
Current Status of Endoplasmic Reticulum Stress in Type II Diabetes

 , ,
, ,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. History of the Study of the Endoplasmic Reticulum
3. Discovery and Investigation of the ER Stress Mechanism
4. ER Stress Response to Insulin Dysfunction
5. ER Stress Response to Oxidative Stress
6. Therapeutic Potential of Targeting ER Stress
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gioacchini, F.M.; Albera, R.; Re, M.; Scarpa, A.; Cassandro, C.; Cassandro, E. Hyperglycemia and diabetes mellitus are related to vestibular organs dysfunction: Truth or suggestion. A literature review. Acta Diabetol. 2018, 55, 1201–1207. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; Rocha, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: Therapeutic and molecular approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [Green Version]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flamment, M.; Hajduch, E.; Ferre, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-labra, R.; Sáez, P.J.; Subiabre, M.; Silva, L.; Toledo, F. Pre-pregnancy maternal obesity associates with endoplasmic reticulum stress in human umbilical vein endothelium. BBA Mol. Basis Dis. 2018, 1864, 3195–3210. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic reticulum stress in metabolic disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Van De Maele, K.; Devlieger, R.; Gies, I. In utero programming and early detection of cardiovascular disease in the offspring of mothers with obesity. Atherosclerosis 2018, 275, 182–195. [Google Scholar] [CrossRef]
- Guerrero-Hernández, A.; Leon-Aparicio, D.; Chavez-Reyes, J.; Olivares-Reyes, J.A.; DeJesus, S. Endoplasmic reticulum stress in insulin resistance and diabetes. Cell Calcium 2014, 56, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Pardo, F.; Villalobos-labra, R.; Chiarello, D.I.; Salsoso, R.; Toledo, F.; Gutierrez, J.; Leiva, A.; Sobrevia, L. Molecular implications of adenosine in obesity. Mol. Asp. Med. 2017, 55, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Claude, A.; Fullam, E.F. A study of tissue culture cells by electron microscopy. J. Exp. Med. 1945, 81, 233–246. [Google Scholar] [CrossRef]
- Palade, G.E.; Porter, K.R. Studies on the endoplasmic reticulum: I. Its identification in cells in Situ. J. Exp. Med. 1954, 100, 641–656. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palade, G.E. The endoplasmic reteculum. J. Cell Biol. 1956, 2, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Voeltz, G.K. The ER in 3D: A multifunctional dynamic membrane network. Trends Cell Biol. 2011, 21, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
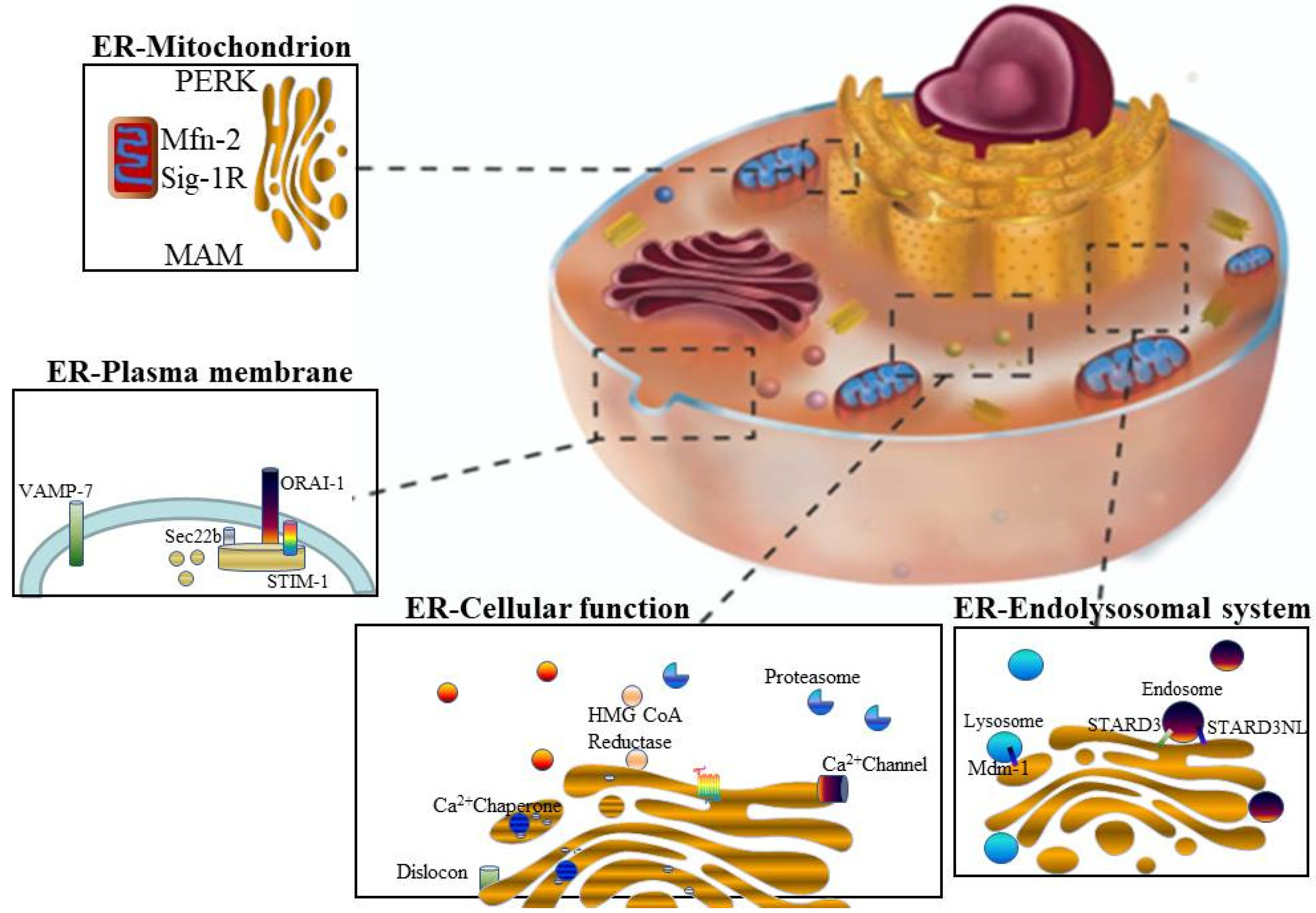
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T. MAM: More than just a house keep. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martinez de Maranon, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications a unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Rovira-Liopis, S.; Banuls, C.; Apostolova, N.; Morillas, C.; Hernandez-Mijares, A.; Rocha, M.; Victor, M.V. Is glycemic control modulating endoplasmic reticulum stress in leukocytes of type 2 diabetic patients. Antioxid. Redox Signal. 2014, 21, 1759–1765. [Google Scholar] [CrossRef]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.D.; Lee, M.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W.; Gonzalez, C.D.; Lee, M.; et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signaling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef]
- Scherz-shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Toulmay, A.; Prinz, W.A. Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Curr. Opin. Cell Biol. 2011, 23, 458–463. [Google Scholar] [CrossRef] [Green Version]
- De Daste, F.; Galli, T.; Tareste, D. Structure and function of longin SNAREs. J. Cell Sci. 2015, 128, 4263–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.A.; Chitwood, P.J.; Phillips, M.J.; Voeltz, G.K. ER contact sites define the position and timing of endosome fission. Cell 2014, 159, 1027–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiu, R.P.C.; Pouyssegur, J.; Pastan, I.R.A. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteins in Rous sarcoma virus-transformed chick embryo fibroblasts. Proc. Natl. Acad. Sci. USA 1977, 74, 3840–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 7–9. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R.B. An Hsp704ike protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986, 46, 291–300. [Google Scholar] [CrossRef]
- Fornace, A.J.; Nebert, D.W.; Hollander, M.C.; Luethy, J.D.; Papathanasiou, M.; Fargnoli, J.; Holbrook, N.J. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol. 1989, 9, 4196–4203. [Google Scholar] [CrossRef] [Green Version]
- Dorner, A.J.; Wasley, L.C.; Kaufman, R.J. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J. Biol. Chem. 1989, 264, 20602–20607. [Google Scholar] [CrossRef]
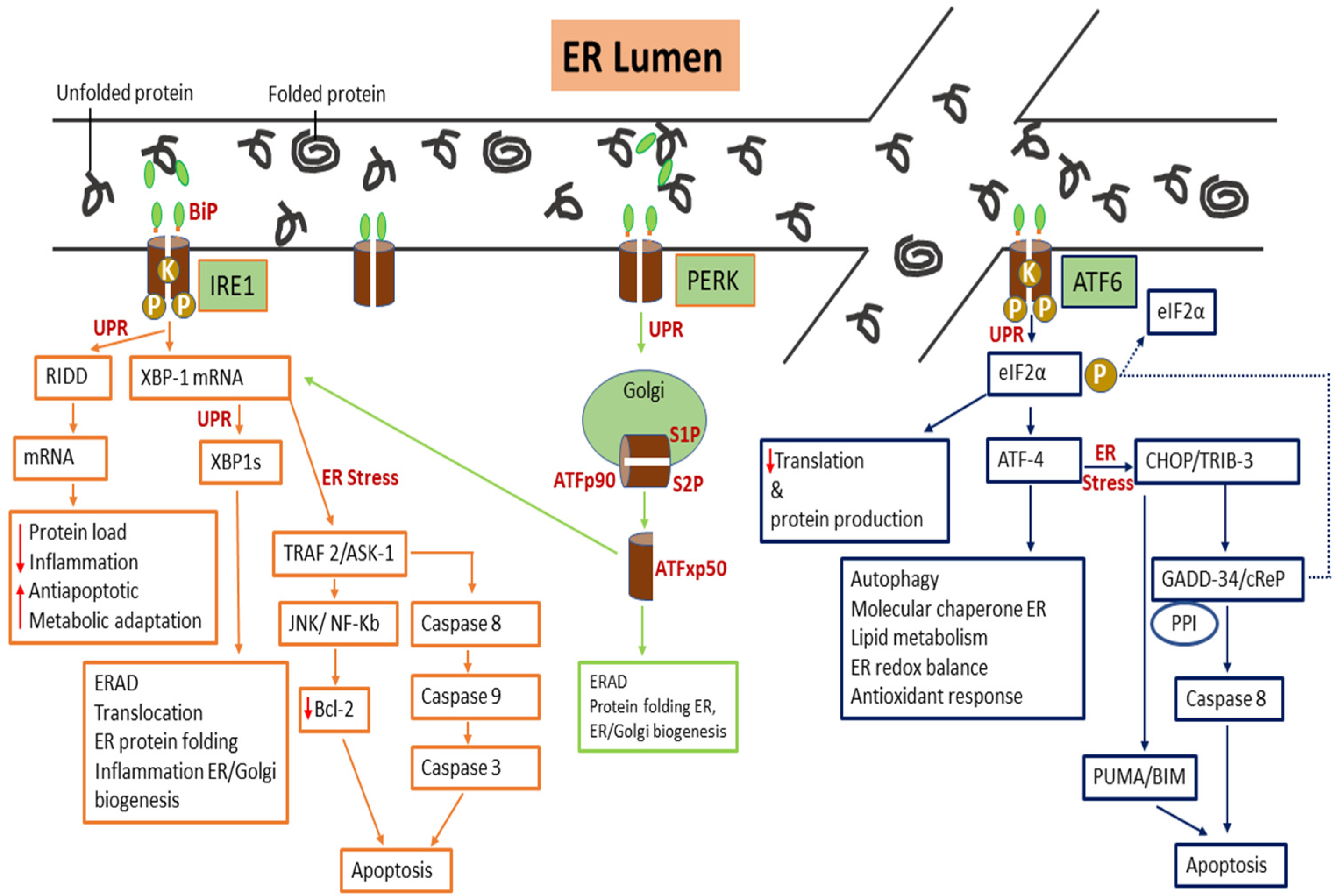
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Kaufman, Z.; Kaufman, R. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-Subunit Kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickhout, J.G.; Lhotak, S.; Hilditch, B.A.; Basseri, S.; Colgan, S.M.; Lynn, E.G.; Carlisle, R.E.; Zhou, J.; Sood, S.K.; Ingram, A.J.; et al. Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FEBS J. 2010, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, L.; Kelly, M.; Coste, A.; Guerrera, I.C.; Fritsch, J. Oxidative stress induces unfolding protein response and inflammation in nasal polyposis. Allergy 2012, 67, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating Stress Signals Through the Stress Sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Sha, B. The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins research papers. Acta Cryst. 2016, 1290–1297. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, K.; Reinhardt, S. ER-stress in Alzheimer’s disease: Turning the scale. Am. J. Neurodegener. Dis. 2013, 2, 247–265. [Google Scholar] [PubMed]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Scheuner, D.; Chen, J.; Kaufman, R.J.; Ron, D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2α) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. USA 2009, 106, 1832–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6 α induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 1, 1626–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.A.O.Y.; Jing, S.H.I.; Lin, Y.U.P.; Qiang, Z.G. The role of endoplasmic reticulum stress-related apoptosis in vascular endothelium pathogenesis. Biomed. Environ. Sci. 2018, 31, 555–559. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.; Yang, L.; Liu, P.; Zhang, Y.; Wang, X. MiR-149 attenuates endoplasmic reticulum stress-induced inflammation and apoptosis in nonalcoholic fatty liver disease by negatively targeting ATF6 pathway. Immunol. Lett. 2020, 222, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.; Gao, T.; Awuapura, N.J.; Ayathamattam, J. Sorcin stimulates activation transcription factor 6 (ATF6) transcriptional activity. bioRxiv. 2020. [Google Scholar] [CrossRef]
- Hong, S.; Lee, J.; Cho, J.H.; Kwon, H.; Park, S.E.; Rhee, E.; Park, C.; Oh, K.; Park, S.; Lee, W. Pioglitazone attenuates palmitate-induced inflammation and endoplasmic reticulum stress in pancreatic β-cells. Endocrinol Metab. 2018, 105–113. [Google Scholar] [CrossRef]
- Hillary, R.F.; Fitzgerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, C. Les filaments basaux des cellules glandulaires. Note Préliminaire 1897, 5, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.R. Observations on a submicroscopic basophilic component of cytoplasm. J. Exp. Med. 1953, 97, 727–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horke, S.; Witte, I.; Wilgenbus, P.; Altenhöfer, S.; Krüger, M.; Li, H.; Förstermann, U. Protective effect of paraoxonase-2 against endoplasmic reticulum stress-induced apoptosis is lost upon disturbance of calcium homoeostasis. Biochem. J. 2008, 416, 395–405. [Google Scholar] [CrossRef] [Green Version]
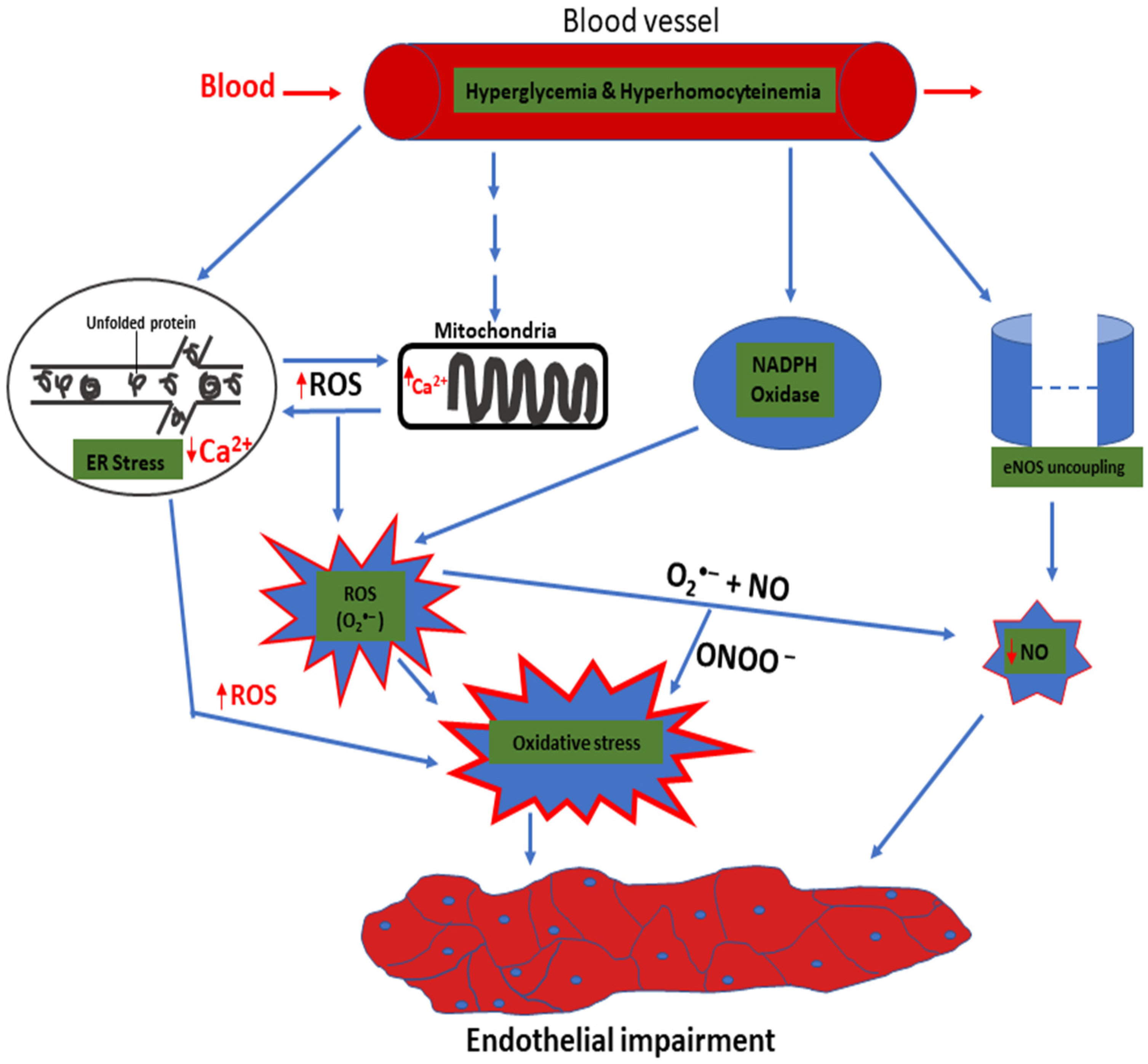
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress. Exp. Diabetes Res. 2012, 2012, 481840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, S.; Mohammed, M.; Azemi, A.K.; Yunusa, I.; Shehu, A.; Mustapha, L.; Wada, Y.; Ahmad, M.H.; Amir, W.; Wan, N.; et al. Potential roles of endoplasmic reticulum stress and cellular proteins implicated in diabesity. Oxid. Med. Cell. Longev. 2021, 2021, 8830880. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, S.; Mohammed, M.; Yunusa, I.; Hanum, A.R.G.; Safiah, S. Potential risks of endoplasmic reticulum stress on vasculopathy in diabetes. Obes. Med. 2020, 19, 100274. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, A.; Lee, N.; Iwakoshi, E.; Ozdelen, G.; Tuncman, C.; Gorgun, L.; Glimcher, G.H. Endoplasmic reticulum stress links obesity, insulin action and type 2 diabetes. Science 2004, 306, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, N.; Marcovecchio, M.L.; Di Silvestre, S.; de Giorgis, T.; Cordone, V.G.; Lanuti, P.; Chiarelli, F.; Bologna, G.; Mohn, A.; Pandolfi, A. Molecular and cellular endocrinology plasma from pre-pubertal obese children impairs insulin stimulated nitric oxide (NO) bioavailability in endothelial cells: Role of ER stress. Mol. Cell. Endocrinol. 2017, 443, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Salvado, L.; Palomer, X.; Barroso, E.; Vazquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Pavithra, K.; Vadivukkarasi, S. Evaluation of free radical scavenging activity of various extracts of leaves from Kedrostis foetidissima (Jacq.) Cogn. Food Sci. Hum. Wellness 2015, 4, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Fernandes, C.; Liu, Y.; Wu, Y.; Wu, H.; Brophy, M.L.; Deng, L.; Song, K.; Wen, A.; Wong, S.; et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diabetes Vasc. Dis. Res. 2016, 14, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlies, D.; Makkinje, M.; Jansens, A.; Braakman, I.; Verkleij, A.J.; Wirtz, K.W.A.; Post, J.A. Oxidation of ER resident proteins upon oxidative stress: Effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid. Redox Signal. 2003, 5, 381–387. [Google Scholar] [CrossRef]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras-sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1 α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C. Redox biology homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.; Bermudez, F.; Acosta, G.; Acosta, A.; Anez, J.; Andara, C.; Leal, E.; Cano, C.; Manuel, V.; Hernandez, R.; et al. Molecular mechanisms of endothelial dysfunction: From nitric oxide synthesis to ADMA inhibition. Am. J. Ther. 2008, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Sipkens, J.A.; Hahn, N.; Van Den Brand, C.S.; Meischl, C.; Smulders, Y.M.; Krijnen, P.A.J.; Stehouwer, C.D.A.; Rauwerda, J.A. Homocysteine-induced apoptosis in endothelial cells coincides with nuclear NOX2 and peri-nuclear NOX4 activity. Cell Biochem. Biophys. 2013, 67, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Taniyama, Y.; Griendling, K.K. Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Marciniak, S.J. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease.2. Protein misfolding and ER stress. Am. J. Physiol. Cell Physiol. 2014, 307, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Kayal, R.A. Diabetic complications and dysregulated innate immunity. Front. Biosci. 2011, 13, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; He, P. fMLP-stimulated release of reactive oxygen species from adherent leukocytes increases microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H365–H372. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Type 2 diabetes—An autoinflammatory disease driven by metabolic stress. BBA Mol. Basis Dis. 2018, 1864, 3805–3823. [Google Scholar] [CrossRef] [PubMed]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulman, I.H.; Zhou, M.S. Vascular insulin resistance: A potential link between cardiovascular and metabolic diseases. Curr. Hypertens. Rep. 2009, 11, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Glembotski, C.C. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J. Mol. Cell. Cardiol. 2014, 71, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 2010, 59, 1937–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Zhao, N.; Lewis, M.T.; Chen, X.; Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Bujisic, B.; Martinon, F. IRE1 gives weight to obesity-associated inflammation. Nat. Immunol. 2017, 18, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, M.; Zhu, M.; Gu, J.; Song, J.; Cui, L.; Liu, D.; Ning, Q.; Jia, X.; Feng, L. Paeoniflorin prevents endoplasmic reticulum stress-associated inflammation in lipopolysaccharide-stimulated human umbilical vein endothelial cells: Via the IRE1α/NF-κB signaling pathway. Food Funct. 2018, 9, 2386–2397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, J. Free radical biology & medicine thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation. Free Radic. Biol. Med. 2011, 51, 2172–2184. [Google Scholar] [CrossRef] [Green Version]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Tong, Q.; Wu, L.; Jiang, T.; Ou, Z.; Zhang, Y.; Zhu, D. Inhibition of endoplasmic reticulum stress-activated IRE1α-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of parkinson’s disease. Eur. J. Pharmacol. 2016, 776, 106–115. [Google Scholar] [CrossRef]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Preventing β-cell loss and diabetes with calcium channel blockers. Diabetes 2012, 61, 848–856. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Year | Reference | Evolution and Advancement | Findings |
|---|---|---|---|
| 1897 | [62] | Ergastoplasm | First observed with a light microscope and named Ergastoplasm |
| 1945 | [16] | Lace-like reticulum | Lace-like reticulum extended into the thin margin and even into the fine processes of the cell from the denser center. |
| 1953 | [63] | Reticulum, which means “network” | Reticulum (network) was applied to describe this fabric of membranes |
| 1954 | [17] | Endoplasmic reticulum (RER and SER) | ER as a network of cavities enlarges into relatively vast and flattened vesicles described as cisternae. |
| 1977 | [35] | GRPs | Glucose-regulated proteins (GRP-78 and GRP-95) because the amount of the proteins is influenced by the presence or absence of glucose. These proteins play an important role in regulating glucose utilization in cultured cells. |
| 1983 | [36] | BiP | At least some H-chains are bound to a protein other than L-chain. Here, we show that the protein (which we term immunoglobulin heavy-chain binding protein, BiP) binds noncovalently to free IgH, but not to IgH associated with IgL. |
| 1986 | [37] | GRP and BiP were found to be the same | Identical with two previously described proteins: GRP78, whose synthesis is induced by glucose starvation, and BiP, which is bound to immunoglobulin heavy chains in pre-B cells. |
| 1989 | [39] | GRP and BiP were associated with misfolded and unfolded proteins | Increased factor VIII synthesis was correlated with an 80-fold increase in GRP78 mRNA and a 10-fold increase in GRP94 mRNA. |
| 1993 | [40] | IRE1 | IRE1 is essential for cell viability under stress conditions that cause unfolded proteins to accumulate in ER. IRE1 encodes a transmembrane serine/threonine kinase that we propose transmits the unfolded protein signal. IRE1 is also required for inositol prototrophy. |
| 1998, 1999 | [44,45] | PERK and ATF6 | PERK, a gene encoding type I transmembrane ER-resident protein, has a lumenal domain similar to the ER-stress-sensing lumenal domain of the ER-resident kinase IRE1 and a cytoplasmic portion that contains a protein-kinase domain most similar to that of the known eIF2α kinases, PKR and HRI. In response to various environmental stresses, eukaryotic cells downregulate the protein synthesis through phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α). In mammals, the phosphorylation is carried out by eIF2α kinases PKR and HRI. |
| 2001 | [42] | XBP1 | The transcription factor XBP1, a target of ATF6, as a mammalian substrate of such an unconventional mRNA splicing system showed that only the spliced form of XBP1 can activate UPR efficiently. |
| 2006 | [21] | Organized ER into a membrane structure | ER has distinct morphological domains composed of sheets and tubules, which differ in their characteristic membrane curvature. |
| 2008 | [64] | ER stress | Paraoxonase-2 (PON2) is a ubiquitously expressed antioxidative protein primarily found in ER. PON2 overexpression provides significant resistance to ER-stress-induced caspase-3 activations. |
| 2012 | [6,65] | ER stress linked to diabetes | Both chronic hyperglycemia and hyperlipidemia, known as critical causative factors of type II diabetes, disrupt ER homeostasis to induce unresolvable UPR activation and β-cell death. ER stress can be an essential contributor to diabetes-related vascular complications. |
| 2019 | [24,66] | ER stress linked to diabetes | The implication of mitochondria in insulin release and the exposure of pancreatic β-cells to hyperglycemia make them especially susceptible to oxidative stress and mitochondrial dysfunction. ER stress response is now recognized as a converging molecular link that connects insulin resistance, lipid metabolism disturbances, cell death, and oxidative stress to endothelial dysfunction. |
| 2020, 2021 | [67,68] | ER stress linked to diabetes | Chronic hyperglycemia, hyperinsulinemia, increased proinflammatory cytokines, and free fatty acids found in diabesity can lead to ER stress. The inflammatory response to the damage induced by hyperglycemia and ROS becomes chronic as diabetes progresses and constitutes the leading cause of vascular complications. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustapha, S.; Mohammed, M.; Azemi, A.K.; Jatau, A.I.; Shehu, A.; Mustapha, L.; Aliyu, I.M.; Danraka, R.N.; Amin, A.; Bala, A.A.; et al. Current Status of Endoplasmic Reticulum Stress in Type II Diabetes. Molecules 2021, 26, 4362. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144362
Mustapha S, Mohammed M, Azemi AK, Jatau AI, Shehu A, Mustapha L, Aliyu IM, Danraka RN, Amin A, Bala AA, et al. Current Status of Endoplasmic Reticulum Stress in Type II Diabetes. Molecules. 2021; 26(14):4362. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144362
Chicago/Turabian StyleMustapha, Sagir, Mustapha Mohammed, Ahmad Khusairi Azemi, Abubakar Ibrahim Jatau, Aishatu Shehu, Lukman Mustapha, Ibrahim Muazzamu Aliyu, Rabi’u Nuhu Danraka, Abdulbasit Amin, Auwal Adam Bala, and et al. 2021. "Current Status of Endoplasmic Reticulum Stress in Type II Diabetes" Molecules 26, no. 14: 4362. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144362