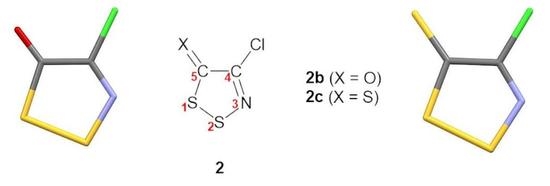
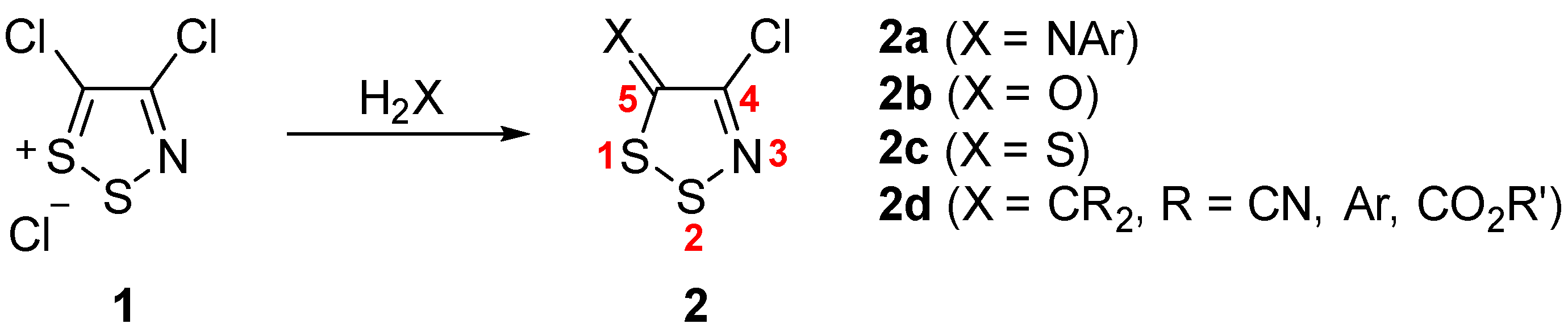
2.1. Intramolecular Geometry
Crystals of the dithiazolone
2b were grown by sublimation under a static vacuum (1.6 Pa) at 30 °C. Dithiazolethione
2c demonstrated polymorphism: polymorphs
2c–α and
2c–β were obtained by slow evaporation of concentrated solutions in pentane and benzene, respectively. Suitable single crystals of dithiazoles
2b,
2c–α and
2c–β were then loaded on a goniometer and their crystal structure and solid-state packing were determined at 100 K by single-crystal X-ray diffractometry (
Table S1 in Supplementary Information). Below, IUPAC numbering (not a crystallographic one) is used to assist the comparison between the new 5
H-1,2,3-dithiazoles reported herein and those reported in the literature.
All three dithiazoles are planar. The maximum deviation of the S and N atoms from the plane of the five-membered rings are 0.020 and 0.027 Å, respectively. Their intramolecular geometrical parameters are similar to each other (
Table 1) and comparable to those reported for 4-benzoyl-5
H-1,2,3-dithiazol-5-one (
3) [
37], 4-[
N-(2-chloroethyl)piperazin-1-yl]-5
H-1,2,3-dithiazole-5-thione (
4) [
38] and 4-phenyl-5
H-1,2,3-dithiazole-5-thione polyiodide S–I
+–S complex (
5) [
39] (
Table S2 in Supplementary Information).
The endocyclic S1–S2 bond lengths are 2.0533(7), 2.066(2), 2.058(4)-2.075(4) Å for
2b,
2c–α and
2c–β, respectively (
Table 1) and are longer than the S–S bond length 2.034(2) Å reported for Appel’s salt
1 [
40]. The short S–S bond in Appel’s salt
1 reflects the significant bond and charge delocalization aiming to achieve a greater aromaticity. Our calculations (
Section 2.3.3) indicate that Appel’s salt cation
1 is more delocalized and more aromatic then either the dithiazolone
2b or dithiazolethione
2c. The angles at the S atoms range from 93.96(6)° (C5–S1–S2 for
2b) to 98.23(7)° (S1–S2–N3 for
2b), all < 120°, are comparable to the analogous angles in previously reported 1,2,3-dithiazol-5-ones/thiones (
Table S2 in Supplementary Information).
The C4–N3 bond lengths are 1.269(3), 1.283(6) and 1.23(1)–1.32(1) Å for
2b,
2c–α and
2c–β, respectively (
Table 1). These have a pronounced double bond character (1.28 Å) [
41] indicating significant localization inside the ring. The S2–N3–C4 internal angles are 115.9(1), 116.2(3) and 114.6(7)-119.6(8)° for 2
b,
2c–α and
2c–β, respectively and are typical of
sp2-hybridized imine N [
41]. The S2–N3 bonds lengths are 1.645(2), 1.642(4) and 1.612(9)–1.663(9) Å for
2b,
2c–α and
2c–β, respectively.
The C4–C5 bond lengths are 1.476(2), 1.470(6) and 1.45(2)–1.51(1) Å for
2b,
2c–α and
2c–β, respectively, and are intermediate between typical aromatic C–C bonds (1.41 Å) and C–C single bonds (1.54 Å) [
41]. Similar C–C bond lengths have been reported for 1,2,3-dithiazol-5-ones/thiones
3–5 (
Table S2 in Supplementary Information).
The C5–O bond length 1.208(2) Å in dithiazolone
2b is similar to that reported for dithiazolone
3 [
37] and is typical of a C=O double bond (1.21 Å) [
41]. The angles around C5 (
Table 1) support a
sp2-hybridized C of a carbonyl group [
41]. The endocyclic C4–C5–S1 angle of 108.7(1)° is narrower and accounts for the five-membered ring strain. The other two C4–C5–O 126.6(2)° and S1–C5–O 124.7(1)° are wider with the one next to the Cl being slightly larger possibly due the steric interactions between the lone pairs of Cl and O atoms.
The C5–X (X=S) bond lengths in
2c–α 1.639(4) Å and
2c–β 1.61(1)–1.65(1) Å are typical C=S double bonds (1.61 Å) [
41]. While these bond lengths are similar to the C5–S bond length 1.657(2) Å in dithiazole
4 [38], the C5–S bond lengths 1.693(6)–1.694(6) Å in the polyiodide complex of 4-phenyl-5
H-1,2,3-dithiazole-5-thione (
5) are slightly longer owing to the delocalization across the S–I
+–S bridge [
39]. The angles around C5 in
2c–α and
2c–β (
Table 1) deviate from the expected value of 120° for a
sp2-hybridized C atom but are typical of thiones with one angle narrower and the other two wider, e.g., C4–C5–S1 108.7(3)°, C4–C5–X 126.9(3)° and S1–C5–X 124.3(2)° for polymorph
2c–α.
While the bond length of C5=O 1.208(2) Å in the dithiazolone
2b is significantly different than the bond length of C5=S 1.639(4) Å in thione
2c–α, the bond angles around C5 are surprisingly similar (
Table 1); C4–C5–S1 108.7(1)°, C4–C5–O 126.6(2)°, S1–C5–O 124.7(1)° for dithiazolone
2b vs. C4–C5–S1 108.7(3)°, C4–C5–S 126.9(3)° and S1–C5–S 124.3(2)° for thione
2c–α.
2.2. Crystal Packing and Short Contacts
1,2,3-Dithiazol-5-ones/thiones
2b and
2c–α crystal pack in the highly symmetrical
Pbca space group with eight symmetry operators in operation, primarily a series of 2-fold screw axis and glide planes (
Table S3 in Supplementary Information). The second polymorph of dithiazolethione
2c–β is of lower symmetry (
P-1) with only two symmetry operators in effect (identity and inversion).
There is a rich network of structure-directing intermolecular interactions in the crystal packing of dithiazoles
2b,
2c–α and
2c–β (
Figure 1,
Figure 2 and
Figure 3). These mainly electrostatic interactions optimize contacts between electronegative and electropositive regions in neighboring molecules. Inside these five-membered rings the S–N and C–N bonds are polar due to the difference in electronegativity of their atoms; 2.58 for S and 3.04 for N and 2.55 for C. The S–N and C–N bonds should therefore be considered polarized in the sense of S
δ+…N
δ− and C
δ+…N
δ−. It is expected that the location of two electropositive S atoms next to each other will create a strong electropositive region near the S atoms (
Table 2). The presence of lone pairs on the N, Cl and on O and S atoms of the C=O carbonyl and C=S thione groups create pockets of electronegative regions. To better understand the electrostatic contribution to bonding we calculated the molecular electrostatic potential maps (MEP) for dithiazol-5-ones/thiones
2b and
2c at the B3LYP/def2-TZVPD level of theory (
Table 2); red color corresponds to a maximum negative charge value of −3.0 × 10
−2 esu, i.e., electronegative character, while blue color corresponds to a maximum positive charge value of 3.0 × 10
−2 esu, i.e., electropositive character.
The MEP for
2b and
2c are as expected blue near the endocyclic electropositive S atoms and red in the vicinity of N, Cl, O, S where the lone pairs of these atoms are located, and a build-up of partial negative charge is expected. Consequently, close intermolecular contacts between the endocyclic S and the rest of the electronegative atoms (N, Cl, O and exocyclic S) should be electrostatically favorable. It should be noted that the exocyclic S atom in
2c has areas that are red, i.e., negatively charged, where the lone pairs are expected to reside and an area in the center of the atom along the C=S bond axis that is green. Our calculations on Fukui functions (
Section 2.3.2) predict an ambivalent chemical behavior which shows the thione S atom to be a site for both nucleophilic and electrophilic chemistry.
Intermolecular interactions are usually considered to be contacts considerably shorter than the sum of the van der Waals radii (∑
VDW) of the atoms participating in these contacts. They are usually 8–20% shorter than the sum of the equilibrium radii [
42]. For some atoms van der Waals radii exhibit significant anisotropy. N and O atoms have almost spherical shapes but for S and Cl the ellipticity and therefore the anisotropy increases [
43]. For these anisotropic atoms the minor radii (minor axis ca. 0° or 180°) correspond to contacts close to the plane of the molecule and the major radii (major axis ca. 90°) for contacts perpendicular to the molecular plane [
43]. The sum of the minor and major van der Waals radii (∑
VDW) of the intermolecular contacts present in the crystal packing of
2b,
2c–α and
2c–β are 3.20, 3.14, 3.18 and 3.20 Å (minor) and 3.63, 3.57, 3.81 and 4.06 Å (major) for S
δ+…N
δ−, S
δ+…O
δ−, S
δ+…Cl
δ− and S
δ+…S
δ− (endocyclic S to exocyclic thione S), respectively. For the discussion below, we provide both the sum of the minor and major van der Waals radii (∑
VDW) since the angle of the intermolecular atom approach is somewhere along the 0–180° range.
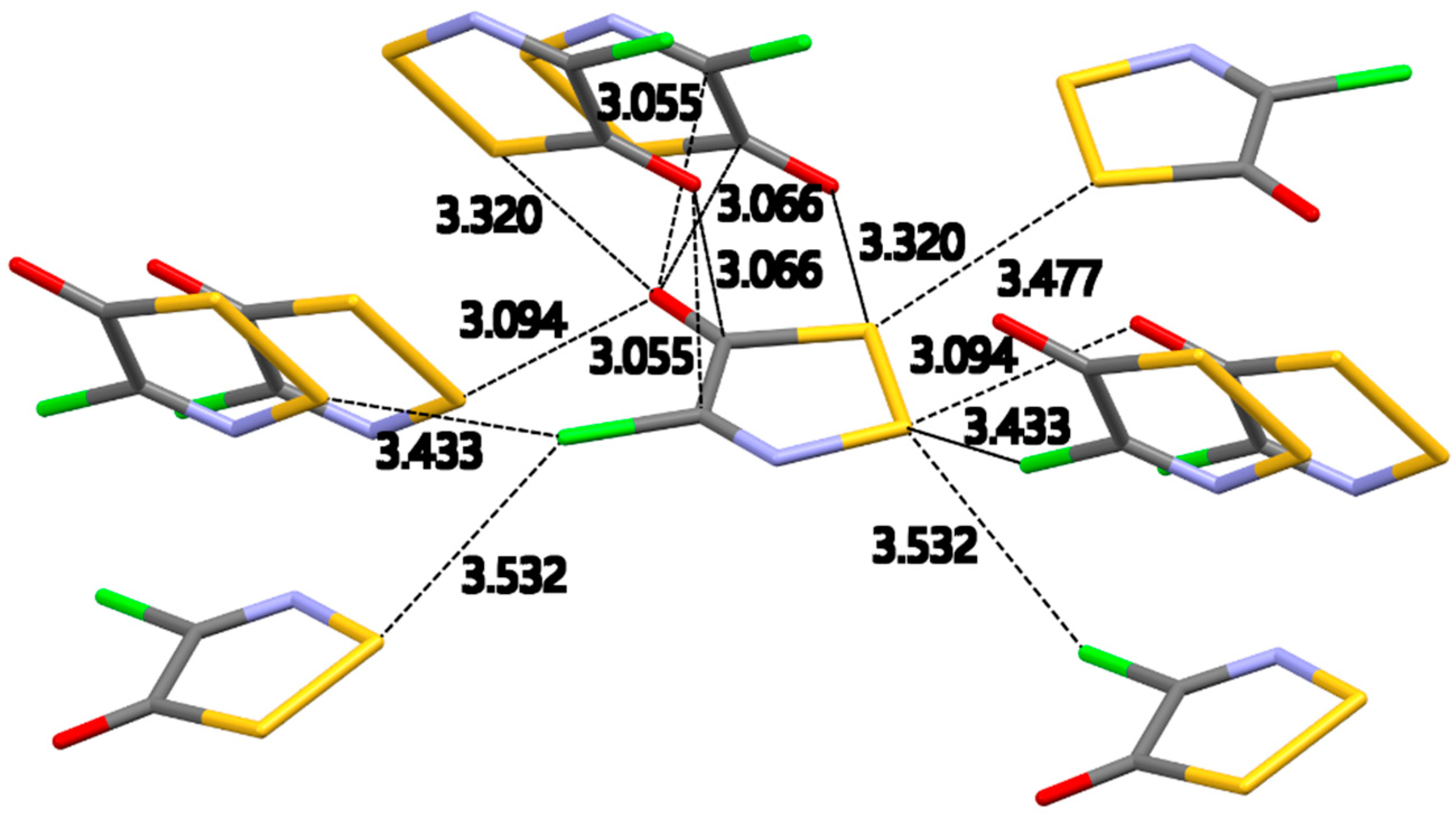
2.2.1. 4-Chloro-5H-1,2,3-dithiazol-5-one (2b)
Dithiazolone
2b crystallized in the
Pbca space group with one molecule in the asymmetric unit cell.
Figure 1 shows all the intermolecular interactions expanded for the central molecule in the asymmetric unit cell. This crystal packing is dominated by S…O and S…Cl intermolecular contacts. The lack of any S…N interactions is surprising but not unexpected as the S…O and S…Cl interactions are potentially stronger and structure directing. The two S…O interactions 3.094(1) Å [∠S–S…O, 89.53(3)°] and 3.320(1) Å [∠S–S…O, 175.00(3)°] are well within the ∑VDW [3.14 Å (minor), 3.57 Å (major)].
There are two S…Cl intermolecular interactions 3.4528(8) Å [∠S–S…Cl, 151.01(3)°] and 3.5316(7) Å [∠S–S…Cl, 130.43(3)°] well within the ∑
VDW [3.18 Å (minor), 3.81 Å (major)]. These interactions (
Figure 1) are longer than the ionic S…Cl interactions previously reported for Appel’s salt
1 [
40].
A triangular and near symmetrical approach of the oxygen atom on top of the C–C bond creates two C…O contacts 3.055(2) Å [∠C–O…C, 119.2(1)°] and 3.066(2) Å [∠C–O…C, 117.7(1)°]. Each carbon atom of the C–C bond has a significant partial positive charge stemming from the polarization of the C–Cl and C=O bonds due to the difference in electronegativity (C
δ+–Cl
δ−, C
δ+=O
δ−). The location of the oxygen atom is, therefore, ideal to create a bifurcated set of C
δ+…O
δ− contacts (
Figure S1a in Supplementary Information). These C…O contacts are within the ∑
VDW [3.25 Å (minor), 3.67 Å (major)] [
44].
The network of intermolecular contacts for dithiazolone 2b is concluded with a S…S contact 3.4771(7) Å [∠C–S…S, 149.75(6)°] between two endocyclic S atoms from neighboring molecules. This represents a short contact due to the proximity of the S atoms. Both atoms are expected to have a partial positive charge Sδ+ but since the electron cloud around S is highly polarizable this could also be an electrostatic interaction.
Molecules of the dithiazolone
2b stack along the
c-axis (
Figure S1a in Supplementary Information) to form one dimensional (1D) columns. Inside these 1D columns the molecules are connected via S…O 3.320(1) Å and C…O 3.055(2)–3.066(2) Å contacts and arrange in a near herringbone pattern with successive plane angles of 71.36° and 73.46°. Neighboring columns are oriented in a face-to-tail manner and connect via S…O 3.094(1) Å and C…Cl 3.5316 (7) Å contacts to form ladders along the
b-axis (
Figure S1a in Supplementary Information). Two parallel chains with the same direction (
Figure S1b in Supplementary Information) form a ribbon along the
bc-diagonal. An antiparallel ribbon completes the 2D sheet along the
bc-diagonal.
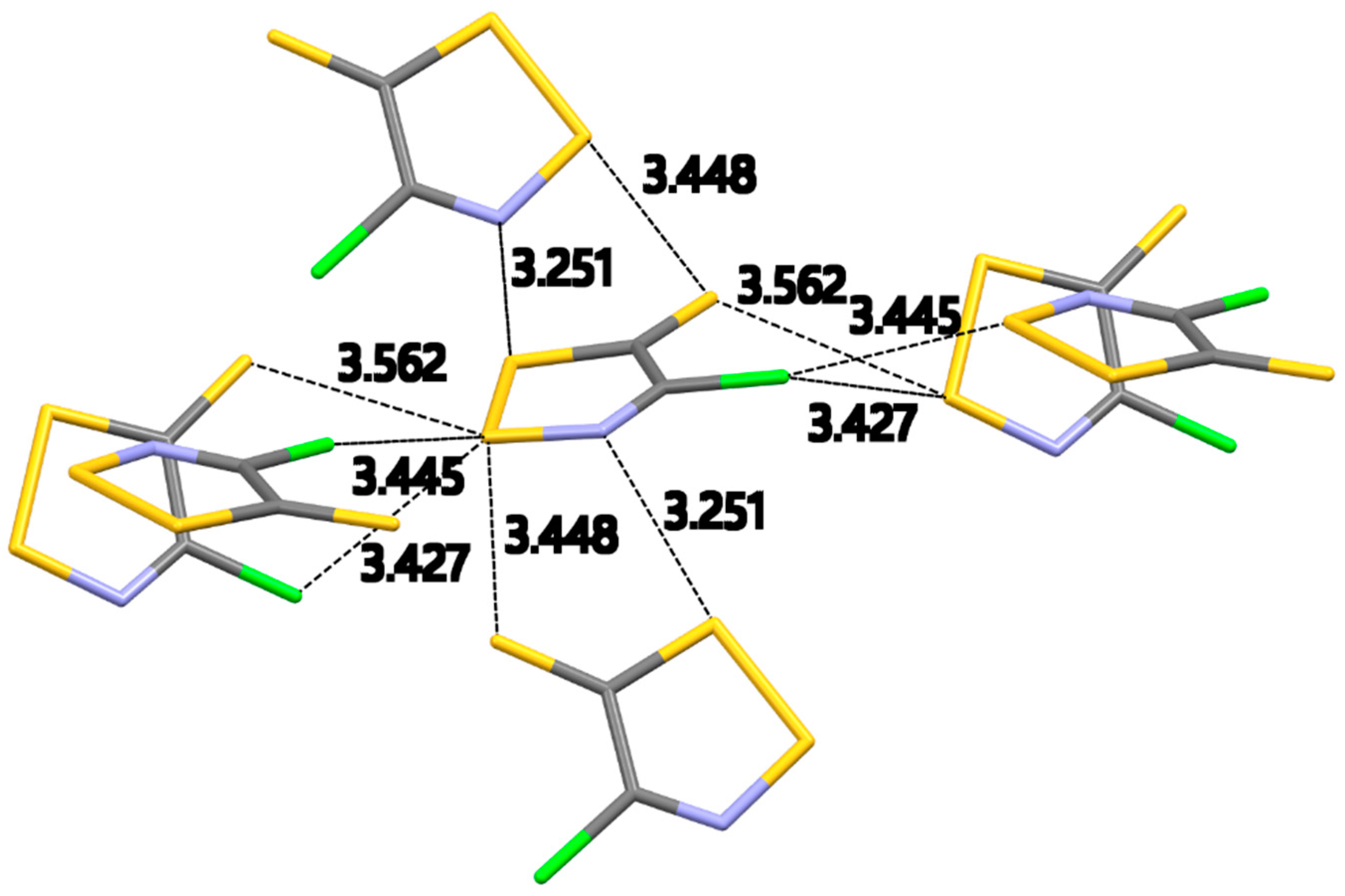
2.2.2. 4-Chloro-5H-1,2,3-dithiazole-5-thione (2c–α)
The
α-phase of dithiazolethione
2c–α crystallized in the
Pbca space group with one molecule in the asymmetric unit cell.
Figure 2 shows all the intermolecular interactions expanded for the central molecule in the asymmetric unit cell. This crystal packing is dominated by S…Cl and S…S intermolecular contacts. In the absence of strong structure directing S…O contacts, S…N interactions appear in the crystal packing. There is only one S…N interaction 3.251(4) Å [∠S–S…N, 166.29(9)°] well within the ∑
VDW [3.20 Å (minor), 3.63 Å (major)].
The crystal packing of S,N-rich heterocycles is dominated by the presence of short S…N intermolecular contacts. S-N bonds are strongly polar (Sδ+…Nδ−) and therefore electrostatically favored.
The planar geometry of this molecule results in a short intramolecular S…N contact of 2.597(2) Å, well below the minor ∑
VDW (3.20 Å). Neighboring molecules form centrosymmetric pairs linked by short S…N interactions of 3.035(2) Å. No S…N contacts appear in the reported crystal structures of the benzoyldithiazolone
3 and dithiazolethiones
4 and
5 [
37,
38,
39].
There are two S…Cl intermolecular interactions of 3.427(1) Å [∠S–S…Cl, 109.88(5)°] and 3.445(1) Å [∠S–S…Cl, 140.66(6)°] well within the ∑VDW [3.18 Å (minor), 3.81 Å (major)]. This is a near triangular interaction of the same covalently bound Cl to two S atoms of neighboring dithiazoles (
Figure 2). The distances of the S…Cl contacts in
2c–α are similar to those in dithiazolone
2b. It should be noted that while dithiazolethione
4 has a terminal C–Cl bond, no S…Cl contacts appear in its crystal packing [
38]. Instead, there is a highly symmetrical bifurcated S…S contact of 3.3894(7) and 3.3143(8) Å between the exocyclic C=S sulfur atom and the endocyclic S–S atoms [
38]. The thione C=S bond is weakly polar and easily polarizable as sulfur is slightly more electronegative than carbon (2.58 for S vs. 2.55 for C) and is therefore expected to bear partial negative charge. The S
δ+…S
δ− contacts between the endocyclic S–S atoms and the exocyclic thione S atom in compound
4 are partially of electrostatic nature. These S…S contacts are also present in the crystal structure of
2c–α albeit with a different geometry. The bifurcation of the thione S atom extends on S atoms of S–S bonds in neighboring rings (
Figure 2). These S…S contacts of 3.562(2) Å [∠N–S…S, 159.8(2)°] and 3.448(2) Å [∠S–S…S, 153.43(6)°] are similar to the ones previously reported in compound
4. The S…S contact for
2c–α is 3.328(1) Å [∠C–S…S, 85.8(1)°] between two endocyclic S atoms from neighboring molecules. A similar interaction appears in the crystal packing of dithiazolone
2b.
Molecules of the dithiazolethione
2c–α form zig-zag chains along the
a-axis (
Figure S2a in Supplementary Information). Inside these chains successive molecules are arranged sideways with opposite ends and are connected via two short S…N and S…S contacts of 3.251(4) and 3.448(2) Å, respectively. The zig-zag chains pack parallel along the
b-axis (
Figure S2b in Supplementary Information) and are connected via S…S contacts of 3.328(1) Å to form a tight brick wall of dithiazolethiones without any voids. The packing along the
c-axis (
Figure S2c in Supplementary Information) resembles those in the crystal packing of dithiazolone
2b. Chains along the
c-axis are formed by molecules that are organized in a head-to-tail orientation and are connected by S…S and S…Cl contacts of 3.562(2) and 3.427(1) Å, respectively. Two chains, linked by S…Cl contact of 3.445(1) Å, run parallel across
c-axis to form a ribbon. Neighboring ribbons run antiparallel to each other and are connected by S…S and S…N contacts of 3.448(2) and 3.251(4) Å, respectively, to form a 2D sheet (
Figure S2c in Supplementary Information).
2.2.3. 4-Chloro-5H-1,2,3-dithiazole-5-thione (2c–β)
The
β-phase of dithiazolethione
2c–β crystallized in the
P-1 space group with six molecules in the asymmetric unit cell and three crystallographically independent trimers.
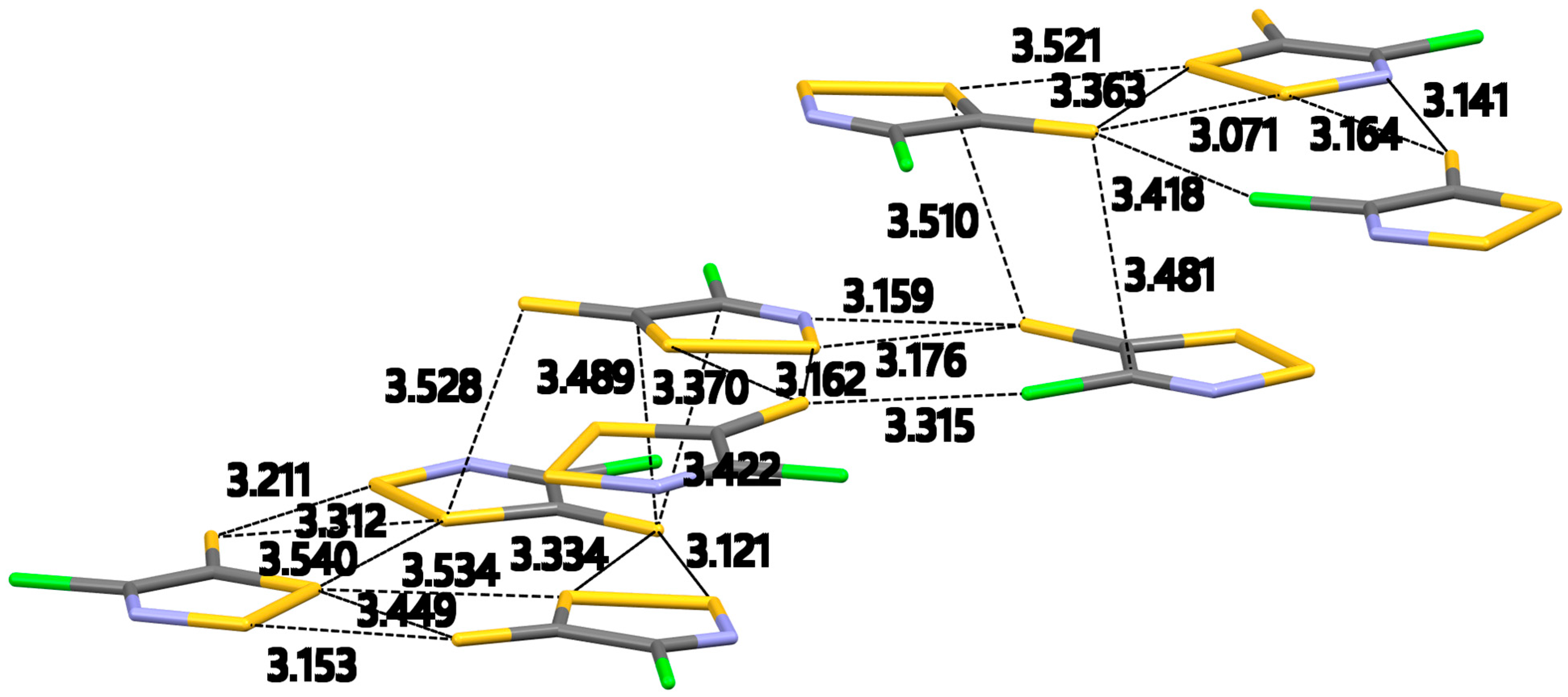
Figure 3 shows the formation of supramolecular triangles and all the intermolecular interactions expanded for the molecules in the asymmetric unit cell. This crystal packing is dominated by S…Cl, S…N and S…S contacts. Inside the supramolecular triangles, molecules of
2c–β are arranged around a non-crystallographic three-fold axis and are connected by a series of in-plane contacts (
Figure 3). The supramolecular triangles pack next to each other to form a planar infinite 2D sheet (
Figure S3a in Supplementary Information). The contacts inside and between the triangles are not of equal length due to the low symmetry of the crystal. While each triangle has the same number and type of contacts, their length varies.
The thione S atoms in
2c–β form bifurcated contacts with the S–S atoms of the dithiazole ring (
Figure 3). These S…S contacts are in the range of 3.071(4)–3.370(4) Å [∠C–S…S, 96.7(4)–136.6(4)°] and are well within the ∑
VDW [3.20 Å (minor), 4.06 Å (major)]. While this type of interaction is also seen in
2c–a, the highly polarizable nature of sulfur allows for the formation of interactions between the exocyclic thione S atom and the more electronegative N and Cl atoms. This type of interaction originates from the positive
σ hole of the S atom. In the C=S bond, some of the electronic charge of the S atom is polarized toward the bond region, leading to a redistribution of electronic density from its outer region (along the extension of the bond) on its equatorial sides [
45]. Therefore, negative electrostatic potential is developed around the sites of the S atom while its outer portion along the C=S bond becomes more positive (
σ hole). This is evident from the MEP of dithiazolethione
2c (
Table 2) where red areas of negative charge density are located on the sides of the S atom and a green area in the outer region of the bond axis. This
σ hole on the thione S atom is potentially responsible for the formation of the S…N and S…Cl interactions. The S…N interactions in
2c–β are formed by the exocyclic thione S atom instead of the endocyclic S–S atoms like in
2c–a. These S…N interactions are in the range of 3.14(1)–3.31(1) Å [∠C–S…N, 163.4(4)–157.8(4)°] and well within the ∑VDW [3.20 Å (minor), 3.63 Å (major)]. The exocyclic thione S atom also formed an interaction with a Cl atom. This S…Cl contact is in the range of 3.315(4)–3.418(4) Å [∠C–S…Cl, 150.3(4–153.4(4)°]. The set of close intermolecular interactions in
2c–β is completed with a Cl…Cl contact of 3.337(4)–3.342(4) Å [∠C–Cl…Cl, 164.3(4)–164.5(4)°] not present in the crystal packing of
2c–a or compounds
2b,
3–5. This interaction might be an outcome of the proximity of the dithiazolethiones.
The 2D sheets formed by the supramolecular triangles, pack parallel to the
ac-diagonal and while well separate from each other (
Figure S3b in Supplementary Information) they are connected via short S…S and S…C contacts of 3.479(4)–3.528(4) Å and 3.42(1)–3.49 (1) Å, respectively.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








