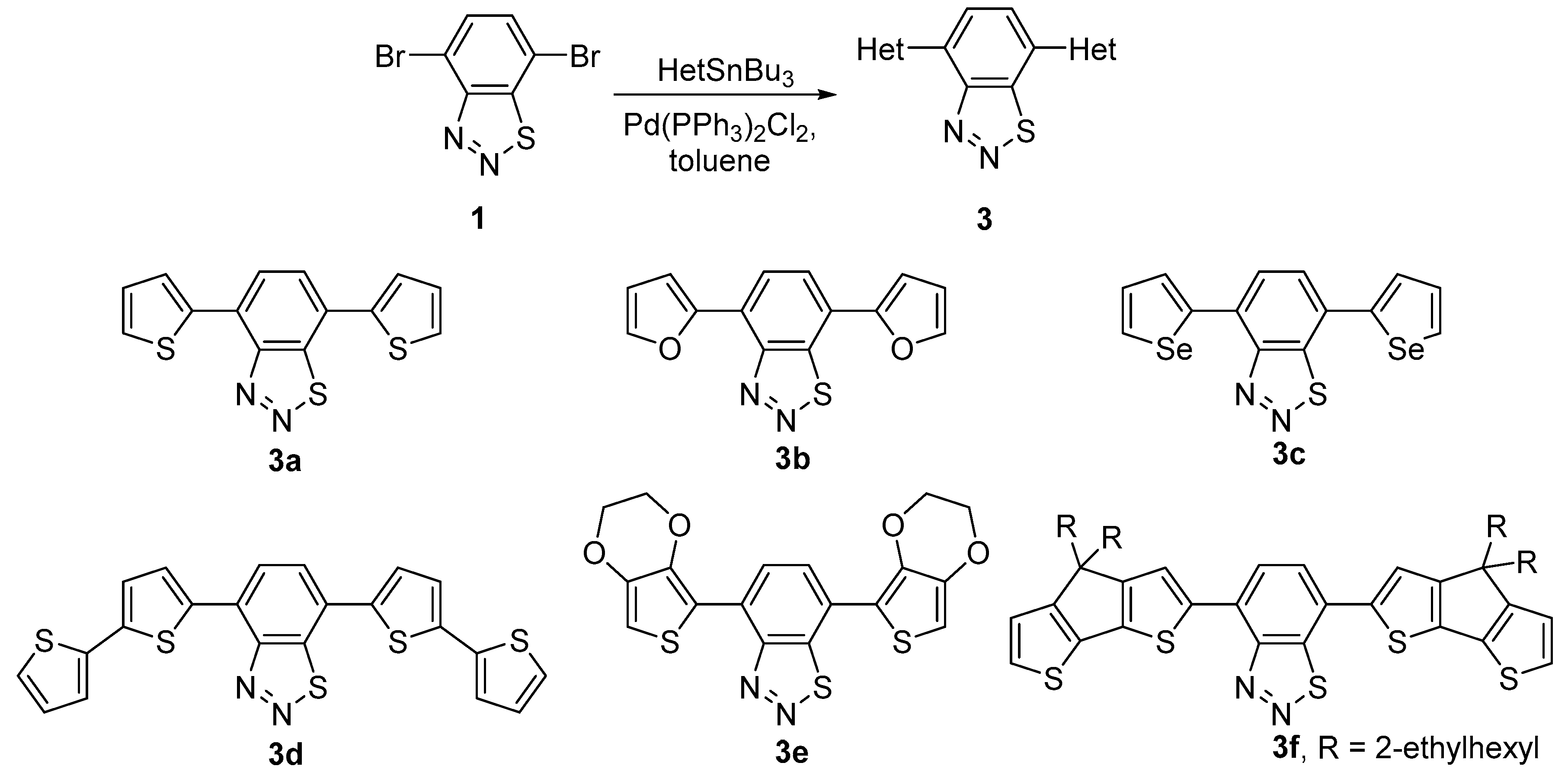
2.2. Cross-Coupling Reactions of 4,7-Dibromobenzo[d][1,2,3]thiadiazole
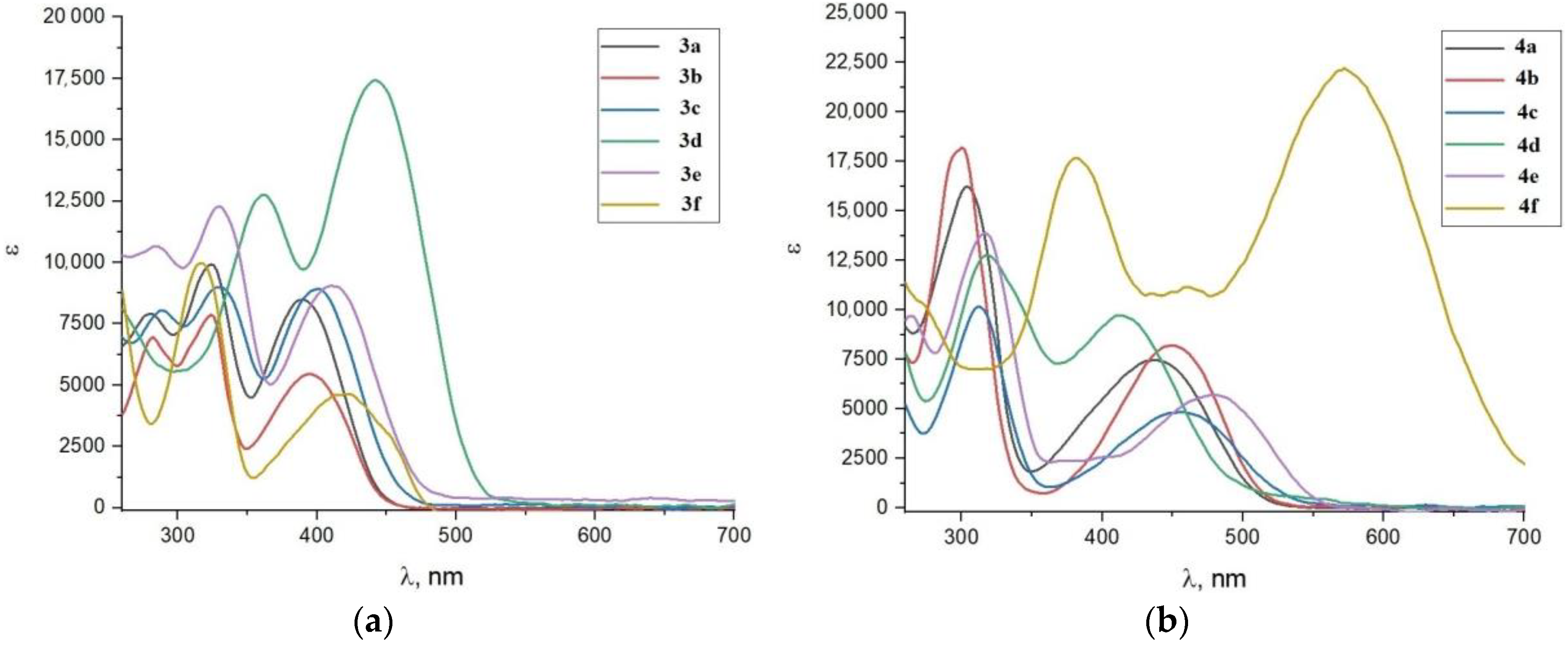
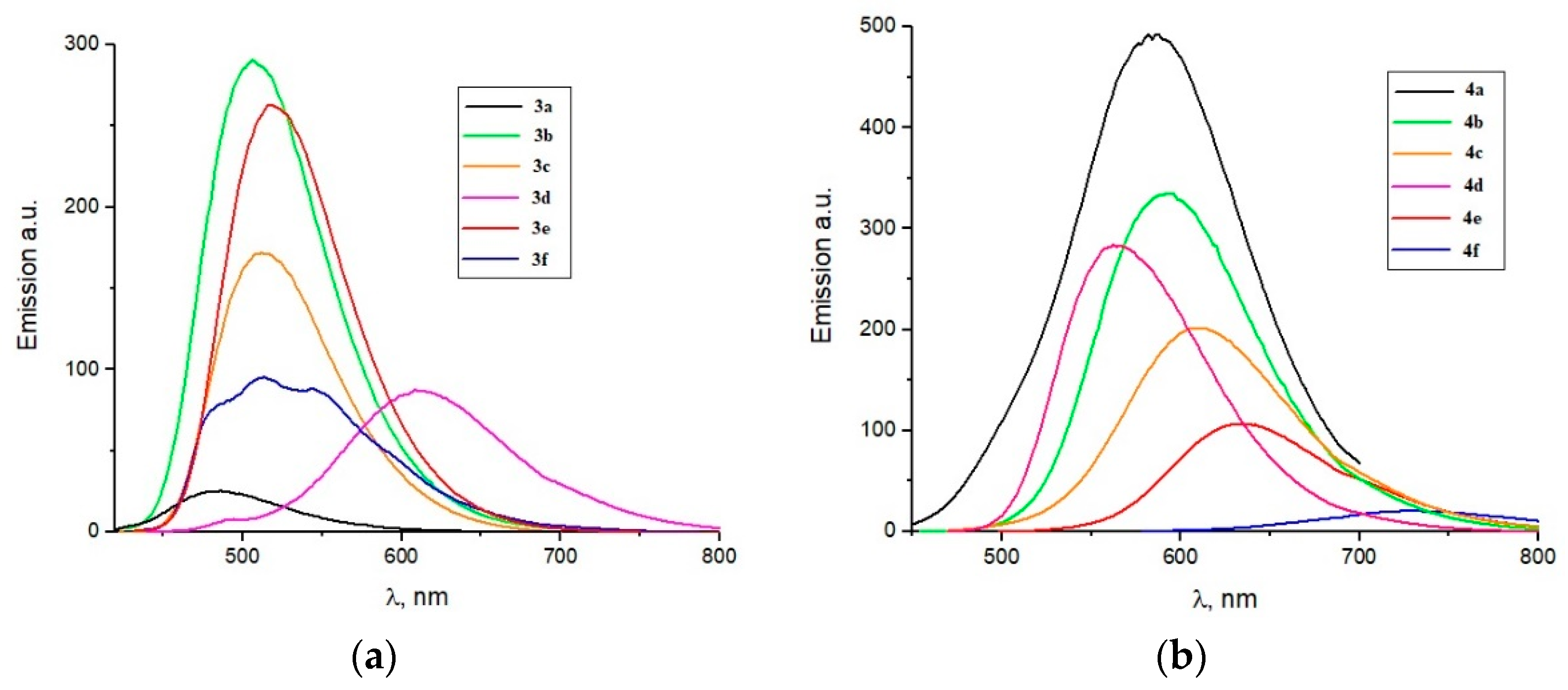
2.3. Optical Properties
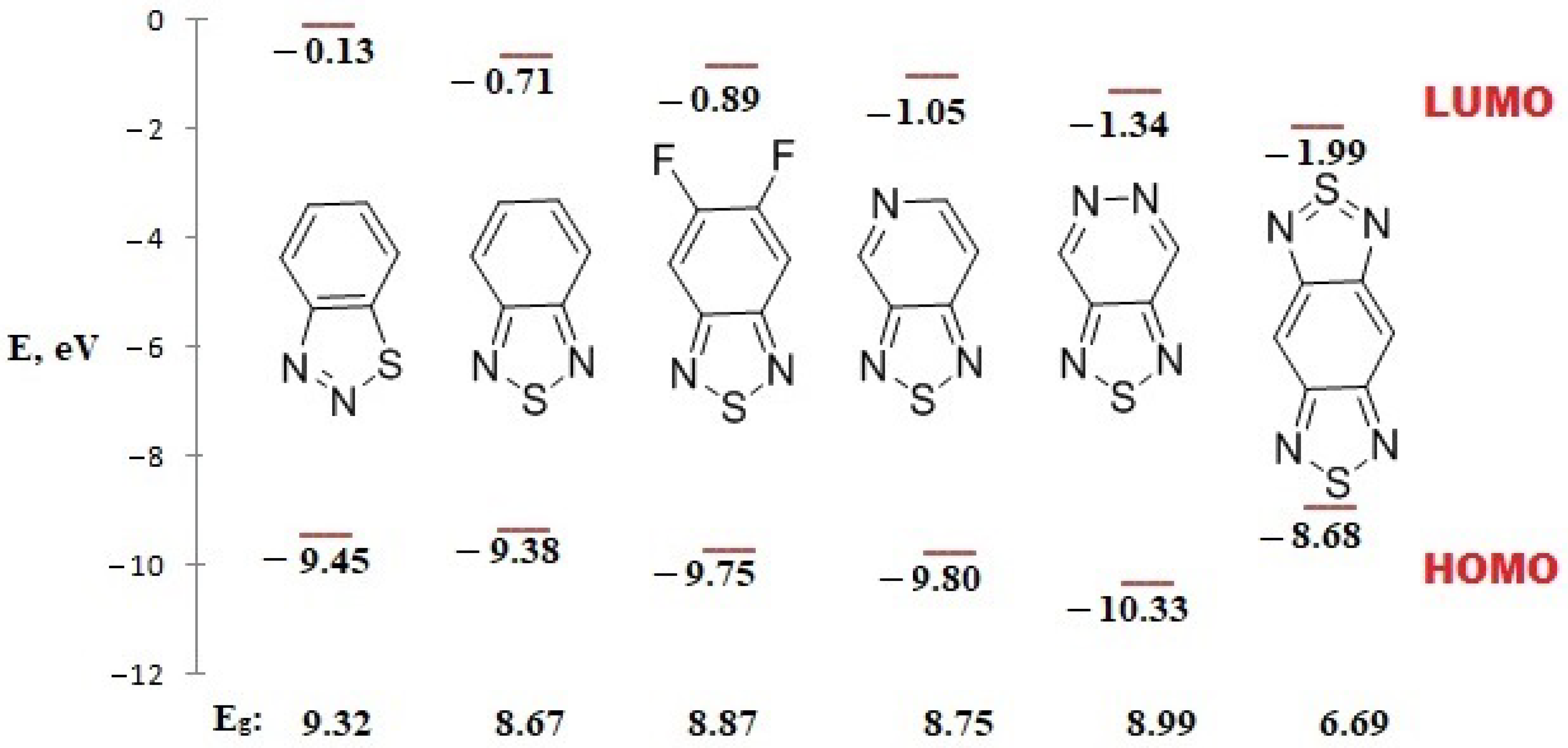
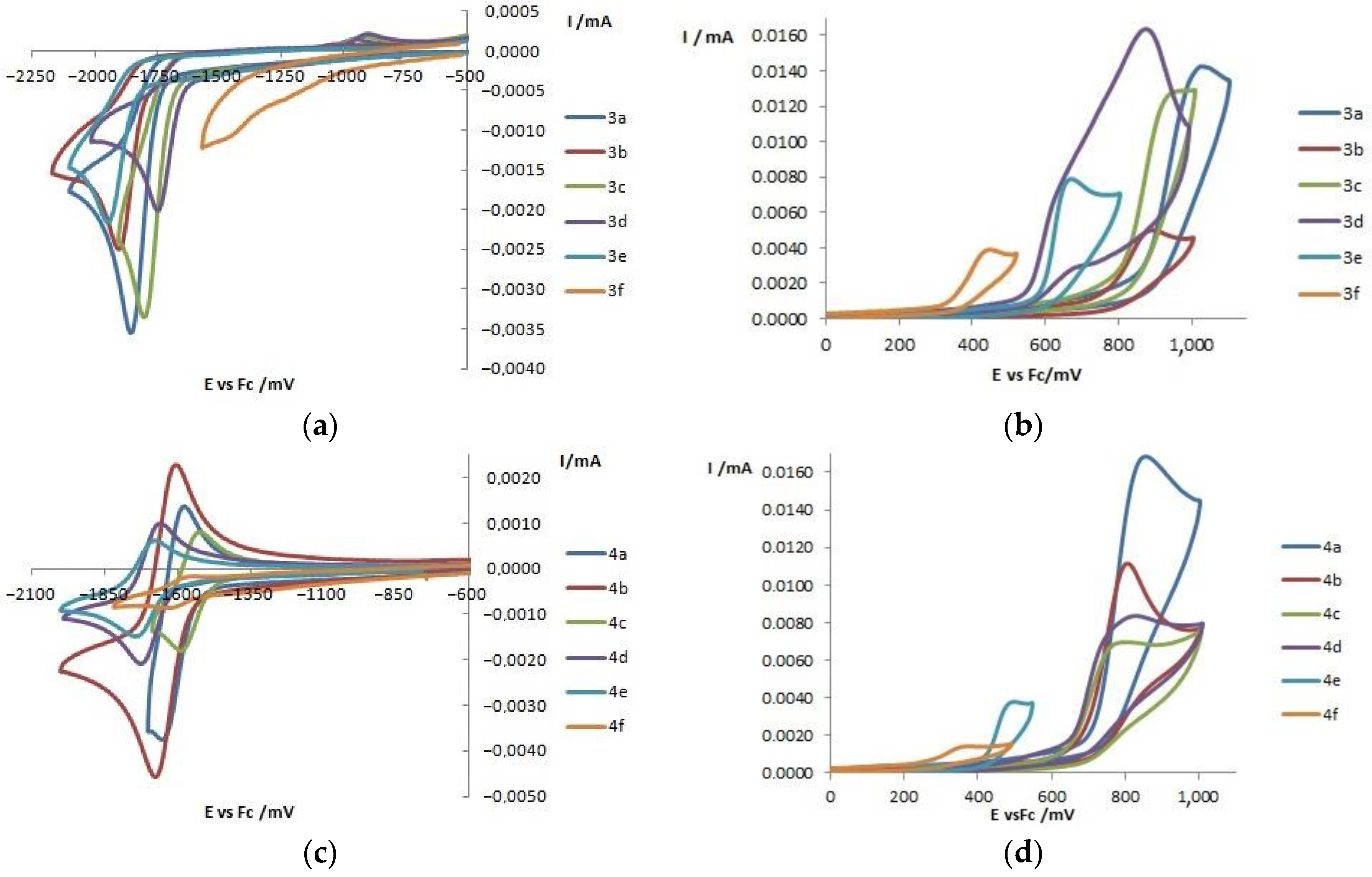
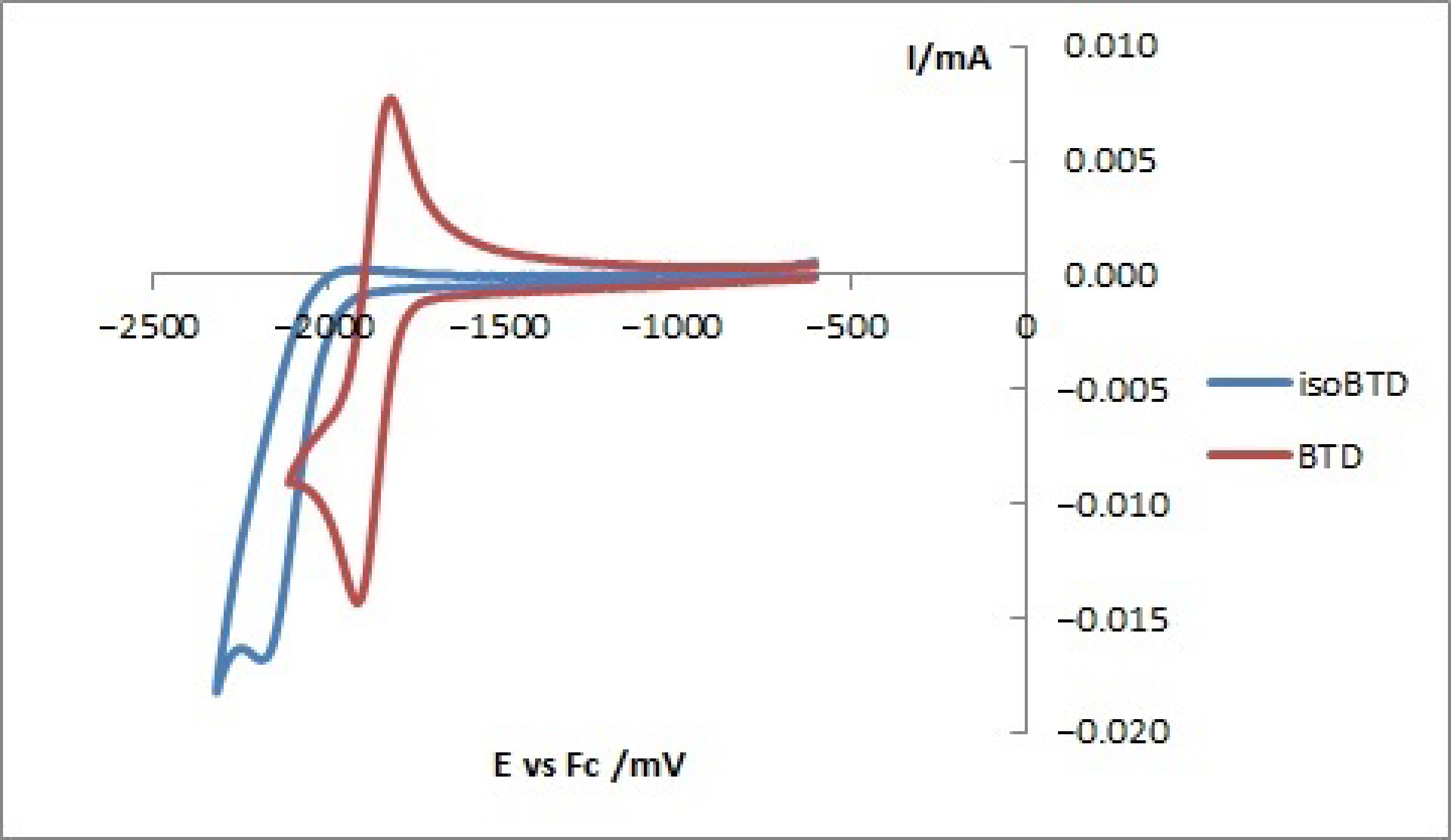
2.4. Electrochemical Properties
3. Experimental Section
3.1. Materials and Reagents
3.2. Analytical Instruments
3.3. Computational Details
3.4. Synthesis of 4,7-Dibromobenzo[d][1,2,3]thiadiazole 1
3.5. General Procedure for the Synthesis of Compounds 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Parker, T.C.; Patel, D.G.; Moudgil, K.; Barlow, S.; Risko, C.; Brédas, J.-L.; Reynolds, J.R.; Marder, S.R. Heteroannulated acceptors based on benzothiadiazole. Mater. Horiz. 2015, 2, 22–36. [Google Scholar] [CrossRef]
- Knyazeva, E.A.; Rakitin, O.A. Influence of structural factors on the photovoltaic properties of dye-sensitized solar cells. Russ. Chem. Rev. 2016, 85, 1146–1183. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Lapis, A.A.M.; da Silva Júnior, E.N.; Dupont, J. 2,1,3-Benzothiadiazole and derivatives: Synthesis, properties, reactions, and applications in light technology of small molecules. J. Eur. J. Org. Chem. 2013, 2, 228–255. [Google Scholar] [CrossRef]
- Knyazeva, E.A.; Rakitin, O.A. 4,7-Dibromo-substituted 2,1,3-benzothia(selena,oxa)diazoles and [1,2,5]thia(selena)diazolo[3,4-c]pyridines as building blocks in solar cells components (microreview). Chem. Heterocycl. Compd. 2017, 53, 855–857. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Knyazeva, E.A.; Rakitin, O.A. Recent Developments in the Synthesis and Applications of 1,2,5-Thia- and Selenadiazoles. A Review. Org. Prep. Proc. Int. 2014, 46, 475–544. [Google Scholar] [CrossRef]
- Rakitin, O.A.; Zibarev, A.V. Recent Progress in Synthesis and Applications of 5-Membered Chalcogen-Nitrogen π-Heterocycles with Three Heteroatoms. Asian J. Org. Chem. 2018, 7, 2397–2416. [Google Scholar] [CrossRef]
- Rakitin, O.A. Fused 1,2,5-thia- and 1,2,5-selenadiazoles: Synthesis and application in materials chemistry. Tetrahedron Lett. 2020, 61, 152230–152241. [Google Scholar] [CrossRef]
- Rakitin, O.A. Recent developments in the synthesis of 1,2,5-thiadiazoles and 2,1,3-benzothiadiazoles. Synthesis 2019, 51, 4338–4347. [Google Scholar] [CrossRef]
- Chen, Z.; Brown, J.; Drees, M.; Seger, M.; Hu, Y.; Xia, Y.; Boudinet, D.; McCray, M.; Delferro, M.; Marks, T.J.; et al. Benzo[d][1,2,3]thiadiazole (isoBT): Synthesis, structural analysis, and implementation in semiconducting polymers. Chem. Mater. 2016, 28, 6390–6400. [Google Scholar] [CrossRef]
- Gudim, N.S.; Knyazeva, E.A.; Obruchnikova, N.V.; Rakitin, O.A.; Popov, V.V. 4-(7-Bromobenzo[d][1,2,3]thiadiazol-4-yl)morpholine. Molbank 2021, 2021, M1202. [Google Scholar] [CrossRef]
- Çakal, D.; Boztaş, Y.; Akdag, A.; Önal, A.M. Investigation of fluorine atom effect on benzothiadiazole acceptor unit in donor acceptor donor systems. J. Electrochem. Soc. 2019, 166, G141–G147. [Google Scholar] [CrossRef]
- Çakal, D.; Ercan, Y.E.; Önal, A.M.; Cihaner, A. Effect of fluorine substituted benzothiadiazole on electro-optical properties of donor-acceptor-donor type monomers and their polymers. Dyes Pigm. 2020, 182, 108622. [Google Scholar] [CrossRef]
- Tam, T.L.; Li, H.; Wei, F.; Tan, K.J.; Kloc, C.; Lam, Y.M.; Mhaisalkar, S.G.; Grimsdale, A.C. One-pot synthesis of 4,8-dibromobenzo-[1,2-c;4,5-c′]bis[1,2,5]thiadiazole. Org. Lett. 2010, 12, 3340–3343. [Google Scholar] [CrossRef]
- Yamashita, Y.; Ono, K.; Tomura, M.; Tanaka, S. Synthesis and properties of benzobis(thiadiazole)s with nonclassical π-electron ring systems. Tetrahedron 1997, 53, 10169–10178. [Google Scholar] [CrossRef]
- Knyazeva, E.A.; Wu, W.; Chmovzh, T.N.; Robertson, N.; Woollins, J.D.; Rakitin, O.A. Dye-sensitized solar cells: Investigation of D-A-π-A organic sensitizers based on [1,2,5]selenadiazolo[3,4-c]pyridine. Sol. Energy 2017, 144, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Chmovzh, T.N.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Amelichev, S.A.; Rakitin, O.A. Synthesis of 4,7-dibromo derivative of ultrahigh electron-deficient [1,2,5]thiadiazolo[3,4-d]pyridazine heterocycle and its cross-coupling reactions. Eur. J. Org. Chem. 2018, 41, 5668–5677. [Google Scholar] [CrossRef]
- Knyazeva, E.A.; Chmovzh, T.N.; Ustimenko, O.O.; Chkhetiani, G.R.; Paleva, I.S.; Konstantinova, L.S.; Mikhal’chenko, L.V.; Rakitin, O.A. Suzuki cross-coupling reactions of 4,7-dibromo[1,2,5]selenadiazolo[3,4-c]pyridine–a path to new solar cell components. Chem. Heterocycl. Comp. 2017, 53, 608–614. [Google Scholar] [CrossRef]
- Leventis, A.; Chmovzh, T.N.; Knyazeva, E.A.; Han, Y.; Heeney, M.J.; Rakitin, O.A.; Bronstein, H. A novel low-bandgap pyridazine thiadiazole-based conjugated polymer with deep molecular orbital levels. Polym. Chem. 2020, 11, 581–585. [Google Scholar] [CrossRef]
- Homberger, K.R.; Berger, D.M.; Chen, X.; Crew, A.P.; Dong, H.; Gupta, R.C.; Kleinberg, A.; Li, A.-H.; Ma, L.; Mulvihill, M.J.; et al. 7-Aminofuropyridine Derivatives. WO2011/100502. U.S. Patent 8,378,104, 19 February 2013. [Google Scholar]
- Mancilha, F.S.; Neto, B.A.D.; Lopes, A.S.; Moreira, P.F., Jr.; Quina, F.H.; Gonçalves, R.S.; Dupont, J. Are molecular 5,8-π-extended quinoxaline derivatives good chromophores for photoluminescence applications? Eur. J. Org. Chem. 2006, 2006, 4924–4933. [Google Scholar] [CrossRef]
- Sivakamasundari, S.; Ganesan, R. Kinetics and mechanism of the bromination of aromatic compounds by N-bromosuccinimide in solution. Int. J. Chem. Kinet. 1980, 12, 837–850. [Google Scholar] [CrossRef]
- Matsidik, R.; Martin, J.; Schmidt, S.; Obermayer, J.; Lombeck, F.; Nübling, F.; Komber, H.; Fazzi, D.; Sommer, M. C-H arylation of unsubstituted furan and thiophene with acceptor bromides: Access to donor-acceptor-donor-type building blocks for organic electronics. J. Org. Chem. 2015, 80, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Alam, F.; Dutta, V.; Jacob, J. Synthesis and photovoltaic studies on novel fluorene based cross-conjugated donor-acceptor type polymers. Org. Electron. 2017, 40, 42–50. [Google Scholar] [CrossRef]
- Yang, S.; Hong, S.H. Palladium-catalyzed direct C-H arylation of arenes promoted by quaternary ammonium salt. Asian J. Org. Chem. 2020, 9, 1846–1851. [Google Scholar] [CrossRef]
- Budy, S.M.; Suresh, S.; Spraul, B.K.; Smith, D.W., Jr. High-temperature chromophores and perfluorocyclobutyl copolymers for electro-optic applications. J. Phys. Chem. C 2008, 112, 8099–8104. [Google Scholar] [CrossRef]
- Xia, P.F.; Feng, X.J.; Lu, J.; Tsang, S.-W.; Movileanu, R.; Tao, Y.; Wong, M.S. Donor-acceptor oligothiophenes as low optical gapchromophores for photovoltaic applications. Adv. Mater. 2008, 20, 4810–4815. [Google Scholar] [CrossRef]
- Qin, P.; Zhu, H.; Edvinsson, T.; Boschloo, G.; Hagfeldt, A.; Sun, L. Design of an organic chromophore for p-type dye-sensitized solar cells. J. Am. Chem. Soc. 2008, 130, 8570–8571. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, M.; Hu, J.; Wang, X.; Jie, J.; Guo, Q.; Chen, C.; Xia, A. Ultrafast investigation of intramolecular charge transfer and solvation dynamics of tetrahydro[5]-helicene-based imide derivatives. Sci. Rep. 2016, 6, 24313. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Ishpal; Dhawan, S.K. Tuning of EMI shielding properties of polypyrrole nanoparticles with surfactant concentration. Synth. Met. 2012, 162, 1471–1477. [Google Scholar] [CrossRef]
- Cardona, C.M.; Li, W.; Kaifer, A.E.; Stockdale, D.; Bazan, G.C. Electrochemical considerations for determining absolute frontier orbital energy levels of conjugated polymers for solar cell applications. Adv. Mater. 2011, 23, 2367–2371. [Google Scholar] [CrossRef]
- Bujak, P.; Kulszewicz-Bajer, I.; Zagorska, M.; Maurel, V.; Wielgusa, I.; Pron, A. Polymers for electronics and spintronics. Chem. Soc. Rev. 2013, 42, 8895–8999. [Google Scholar] [CrossRef]
- Çakal, D.; Cihaner, A.; Önal, A.M. Synthesis and electropolymerization of thieno[3,4-c]pyrrole-4,6-dione based donor-acceptor-donor type monomers. J. Electroanal. Chem. 2020, 862, 114000. [Google Scholar] [CrossRef]
- Jordis, U.; Rudolf, M. Conversion of cyclic trithiocarbonates to thioacetals, including 1,3-dithiane, by reduction with diisobutylaluminium hydride (dibal). Phosphorus Sulfur Related Elem. 1984, 19, 279–283. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, W.; Xu, H.; Li, B.; Cao, X.; Zhang, H. Synthesis and characterization of planar five-ring-fused dithiophene-dione. Chin. J. Chem. 2012, 30, 681–688. [Google Scholar] [CrossRef]
- Melucci, M.; Barbarella, G.; Sotgiu, G. Solvent-free, microwave-assisted synthesis of thiophene oligomers via suzuki coupling. J. Org. Chem. 2002, 67, 8877–8884. [Google Scholar] [CrossRef]
- Istanbulluoglu, C.; Göker, S.; Hizalan, G.; Hacioglu, S.O.; Udum, Y.A.; Yildiz, E.D.; Cirpan, A.; Toppare, L. Synthesis of a benzotriazole bearing alternating copolymer for organic photovoltaic applications. New J. Chem. 2015, 39, 6623–6630. [Google Scholar] [CrossRef]
- Akpınar, H.; Balan, A.; Baran, D.; Ünver, E.K.; Toppare, L. Donor–acceptor–donor type conjugated polymers for electrochromic applications: Benzimidazole as the acceptor unit. Polymer 2010, 51, 6123–6131. [Google Scholar] [CrossRef]
- Gao, S.; Balan, B.; Yoosaf, K.; Monti, F.; Bandini, E.; Barbieri, A.; Armaroli, N. Highly efficient luminescent solar concentrators based on benzoheterodiazole dyes with large Stokes’ shifts. Chem. Eur. J. 2020, 26, 11013–11023. [Google Scholar] [CrossRef]
- Kawabata, K.; Takeguch, M.; Goto, H. Optical activity of heteroaromatic conjugated polymer films prepared by asymmetric electrochemical polymerization in cholesteric liquid crystals: Structural function for chiral induction. Macromolecules 2013, 46, 2078–2091. [Google Scholar] [CrossRef]
- Kim, B.; Yeom, H.R.; Yun, M.H.; Kim, J.Y.; Yang, C. A selenophene analogue of PCDTBT: Selective fine-tuning of lumo to lower of the bandgap for efficient polymer solar cells. Macromolecules 2012, 45, 8658–8664. [Google Scholar] [CrossRef]
- Steiberger, S.; Mishra, A.; Reinold, E.; Müller, C.M.; Uhrich, C.; Pfeiffer, M.; Bäuerle, P. A-D-A-D-A-type oligothiophenes for vacuum-deposited organic solar cells. Org. Lett. 2011, 13, 90–93. [Google Scholar] [CrossRef]
- Wong, W.Y.; Wang, X.Z.; Zhang, H.-L.; Cheung, K.-Y.; Fung, M.-K.; Djurisic, A.B.; Chan, W.-K. Synthesis, characterization and photovoltaic properties of a low-bandgap platinum(II) polyyne functionalized with a 3,4-ethylenedioxythiophene-benzothiadiazole hybrid spacer. J. Organomet. Chem. 2008, 693, 3603–3612. [Google Scholar] [CrossRef]
- Lu, B.-S.; Zhang, Y.; Hu, T.-Y.; Ma, Y.-F.; Zhu, Y.-N.; Liu, D.-W.; Zhang, Z.-Q.; Wang, E.; Ma, W.; Zhang, H.-L. Non-fullerene acceptors based on multiple non-covalent interactions for low cost and air stable organic solar cells. Org. Electron. 2021, 93, 106132. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 2:NBS | Acid | Conc. of 2 in Acid | Conditions | Yield of 1 (%) |
|---|---|---|---|---|---|
| 1 | 1:2.2 | H2SO4 | 0.07 M | 50 °C, 16 h | 81 |
| 2 | 1:4 | H2SO4 | 0.07 M | 50 °C, 6 h | 52 |
| 3 | 1:4 | H2SO4 | 0.14 M | 50 °C, 6 h | 83 |
| 4 | 1:4 | H2SO4 | 0.28 M | 50 °C, 12 h | 33 |
| 5 | 1:8 | H2SO4 | 0.14 M | 50 °C, 6 h | 0 |
| 6 | 1:2.2 | - | - | CCl4, Δ, 12 h | 0 |
| 7 | 1:2.2 | TfOH | 1.14 M | CHCl3, Δ, 12 h | 0 |
| 8 | 1:2.2 | CF3CO2H | 0.28 M | 60 °C, 6 h | 0 |
| 9 | 1:2.2 | TfOH | 0.28 M | 50 °C, 6 h | 84 |
| 10 a | 1:4 | H2SO4 | 0.14 M | 50 °C, 6 h | 80 |
| Entry | X (Equiv.) | Catalyst | Solvent | Base | Temp. (°C) | Time (h) | Yield of 1 (%) |
|---|---|---|---|---|---|---|---|
| Suzuki-Miyaura coupling | |||||||
| 1 | B(OH)2 (2.2) | Pd(PPh3)2 | THF/H2O | K2CO3 | 80 | 6 | 48 |
| 2 | B(OH)2 (2.2) | Pd(dppf)Cl2 | H2O | Et3N | 90 | 7 | 5 |
| 3 | B(OH)2 (2.2) | Pd(PPh3)4 | toluene | K2CO3 | 90 | 7 | 9 |
| 4 | B(OH)2 (2.2) | Pd(PPh3)4 | ethanol/ benzene | Na2CO3 | 80 | 7 | 54 |
| 5 | B(OH)2 (2.2) | Pd(PPh3)4 | dioxane/ H2O | K2CO3 | 90 | 7 | 50 |
| 6 | BPin (2.2) | Pd(PPh3)4 | THF/H2O | K2CO3 | 80 | 6 | 43 |
| 7 | BPin (2.2) | Pd(PPh3)4 | H2O/toluene | K2CO3 | 90 | 8 | 36 |
| Stille coupling | |||||||
| 8 | SnBu3 (2.2) | Pd(PPh3)2Cl2 | toluene | - | 110 | 10 | 60 |
| 9 | SnBu3 (2.2) | Pd(PPh3)2Cl2 | THF | - | 66 | 10 | 52 |
| 10 | SnBu3 (2.2) | Pd(PPh3)2Cl2 | dioxane | - | 100 | 10 | 45 |
| 11 | SnBu3 (2.2) | Pd(PPh3)4 | DMF, toluene | - | 100 | 8 | 54 |
| 12 | SnBu3 (2.2) | Pd(PPh3)4 | toluene | - | 90 | 8 | 59 |
| 13 | SnBu3 (2.2) | Pd2dba3, (t-Bu)3P | THF | - | 85 | 8 | 60 |
| Direct C-H arylation | |||||||
| 14 | H (10) | Pd(OAc)2 | DMA | AcOK | 155 | 3 | 24 |
| 15 | H (10) | Pd(OAc)2, CsF, TBAB | DMA | PivOCs | 80 | 1 | 33 |
| 16 | H (10) | Pd(OAc)2 | DMF | PivOK | 80 | 4 | 12 |
| Dye | λmax1, nm | εmax1, M−1 cm−1 | λmax2, nm | εmax2, M−1 cm−1 | λemis, nm | Stoke’s Shift, nm |
|---|---|---|---|---|---|---|
| 3a | 329 | 9923 | 389 | 8499 | 486 | 97 |
| 3b | 334 | 7867 | 400 | 5455 | 506 | 106 |
| 3c | 334 | 8995 | 413 | 8916 | 512 | 99 |
| 3d | 367 | 12,756 | 455 | 17,425 | 612 | 153 |
| 3e | 334 | 12,282 | 422 | 9046 | 516 | 94 |
| 3f | 320 | 9973 | 434 | 4643 | 544 | 110 |
| 4a | 311 | 16,228 | 434 | 7461 | 586 | 152 |
| 4b | 302 | 18,190 | 442 | 8209 | 594 | 152 |
| 4c | 320 | 10,168 | 461 | 4824 | 612 | 151 |
| 4d | 323 | 12,774 | 430 | 9731 | 562 | 132 |
| 4e | 325 | 13,877 | 488 | 5710 | 632 | 144 |
| 4f | 383 | 17,677 | 569 | 22,211 | 724 | 155 |
| Dye | Eredonset (vs. Fc/Fc+) a, V | Eoxonset (vs. Fc/Fc+) a, V | ELUMO b, eV | EHOMO b, eV | Eg c, eV |
|---|---|---|---|---|---|
| isoBTD | −1.99 | - | −3.11 | - | - |
| 3a | −1.72 | 0.87 | −3.38 | −5.97 | 2.59 |
| 3b | −1.78 | 0.73 | −3.32 | −5.83 | 2.51 |
| 3c | −1.68 | 0.80 | −3.42 | −5.90 | 2.48 |
| 3d | −1.63 | 0.55 | −3.47 | −5.65 | 2.18 |
| 3e | −1.82 | 0.59 | −3.28 | −5.69 | 2.41 |
| 3f | −1.00 | 0.36 | −4.10 | −5.46 | 1.36 |
| BTD | −1.79 | - | −3.31 | - | - |
| 4a | −1.53 | 0.72 | −3.57 | −5.82 | 2.25 |
| 4b | −1.56 | 0.52 | −3.54 | −5.62 | 2.08 |
| 4c | −1.46 | 0.63 | −3.64 | −5.73 | 2.09 |
| 4d | −1.61 | 0.64 | −3.49 | −5.74 | 2.25 |
| 4e | −1.61 | 0.41 | −3.49 | −5.51 | 2.02 |
| 4f | −1.51 | 0.25 | −3.59 | −5.35 | 1.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gudim, N.S.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Popov, V.V.; Obruchnikova, N.V.; Rakitin, O.A. Benzothiadiazole vs. iso-Benzothiadiazole: Synthesis, Electrochemical and Optical Properties of D–A–D Conjugated Molecules Based on Them. Molecules 2021, 26, 4931. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164931
Gudim NS, Knyazeva EA, Mikhalchenko LV, Golovanov IS, Popov VV, Obruchnikova NV, Rakitin OA. Benzothiadiazole vs. iso-Benzothiadiazole: Synthesis, Electrochemical and Optical Properties of D–A–D Conjugated Molecules Based on Them. Molecules. 2021; 26(16):4931. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164931
Chicago/Turabian StyleGudim, Nikita S., Ekaterina A. Knyazeva, Ludmila V. Mikhalchenko, Ivan S. Golovanov, Vadim V. Popov, Natalia V. Obruchnikova, and Oleg A. Rakitin. 2021. "Benzothiadiazole vs. iso-Benzothiadiazole: Synthesis, Electrochemical and Optical Properties of D–A–D Conjugated Molecules Based on Them" Molecules 26, no. 16: 4931. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164931