
Chemoinformatics Analyses of Tau Ligands Reveal Key Molecular Requirements for the Identification of Potential Drug Candidates against Tauopathies



Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Preparation
2.2. 2D similarity Calculations
2.3. Analysis and Comparison of Molecular Descriptors
2.4. Molecular Fragment Analyses
3. Results and Discussion
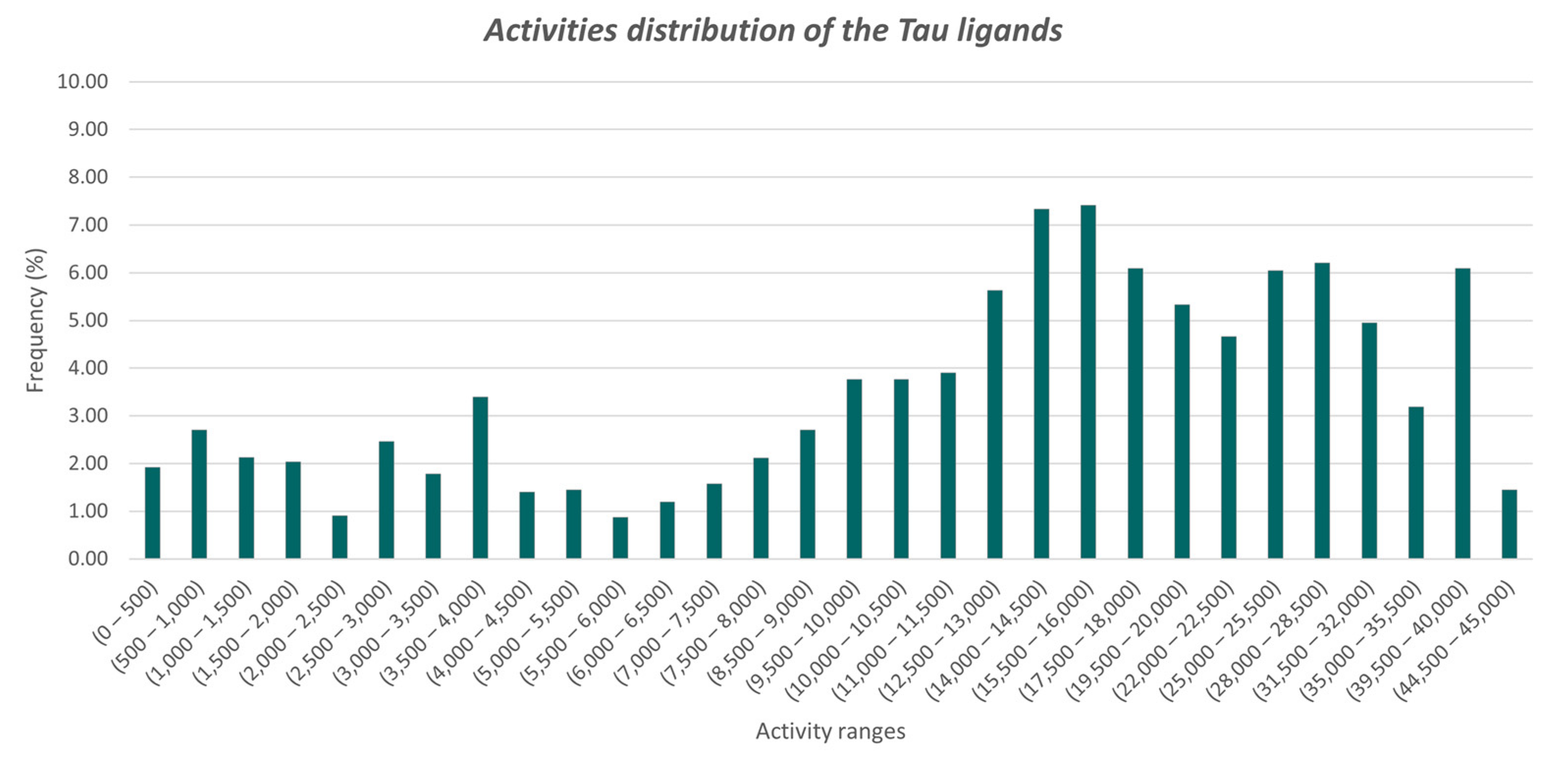
3.1. Dataset Preparation for the Analyses
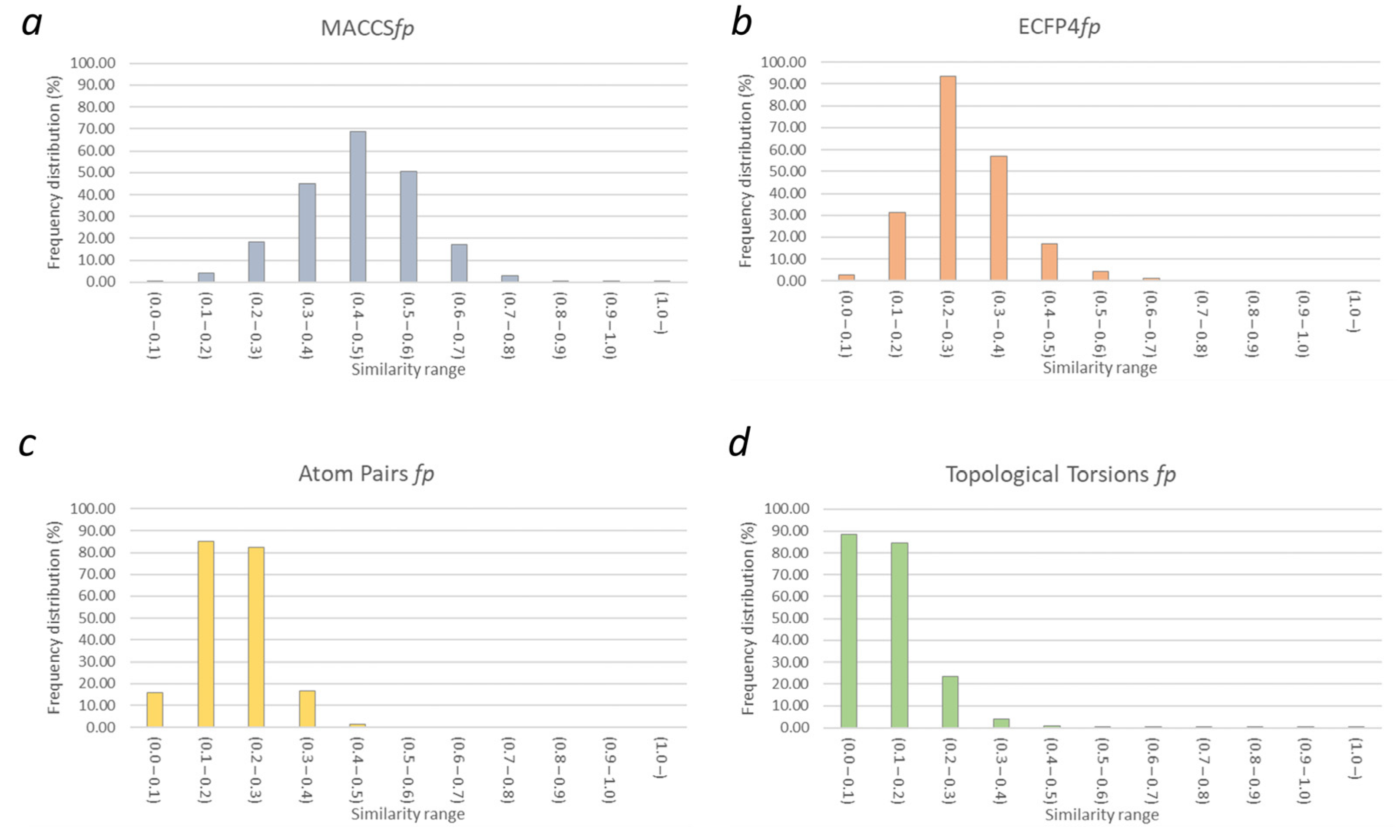
3.2. Similarity Calculations in the Set of Active Compounds
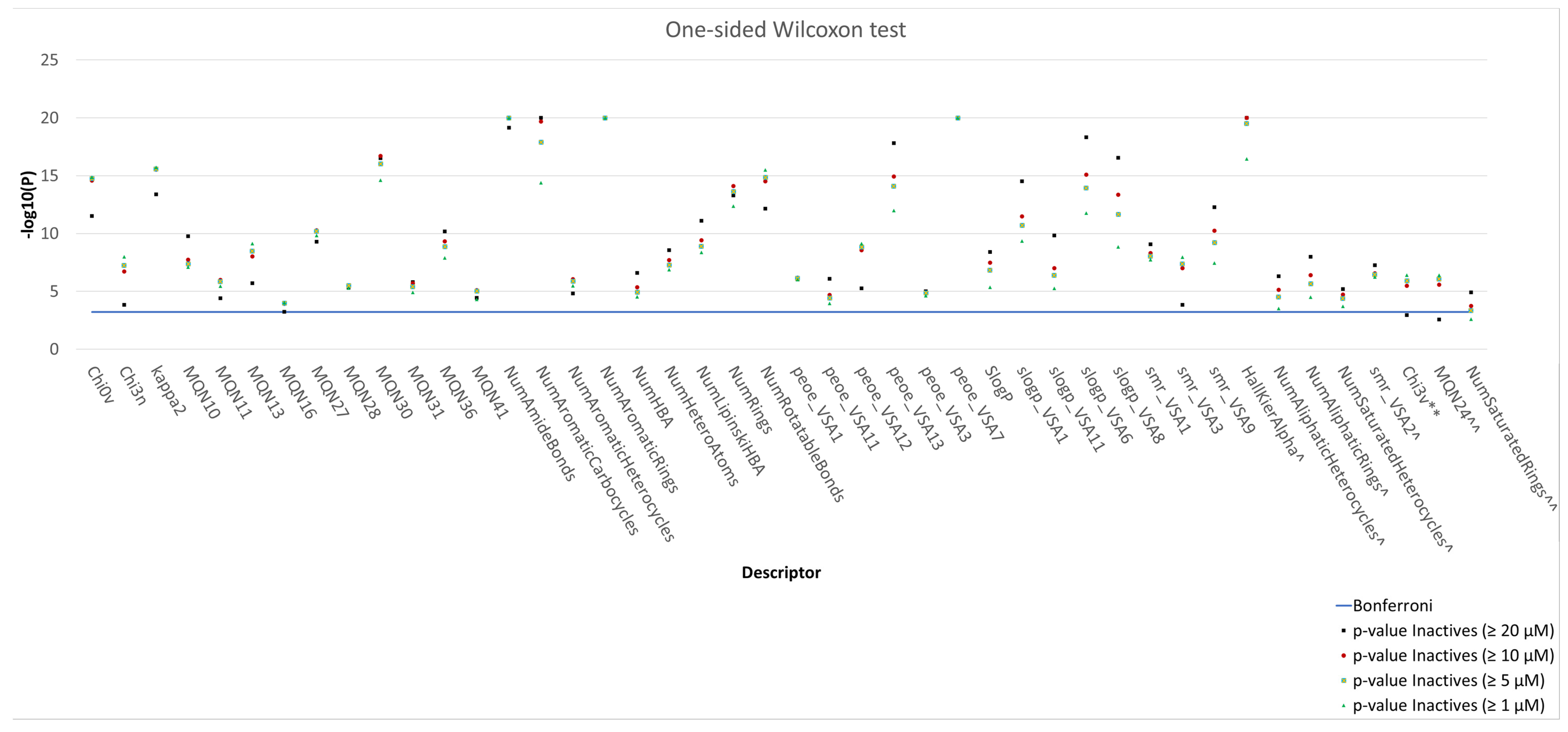
3.3. Analysis and Comparison of Molecular Descriptors
3.4. Analysis of Molecular Fragments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chi, H.; Sang, T.-K.; Chang, H.-Y. Tauopathy. In Cognitive Disorders; Foyaca Sibat, H., Ed.; IntechOpen: Rijeka, Croatia, 2019; pp. 33–63. [Google Scholar]
- Josephs, K.A. Current Understanding of Neurodegenerative Diseases Associated With the Protein Tau. Mayo Clinic Proc. 2017, 92, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and Tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R. Tauopathies: Classification and Clinical Update on Neurodegenerative Diseases Associated with Microtubule-Associated Protein Tau. Intern. Med. J. 2006, 36, 652–660. [Google Scholar] [CrossRef]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Sergeant, N.; Delacourte, A.; Buée, L. Tau Protein as a Differential Biomarker of Tauopathies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1739, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Götz, J. Amyloid-β and Tau—A Toxic Pas de Deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau Pathology and Neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Bakota, L.; Ussif, A.; Jeserich, G.; Brandt, R. Systemic and Network Functions of the Microtubule-Associated Protein Tau: Implications for Tau-Based Therapies. Mol. Cell. Neurosci. 2017, 84, 132–141. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Skrabana, R.; Skrabanova, M.; Csokova, N.; Sevcik, J.; Novak, M. Intrinsically Disordered Tau Protein in Alzheimer’s Tangles: A Coincidence or a Rule? Bratisl. Lek. Listy 2006, 107, 354–358. [Google Scholar]
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [Green Version]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.-M.; Mandelkow, E. Assembly of τ Protein into Alzheimer Paired Helical Filaments Depends on a Local Sequence Motif (306VQIVYK311) Forming β Structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front. Aging Neurosci. 2019, 11, 256. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Trushina, N.I.; Bakota, L. Much More Than a Cytoskeletal Protein: Physiological and Pathological Functions of the Non-Microtubule Binding Region of Tau. Front. Neurol. 2020, 11, 590059. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their (Disordered) Proteomes in Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janning, D.; Igaev, M.; Sündermann, F.; Brühmann, J.; Beutel, O.; Heinisch, J.J.; Bakota, L.; Piehler, J.; Junge, W.; Brandt, R. Single-Molecule Tracking of Tau Reveals Fast Kiss-and-Hop Interaction with Microtubules in Living Neurons. Mol. Biol. Cell 2014, 25, 3541–3551. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and Phosphatases and Tau Sites Involved in Alzheimer Neurofibrillary Degeneration. Eur. J. Neurosci. 2007, 25, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Schoch, K.M.; DeVos, S.L.; Miller, R.L.; Chun, S.J.; Norrbom, M.; Wozniak, D.F.; Dawson, H.N.; Bennett, C.F.; Rigo, F.; Miller, T.M. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron 2016, 90, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Dominguez-Meijide, A.; Vasili, E.; Outeiro, T.F. Pharmacological Modulators of Tau Aggregation and Spreading. Brain Sci. 2020, 10, 858. [Google Scholar] [CrossRef]
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and Mechanism of Action of Tau Aggregation Inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Reddy, P.H. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulic, B.; Pickhardt, M.; Mandelkow, E. Progress and Developments in Tau Aggregation Inhibitors for Alzheimer Disease. J. Med. Chem. 2013, 56, 4135–4155. [Google Scholar] [CrossRef] [PubMed]
- Calcul, L.; Zhang, B.; Jinwal, U.K.; Dickey, C.A.; Baker, B.J. Natural Products as a Rich Source of Tau-Targeting Drugs for Alzheimer’s Disease. Future Med. Chem. 2012, 4, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the Efficacy of Natural Products in Alleviating Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 1321–1329. [Google Scholar] [CrossRef]
- Rastelli, G.; Pellati, F.; Pinzi, L.; Gamberini, M.C. Repositioning Natural Products in Drug Discovery. Molecules 2020, 25, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A Walk through Tau Therapeutic Strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef]
- Li, C.; Götz, J. Tau-Based Therapies in Neurodegeneration: Opportunities and Challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Lunven, L.; Bonnet, H.; Yahiaoui, S.; Yi, W.; Da Costa, L.; Peuchmaur, M.; Boumendjel, A.; Chierici, S. Disruption of Fibers from the Tau Model AcPHF6 by Naturally Occurring Aurones and Synthetic Analogues. ACS Chem. Neurosci. 2016, 7, 995–1003. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective Inhibition of Alzheimer Disease-like Tau Aggregation by Phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Larbig, G.; Pickhardt, M.; Lloyd, D.G.; Schmidt, B.; Mandelkow, E. Screening for Inhibitors of Tau Protein Aggregation into Alzheimer Paired Helical Filaments: A Ligand Based Approach Results in Successful Scaffold Hopping. Curr. Alzheimer Res. 2007, 4, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Necula, M.; Chirita, C.N.; Kuret, J. Cyanine Dye N744 Inhibits Tau Fibrillization by Blocking Filament Extension: Implications for the Treatment of Tauopathic Neurodegenerative Diseases. Biochemistry 2005, 44, 10227–10237. [Google Scholar] [CrossRef]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small Molecule Inhibitors of Aggregation Indicate That Amyloid Beta Oligomerization and Fibrillization Pathways Are Independent and Distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications, Proceedings of the 31st Annual Conference of the Gesellschaft für Klassifikation e.V.; Breisgau, Germany, 7–9 March 2007, Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- OpenEye Toolkits 2020.2.2 OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 20 January 2021).
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 5 June 2020).
- Willett, P. Searching Techniques for Databases of Two- and Three-Dimensional Chemical Structures. J. Med. Chem. 2005, 48, 4183–4199. [Google Scholar] [CrossRef] [PubMed]
- Jasial, S.; Hu, Y.; Vogt, M.; Bajorath, J. Activity-Relevant Similarity Values for Fingerprints and Implications for Similarity Searching. F1000Res 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Kuenemann, M.A.; Labbé, C.M.; Cerdan, A.H.; Sperandio, O. Imbalance in Chemical Space: How to Facilitate the Identification of Protein-Protein Interaction Inhibitors. Sci. Rep. 2016, 6, 23815. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.F. Examination of the Computed Molecular Properties of Compounds Selected for Clinical Development. Biotechniques 2003, 8, 16–20. [Google Scholar] [CrossRef]
- Armstrong, R.A. When to Use the Bonferroni Correction. Ophthalmic Physiol. Opt. 2014, 34, 502–508. [Google Scholar] [CrossRef]
- Degen, J.; Wegscheid-Gerlach, C.; Zaliani, A.; Rarey, M. On the Art of Compiling and Using “Drug-Like” Chemical Fragment Spaces. ChemMedChem 2008, 3, 1503–1507. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAP—Retrosynthetic Combinatorial Analysis Procedure: A Powerful New Technique for Identifying Privileged Molecular Fragments with Useful Applications in Combinatorial Chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef]
- CHOMP 3.1.1.2: OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 20 January 2021).
- Jameson, L.P.; Smith, N.W.; Dzyuba, S.V. Dye-Binding Assays for Evaluation of the Effects of Small Molecule Inhibitors on Amyloid (Aβ) Self-Assembly. ACS Chem. Neurosci. 2012, 3, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a Method to Reveal Structures and Membrane-Interactions of Amyloidogenic Proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 1862–1885. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Teruya, T.; Kawakami, H.; Takahashi, T.; Sugimoto, H. Design and Synthesis of Curcumin Derivatives as Tau and Amyloid β Dual Aggregation Inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 5024–5028. [Google Scholar] [CrossRef]
- Fuse, S.; Matsumura, K.; Fujita, Y.; Sugimoto, H.; Takahashi, T. Development of Dual Targeting Inhibitors against Aggregations of Amyloid-β and Tau Protein. Eur. J. Med. Chem. 2014, 85, 228–234. [Google Scholar] [CrossRef]
- Available online: http://rdkit.blogspot.com/2013/10/fingerprint-thresholds.html (accessed on 5 June 2020).
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Nilakantan, R.; Bauman, N.; Dixon, J.S.; Venkataraghavan, R. Topological Torsion: A New Molecular Descriptor for SAR Applications. Comparison with Other Descriptors. J. Chem. Inf. Comput. Sci. 1987, 27, 82–85. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for Evaluation of Computational Methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Truchon, J.-F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-1: Canvas; Schrödinger, LLC: New York, NY, USA, 2020.
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. In Reviews in Computational Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1991; pp. 367–422. [Google Scholar]
- Hall, L.H.; Kier, L.B. The Nature of Structure-Activity Relationships and Their Relation to Molecular Connectivity. Eur. J. Med. Chem 1977, 12, 307–312. [Google Scholar]
- Nguyen, K.T.; Blum, L.C.; van Deursen, R.; Reymond, J.-L. Classification of Organic Molecules by Molecular Quantum Numbers. ChemMedChem 2009, 4, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, P.; Upadhyay, S.P.; Suo, W.Z.; Singh, V.; Gurung, P.; Lee, E.S.; Sharma, R.; Sharma, M. Chalcone and Its Analogs: Therapeutic and Diagnostic Applications in Alzheimer’s Disease. Bioorganic Chem. 2021, 108, 104681. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-H.; Chiu, Y.-J.; Lin, C.-H.; Lin, C.-Y.; Chao, C.-Y.; Chen, Y.-C.; Yang, S.-M.; Lin, W.; Mei Hsieh-Li, H.; Wu, Y.-R.; et al. Exploration of Multi-Target Effects of 3-Benzoyl-5-Hydroxychromen-2-One in Alzheimer’s Disease Cell and Mouse Models. Aging Cell 2020, 19, e13169. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, L.G.; Pinzi, L.; Facchetti, G.; Bortolini, B.; Christodoulou, M.S.; Binda, C.; Rastelli, G.; Rimoldi, I.; Passarella, D.; Di Paolo, M.L.; et al. Promising Non-Cytotoxic Monosubstituted Chalcones to Target Monoamine Oxidase-B. ACS Med. Chem. Lett. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Dhavale, D.; Kotzbauer, P.; Mach, R.H. Chalcones and Five-Membered Heterocyclic Isosteres Bind to Alpha Synuclein Fibrils in Vitro. ACS Omega 2018, 3, 4486–4493. [Google Scholar] [CrossRef] [Green Version]
- Jeon, K.-H.; Lee, E.; Jun, K.-Y.; Eom, J.-E.; Kwak, S.Y.; Na, Y.; Kwon, Y. Neuroprotective Effect of Synthetic Chalcone Derivatives as Competitive Dual Inhibitors against μ-Calpain and Cathepsin B through the Downregulation of Tau Phosphorylation and Insoluble Aβ Peptide Formation. Eur. J. Med. Chem. 2016, 121, 433–444. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Fingerprints 1 | Number of Identified Pairs (Actives) 2 | % of Identified Pairs (Actives) 3 | Number of Identified Pairs (Inactives) 2 | % of Identified Pairs (Inactives) 3 |
|---|---|---|---|---|
| MACCSfp | 1859 | 0.5 | 27,604 | 0.1 |
| ECFP4fp | 159,125 | 38.8 | 13,460,261 | 30.8 |
| AtomPairsfp | 9093 | 2.2 | 488,407 | 1.1 |
| TopologicalTorsionfp | 42,477 | 10.4 | 2,670,815 | 6.1 |
| Number of Queries per Combination | % of Actives (≤500 nM) 1 | % of Inactives (≥1 µM) 1 | Difference between the % of Actives and Inactives 2 |
|---|---|---|---|
| 1 | 10.8 (98) | 3.2 (1568) | 7.6 (16) |
| 2 | 18.1 (164) | 5.1 (2465) | 13.0 (15) |
| 3 | 22.4 (203) | 6.6 (3189) | 15.8 (16) |
| 4 | 24.2 (219) | 6.7 (3238) | 17.5 (15) |
| 5 | 28.3 (256) | 9.5 (4591) | 18.8 (18) |
| 6 | 30.1 (272) | 10.5 (5074) | 19.6 (19) |
| 7 | 32.7 (296) | 12.3 (5944) | 20.4 (20) |
| 8 | 32.2 (291) | 12.1 (5847) | 20.1 (20) |
| 9 | 33.7 (305) | 13.6 (6572) | 20.1 (22) |
| 10 | 33.8 (306) | 13.9 (6717) | 19.9 (22) |
| 11 | 32.9 (298) | 12.9 (6234) | 20.0 (21) |
| 12 | 33.7 (305) | 13.3 (6427) | 20.4 (21) |
| 13 | 35.0 (317) | 14.3 (6910) | 20.7 (22) |
| 14 | 35.0 (317) | 14.4 (6959) | 20.6 (22) |
| 15 | 35.1 (318) | 14.5 (7007) | 20.6 (22) |
| 16 | 35.7 (323) | 14.8 (7152) | 20.9 (22) |
| 17 | 36.8 (333) | 15.7 (7587) | 21.1 (23) |
| 18 | 36.8 (333) | 15.7 (7587) | 21.1 (23) |
| 19 | 36.8 (333) | 15.6 (7539) | 21.2 (23) |
| 20 | 37.1 (336) | 15.9 (7684) | 21.2 (23) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinzi, L.; Tinivella, A.; Rastelli, G. Chemoinformatics Analyses of Tau Ligands Reveal Key Molecular Requirements for the Identification of Potential Drug Candidates against Tauopathies. Molecules 2021, 26, 5039. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26165039
Pinzi L, Tinivella A, Rastelli G. Chemoinformatics Analyses of Tau Ligands Reveal Key Molecular Requirements for the Identification of Potential Drug Candidates against Tauopathies. Molecules. 2021; 26(16):5039. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26165039
Chicago/Turabian StylePinzi, Luca, Annachiara Tinivella, and Giulio Rastelli. 2021. "Chemoinformatics Analyses of Tau Ligands Reveal Key Molecular Requirements for the Identification of Potential Drug Candidates against Tauopathies" Molecules 26, no. 16: 5039. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26165039