
Synthesis and Evaluation of Novel 1,2,6-Thiadiazinone Kinase Inhibitors as Potent Inhibitors of Solid Tumors

 , , , , and
, , , , and
Abstract
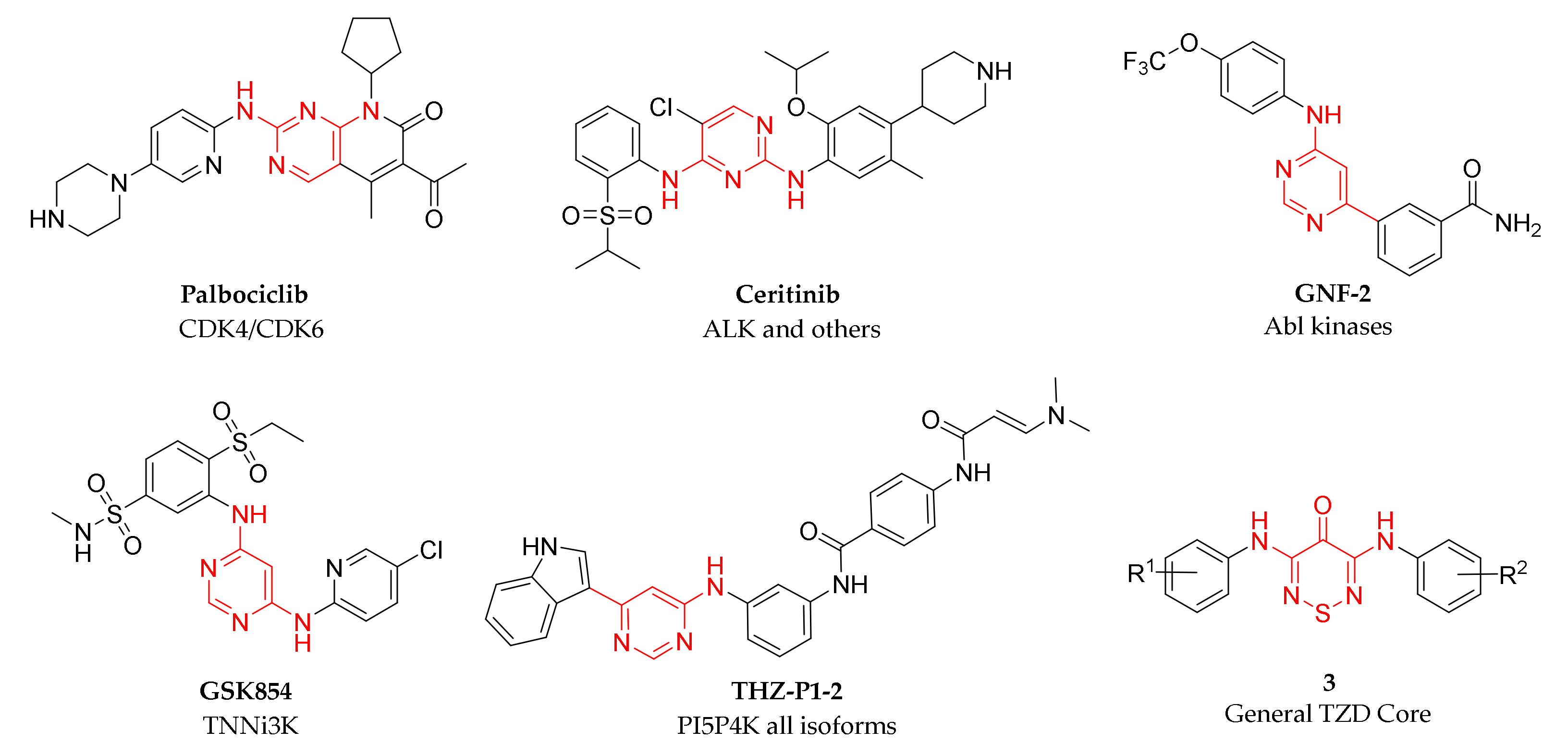
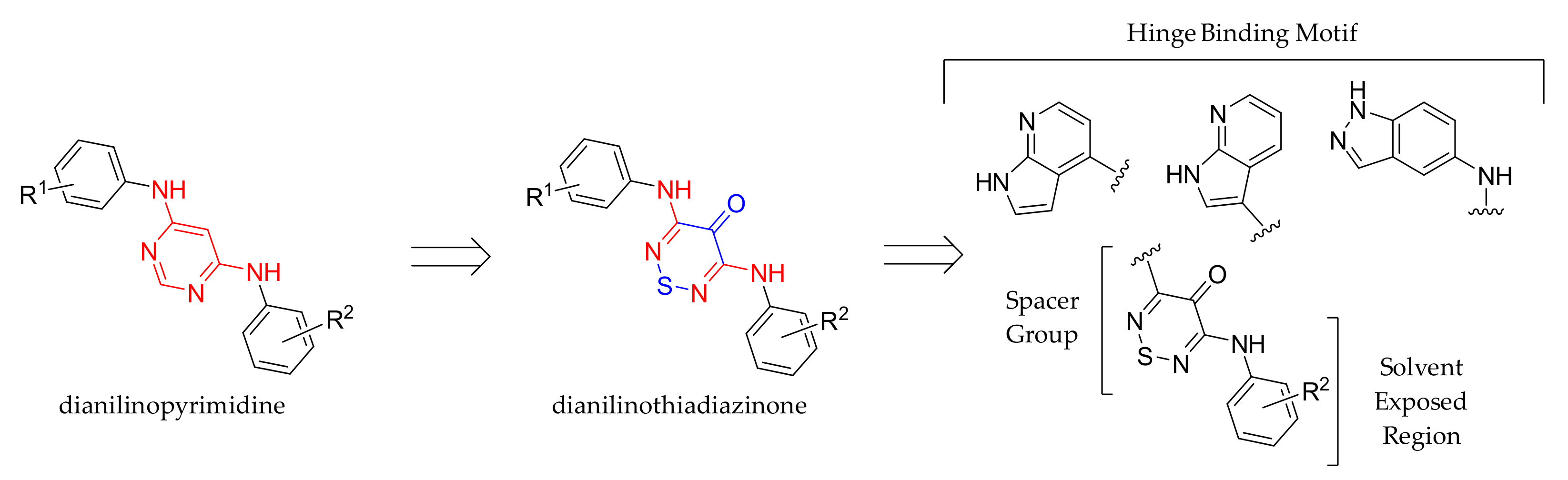
:1. Introduction
2. Results
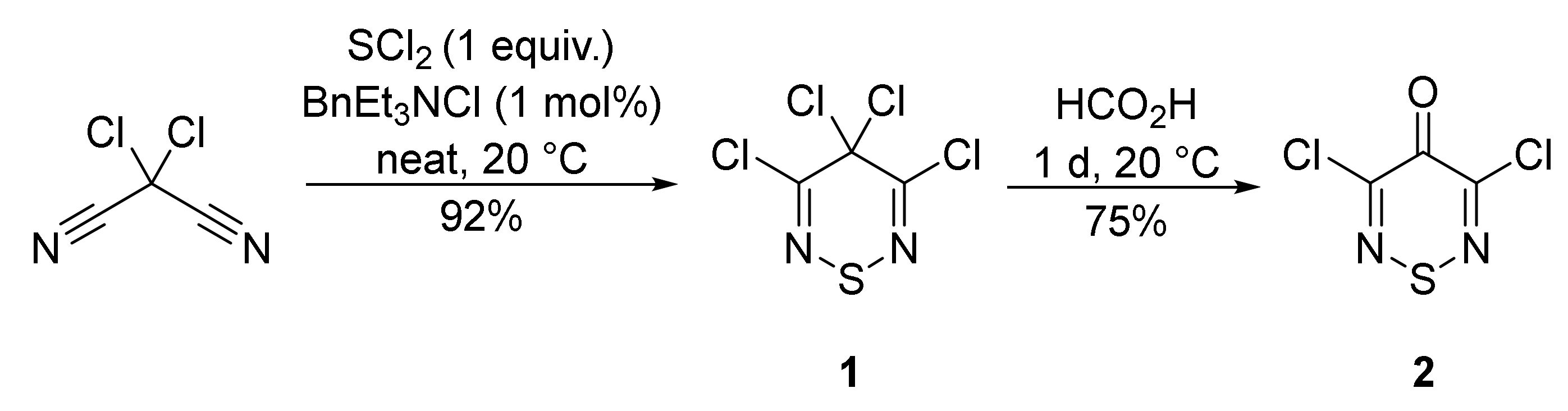
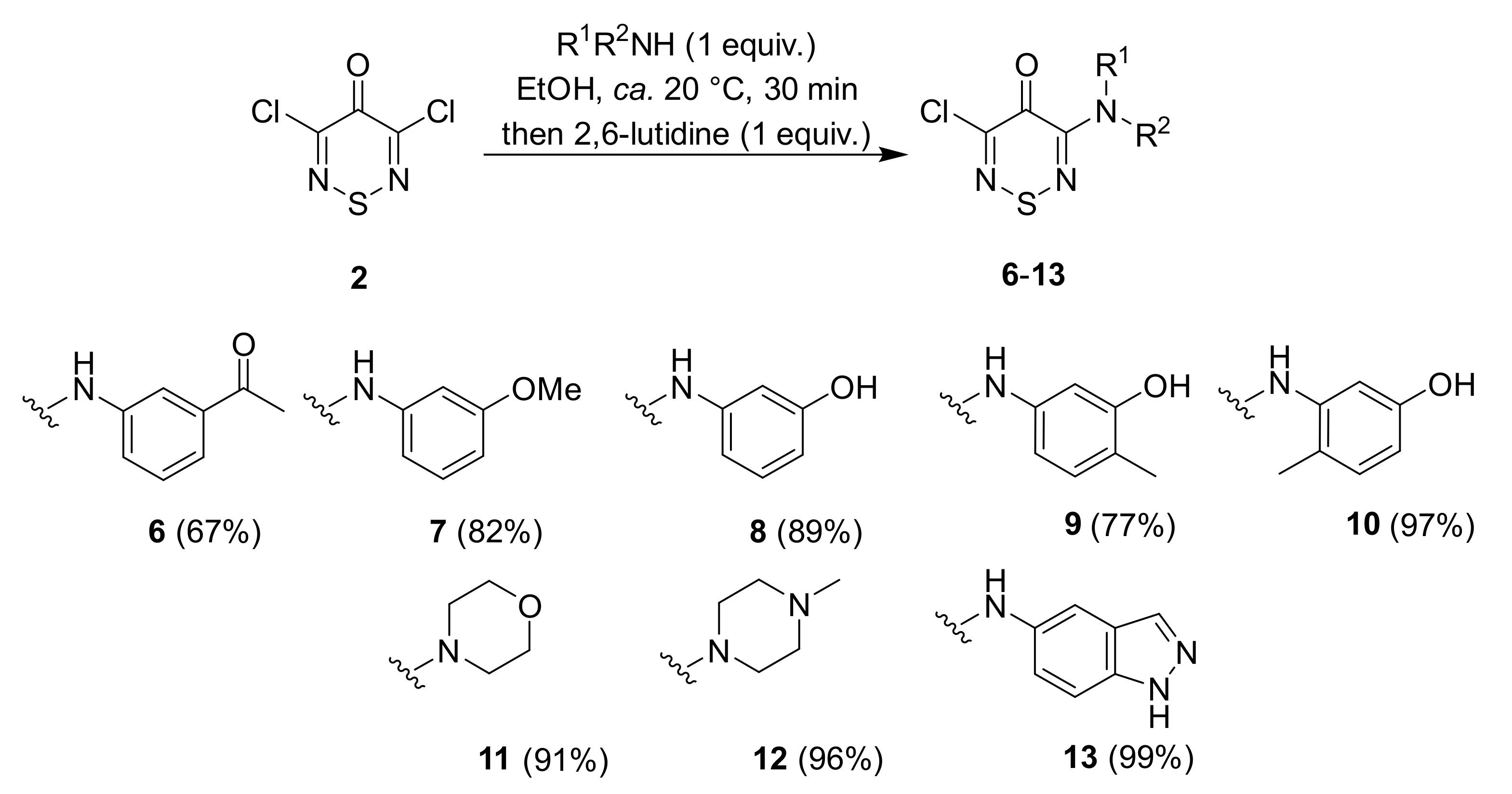
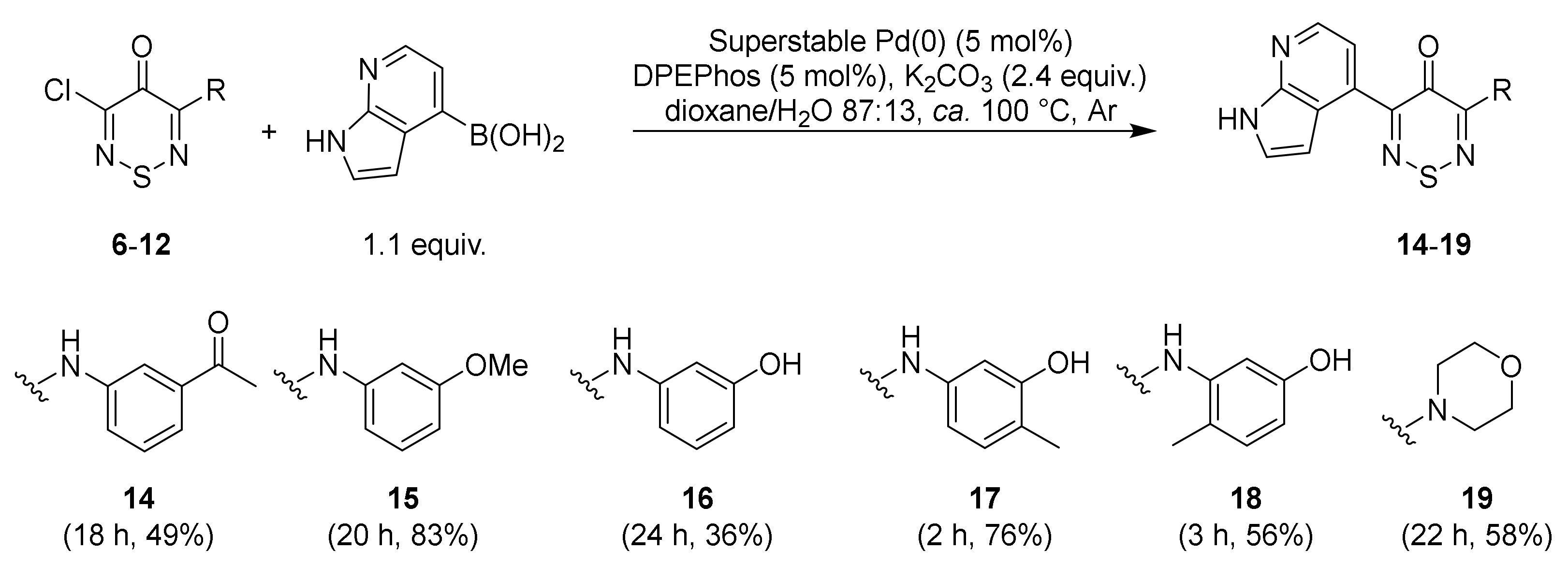
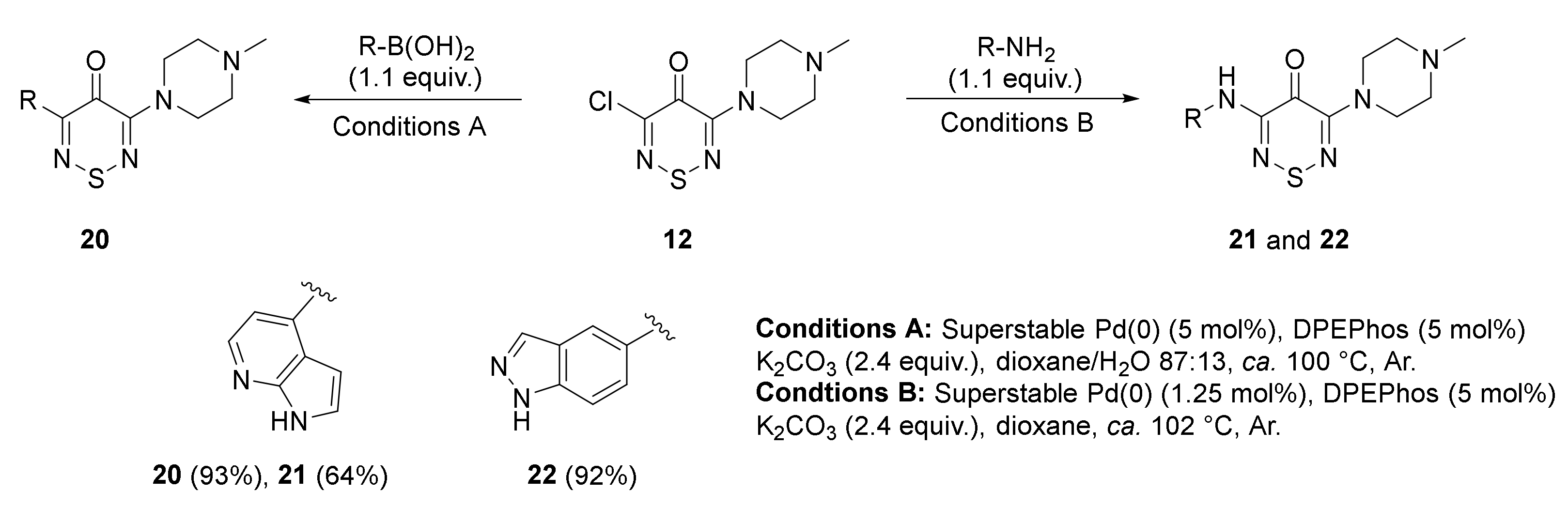
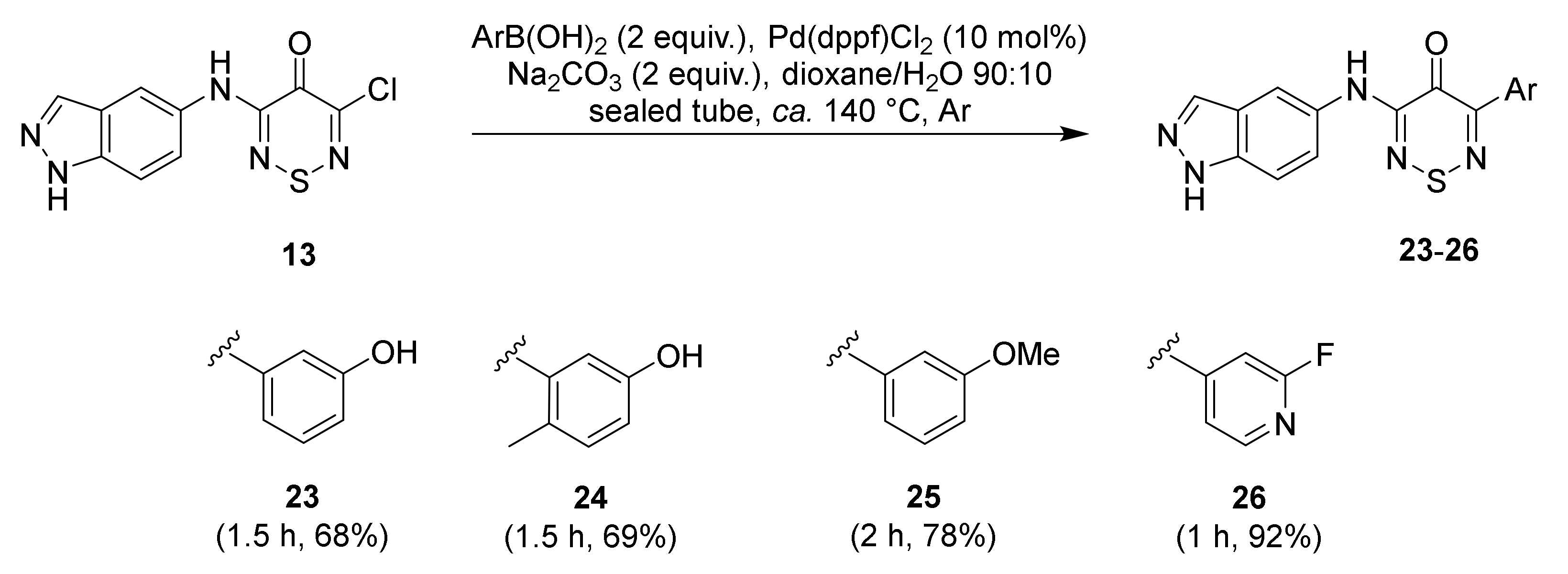
2.1. Synthesis
2.2. Cancer Cell Screening
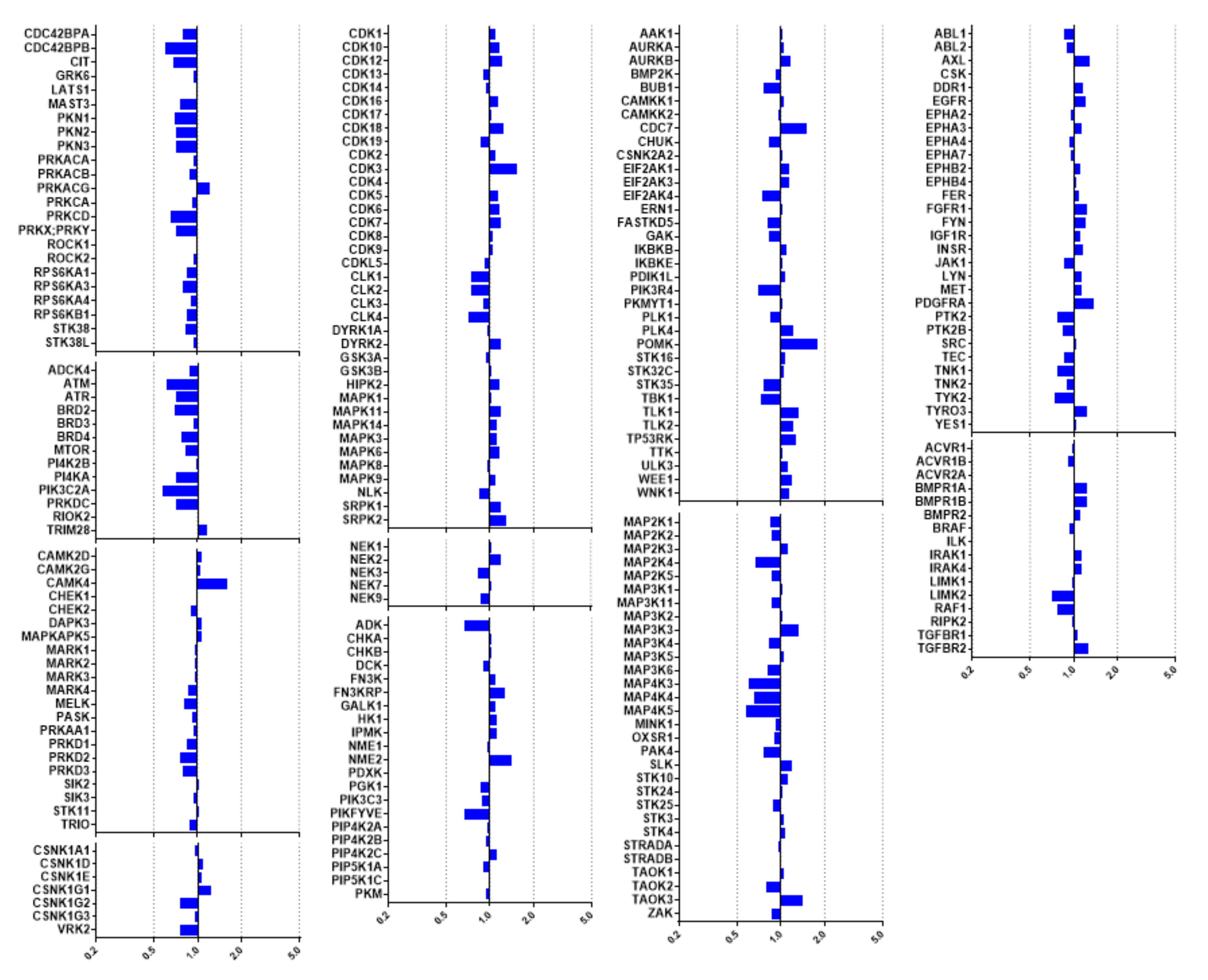
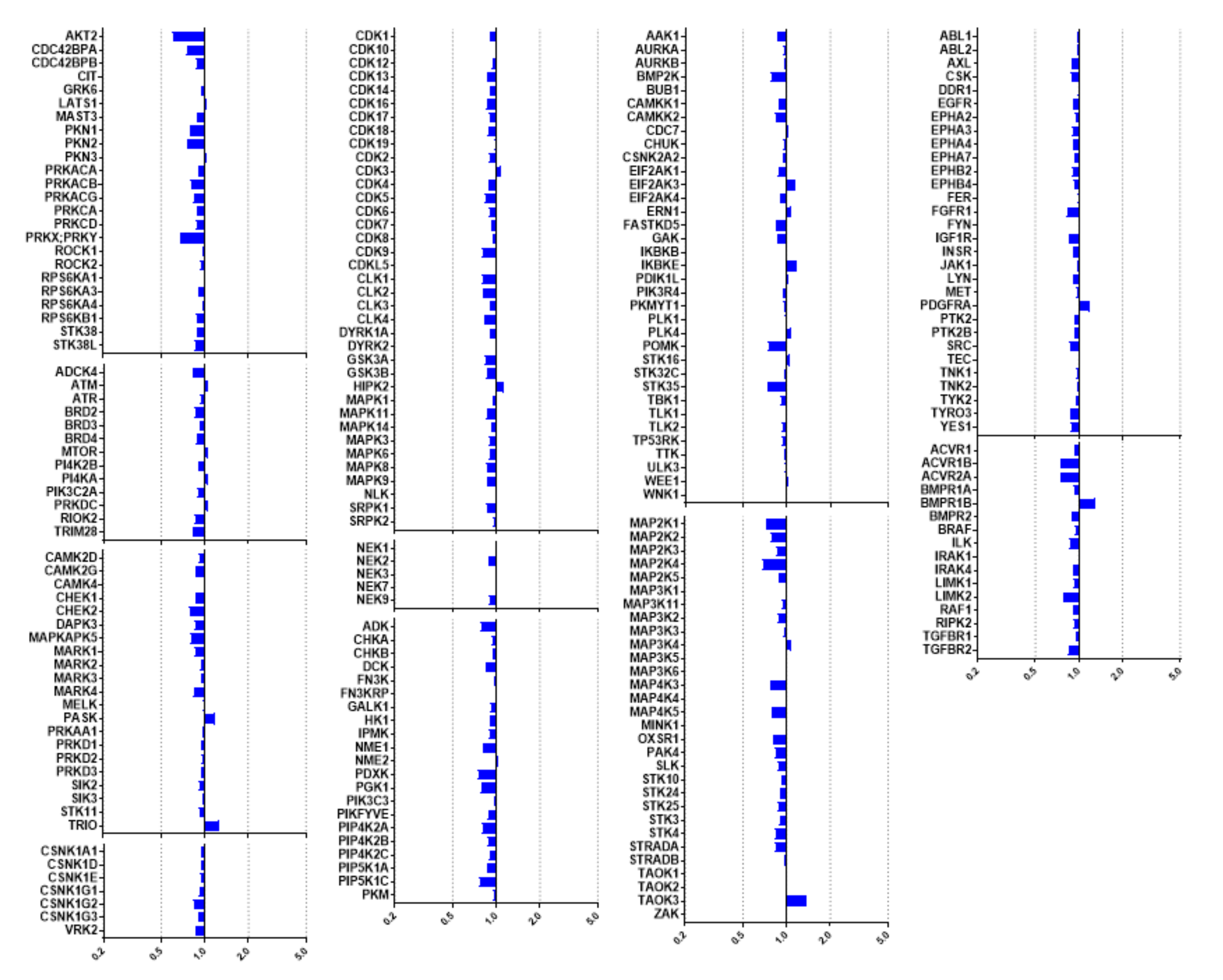
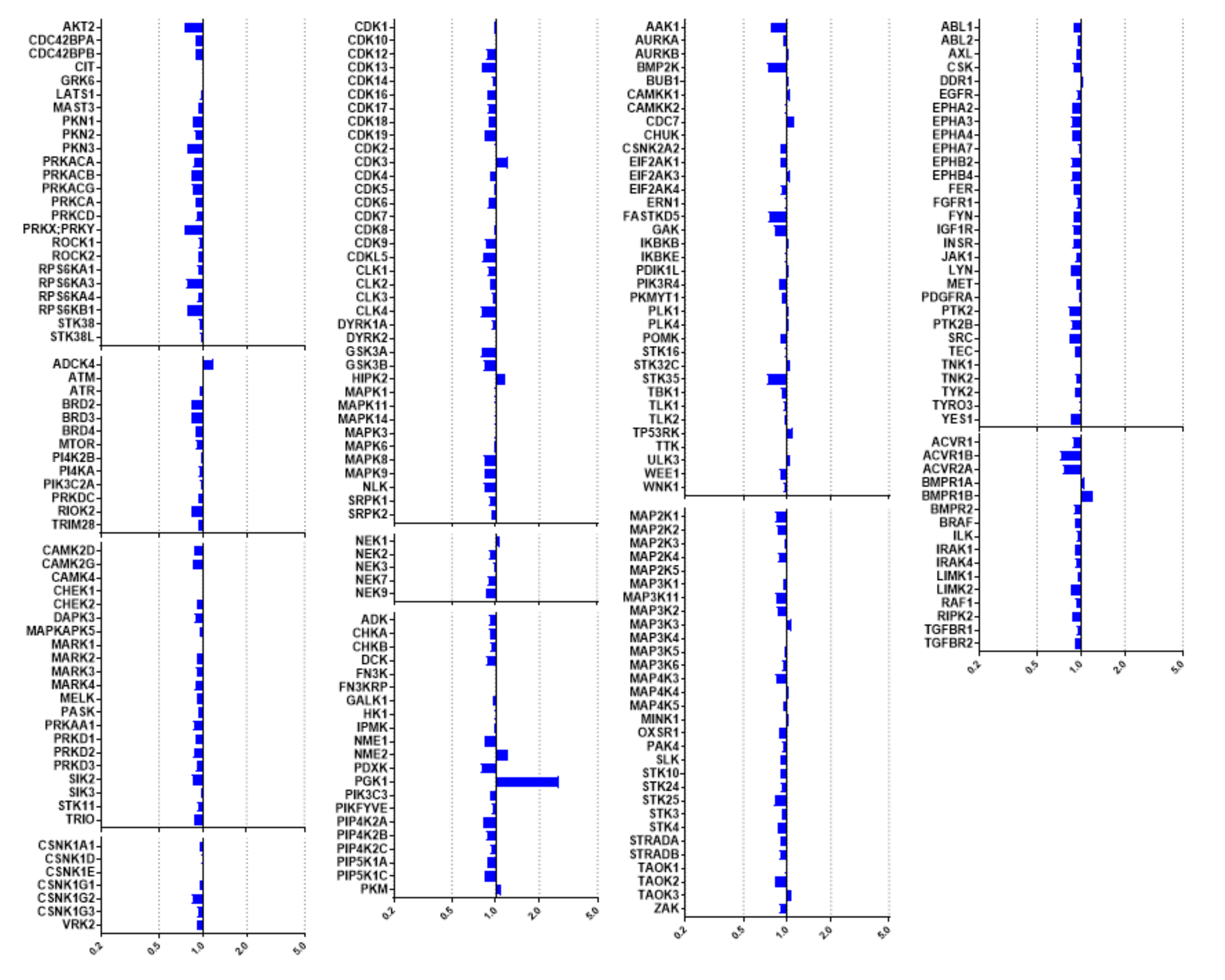
2.3. Kinome Profiling of Thiadiazinones 16, 17, and 26
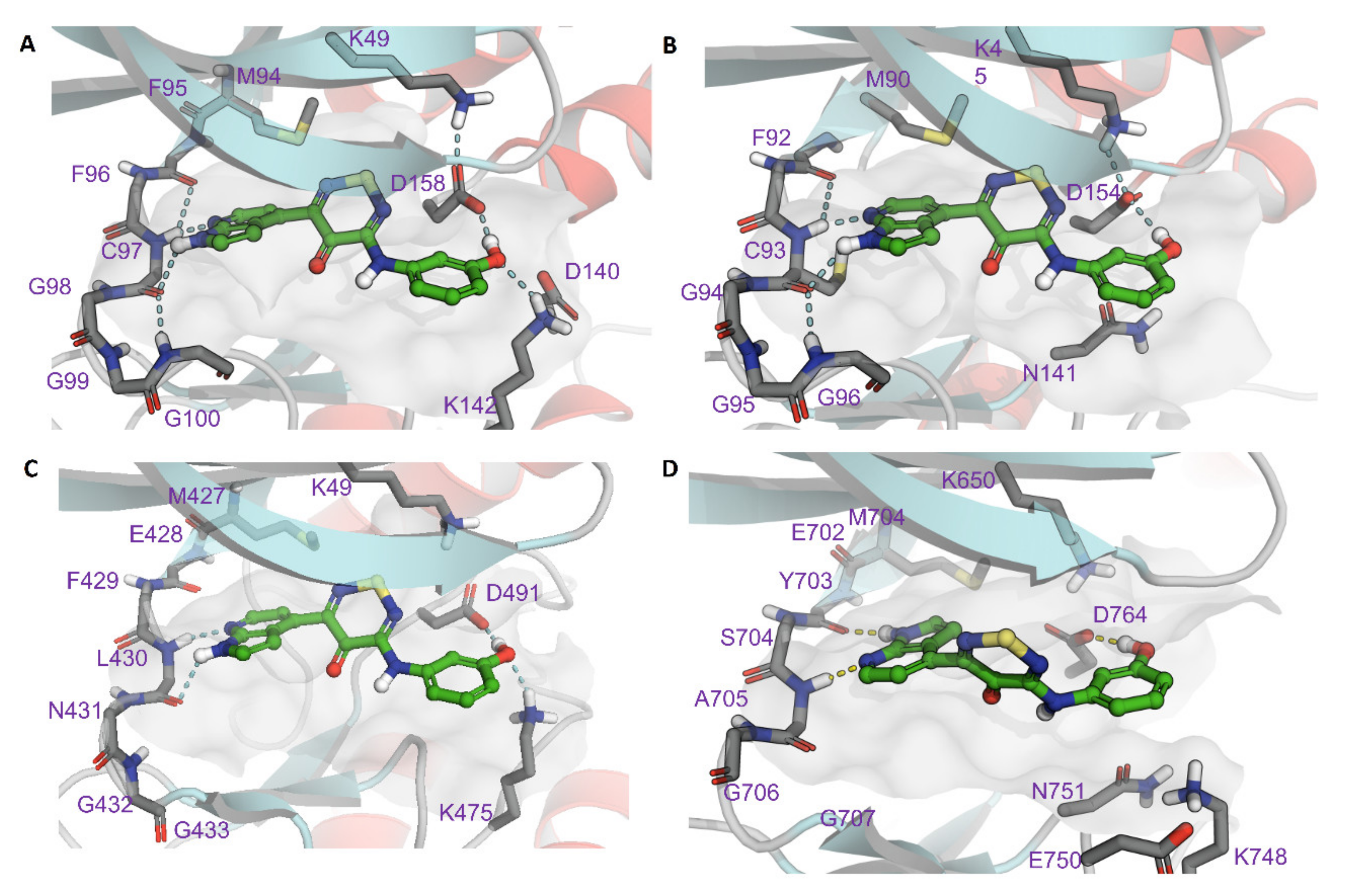
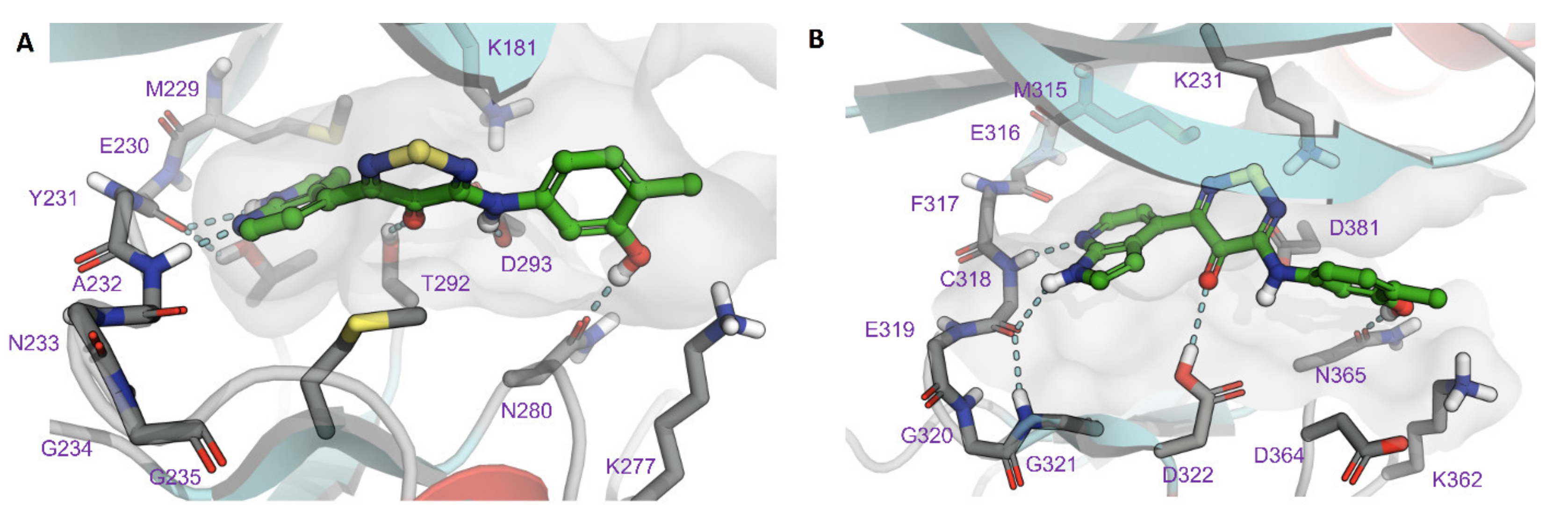
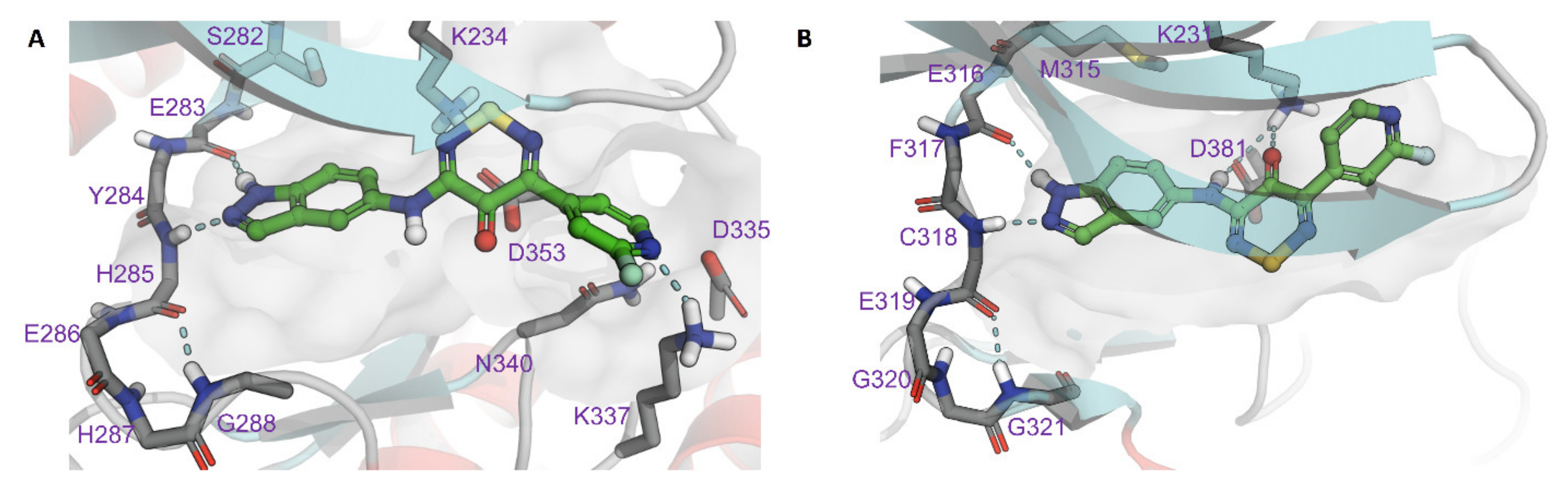
2.4. Modelling of Hits from Kinome Profiling on Thiadiazinones 16, 17, and 26
3. Discussion
4. Materials and Methods
4.1. Cancer Cell Line Screening Panel
4.2. MIBS Profiling Methods
4.3. Molecular Modelling Methods
4.3.1. Ligand Preparation
4.3.2. Protein Preparation
4.3.3. Development of Homology-Based Model
4.3.4. Molecular Docking
4.4. Chemistry Experimental Section
4.4.1. General Methods and Materials
4.4.2. Preparation of 3-Aminosubstituted-4H-1,2,6-thiadiazines
4.4.3. Preparation of 5-Amino-Substituted 3-Arylthiadiazinones
4.4.4. Preparation of 3,5-Diaminosubstituted Thiadiazines
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Boström:, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727. [Google Scholar] [CrossRef]
- Virshup, A.M.; Contreras-García, J.; Wipf, P.; Yang, W.; Beratan, D.N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hou, T. Drug and drug candidate building block analysis. J. Chem. Inf. Model. 2010, 50, 55–67. [Google Scholar] [CrossRef]
- Taylor, R.D.; MacCoss, M.; Lawson, A.D.G. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Lücking, U. Neglected sulfur(vi) pharmacophores in drug discovery: Exploration of novel chemical space by the interplay of drug design and method development. Org. Chem. Front. 2019, 6, 1319–1324. [Google Scholar] [CrossRef] [Green Version]
- Schiesser, S.; Cox, R.J.; Czechtizky, W. The powerful symbiosis between synthetic and medicinal chemistry. Future Med. Chem. 2021, 13, 941–944. [Google Scholar] [CrossRef]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-3-chloro-4H-1,2,6-thiadiazin-4-one antifungal agents. U.S. Patent 4,097,594, 27 June 1978. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-thio-3-chloro-4H-1,2,6-thiadiazin-4-one antifungal agents. U.S. Patent 4,100,281, 1 April 1978. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. 3-Chloro-5-(optionally substituted heterocycloxy)-4H-1,2,6-thiadiazin-4-one antifungal agents. U.S. Patent 4,143,138, 6 March 1979. [Google Scholar]
- Peake, C.J.; Harnish, W.N.; Davidson, B.L. Mono-5-substituted-3-chloro-4H-1,2,6-thiadiazin-4-one antifungal agents. U.S. Patent 4,201,780, 6 May 1980. [Google Scholar]
- Portnoy, R.C. Thiadiazinone plant disease control agents. U.S. Patent 4,497,807, 5 February 1985. [Google Scholar]
- Chochos, C.L.; Kalogirou, A.S.; Ye, T.; Tatsi, E.; Katsouras, A.; Zissimou, G.A.; Gregoriou, V.G.; Avgeropoulos, A.; Koutentis, P.A. 4H-1,2,6-Thiadiazine-containing donor–acceptor conjugated polymers: Synthesis, optoelectronic characterization and their use in organic solar cells. J. Mater. Chem. C 2018, 6, 3658–3667. [Google Scholar] [CrossRef]
- Hermerschmidt, F.; Kalogirou, A.S.; Min, J.; Zissimou, G.A.; Tuladhar, S.M.; Ameri, T.; Faber, H.; Itskos, G.; Choulis, S.A.; Anthopoulos, T.D.; et al. 4H-1,2,6-Thiadiazin-4-one-containing small molecule donors and additive effects on their performance in solution-processed organic solar cells. J. Mater. Chem. C 2015, 3, 2358–2365. [Google Scholar] [CrossRef] [Green Version]
- Gómez, T.; Macho, S.; Miguel, D.; Neo, A.G.; Rodríguez, T.; Torroba, T. Cyclopentathiadiazines, Cyclohepta- and Cyclopentadithiazoles: New Materials and a Rich Heterocyclic Chemistry of Cyclic Enaminonitriles. Eur. J. Org. Chem. 2005, 2005, 5055–5066. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Godoi, P.H.; Couñago, R.M.; Laitinen, T.; Scott, J.W.; Langendorf, C.G.; Oakhill, J.S.; Drewry, D.H.; Zuercher, W.J.; Koutentis, P.A.; et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules 2018, 23, 1221. [Google Scholar] [CrossRef] [Green Version]
- Kristinson, H. Addition of sulfenyl chlorides to the cyanogen bond in activated nitriles. Tetrahedron Lett. 1973, 14, 4489–4490. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. The chemistry of non-S-oxidised 4H-1,2,6-thiadiazines. Targets Heterocycl. Syst. 2018, 22, 82–118. [Google Scholar] [CrossRef]
- Geevers, J.; Trompen, W.P. Synthesis and reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Recl. Trav. Chim. Pays-Bas 1974, 93, 270–272. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Clark, M.J.; Miduturu, C.; Schmidt, A.G.; Zhu, X.; Pitts, J.D.; Wang, J.; Potisopon, S.; Zhang, J.; Wojciechowski, A.; Hann, C.J.J.; et al. GNF-2 Inhibits Dengue Virus by Targeting Abl Kinases and the Viral E Protein. Cell Chem. Biol. 2016, 23, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagnozzi, R.J.; Gatto, G.J.; Kallander, L.S.; Hoffman, N.E.; Mallilankaraman, K.; Ballard, V.L.; Lawhorn, B.G.; Stoy, P.; Philp, J.; Graves, A.P.; et al. Inhibition of the cardiomyocyte-specific kinase TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart. Sci. Transl. Med. 2013, 5, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, J.; Lawhorn, B.G.; Graves, A.P.; Shewchuk, L.; Rivera, K.R.; Jolivette, L.J.; Holt, D.A.; Gatto, G.J.; Kallander, L.S. 4,6-Diaminopyrimidines as Highly Preferred Troponin I-Interacting Kinase (TNNI3K) Inhibitors. J. Med. Chem. 2018, 61, 3076–3088. [Google Scholar] [CrossRef] [PubMed]
- Sivakumaren, S.C.; Shim, H.; Zhang, T.; Ferguson, F.M.; Lundquist, M.R.; Browne, C.M.; Seo, H.S.; Paddock, M.N.; Manz, T.D.; Jiang, B.; et al. Targeting the PI5P4K Lipid Kinase Family in Cancer Using Covalent Inhibitors. Cell Chem. Biol. 2020, 27, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Abdul Azeez, K.R.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.P.; Roth, B.L.; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Drewry, D.H.; Wells, C.I.; Andrews, D.M.; Angell, R.; Al-Ali, H.; Axtman, A.D.; Capuzzi, S.J.; Elkins, J.M.; Ettmayer, P.; Frederiksen, M.; et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS ONE 2017, 12, e0181585. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Palladium Catalyzed C–C Coupling Reactions of 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one. Org. Lett. 2011, 13, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, H.A.; Koutentis, P.A. Synthesis of asymmetric 3,5-diaryl-4H-1,2,6-thiadiazin-4-ones via Suzuki–Miyaura and Stille coupling reactions. Tetrahedron 2012, 68, 7380–7385. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. Pd-catalyzed C-N Coupling of Primary (Het)arylamines with 5-Substituted 3-Chloro-4H-1,2,6-thiadiazin-4-ones. Tetrahedron Lett. 2018, 59, 2653–2656. [Google Scholar] [CrossRef]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bio. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef]
- Dubinina, G.G.; Chupryna, O.O.; Platonov, M.O.; Borisko, P.O.; Ostrovska, G.V.; Tolmachov, A.O.; Shtil, A.A. In silico design of protein kinase inhibitors: Successes and failures. Anticancer Agents Med. Chem. 2007, 7, 171–188. [Google Scholar] [CrossRef]
- Mukherjee, P.; Bentzien, J.; Bosanac, T.; Mao, W.; Burke, M.; Muegge, I. Kinase crystal miner: A powerful approach to repurposing 3D hinge binding fragments and its application to finding novel bruton tyrosine kinase inhibitors. J. Chem. Inf. Model. 2017, 57, 2152–2160. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Fleck, N.; Torrice, C.D.; Crona, D.J.; Grundner, C.; Zuercher, W.J. Anti-tubercular activity of novel 4-anilinoquinolines and 4-anilinoquinazolines. Bioorg. Med. Chem. Lett. 2019, 29, 2695–2699. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Maffuid, K.A.; Laitinen, T.; Torrice, C.D.; Tizzard, G.J.; Crona, D.J.; Zuercher, W.J. Targeting an EGFR Water Network with 4-Anilinoquin(az)oline Inhibitors for Chordoma. ChemMedChem 2019, 14, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Maffuid, K.A.; Koyioni, M.; Torrice, C.D.; Murphy, W.A.; Mewada, H.K.; Koutentis, P.A.; Crona, D.J.; Asquith, C.R.M. Design and evaluation of 1,2,3-dithiazoles and fused 1,2,4-dithiazines as anti-cancer agents. Bioorganic Med. Chem. Lett. 2021, 43, 128078. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Naegeli, K.M.; East, M.P.; Laitinen, T.; Havener, T.M.; Wells, C.I.; Johnson, G.L.; Drewry, D.H.; Zuercher, W.J.; Morris, D.C. Design of a Cyclin G Associated Kinase (GAK)/Epidermal Growth Factor Receptor (EGFR) Inhibitor Set to Interrogate the Relationship of EGFR and GAK in Chordoma. J. Med. Chem. 2019, 62, 4772–4778. [Google Scholar] [CrossRef]
- Gillet, J.; Varma, S.; Gottesman, M.M.J. The clinical relevance of cancer cell lines. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Zhao, F.; Song, H.; Guo, J.; Li, X.; Jiang, X.; Huan, R.; Song, S.; Zhang, Q.; Wang, R.; et al. Structure-based design of 6-chloro-4-aminoquinazoline-2-carboxamide derivatives as potent and selective p21-activated kinase 4 (PAK4) inhibitors. J. Med. Chem. 2018, 61, 265–285. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Tester, R.W.; Luedtke, G.R.; Chakravarty, S.; Mavunkel, B.J.; Perumattam, J.J.; Lu, Q.; Nashashibi, I.; Jung, J.; Hu, J.; et al. Design and synthesis of piperazine-indole p38 alpha MAP kinase inhibitors with improved pharmacokinetic profiles. Bioorg. Med. Chem. Lett. 2010, 20, 828–831. [Google Scholar] [CrossRef]
- Safina, B.S.; Baker, S.; Baumgardner, M.; Blaney, P.M.; Chan, B.K.; Chen, Y.-H.; Cartwright, M.W.; Castanedo, G.; Chabot, C.; Cheguillaume, A.J.; et al. Discovery of novel PI3-kinase δ specific inhibitors for the treatment of rheumatoid arthritis: Taming CYP3A4 time-dependent inhibition. J. Med. Chem. 2012, 55, 5887–5900. [Google Scholar] [CrossRef]
- Talele, T.T. Natural-Products-Inspired Use of the gem-Dimethyl Group in Medicinal Chemistry. J. Med. Chem. 2018, 61, 2166–2210. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Asquith, C.R.M.; Koutentis, P.A. Synthesis of (R) and (S)-3-Chloro-5-(2,4-dimethylpiperazin-1-yl)-4H-1,2,6-thiadiazin-4-ones. Molbank 2020, 2020, M1139. [Google Scholar] [CrossRef]
- Crawford, T.D.; Ndubaku, C.O.; Chen, H.; Boggs, J.W.; Bravo, B.J.; DeLaTorre, K.; Giannetti, A.M.; Gould, S.E.; Harris, S.F.; Magnuson, S.R.; et al. Discovery of selective 4-amino-pyridopyrimidine inhibitors of MAP4K4 using fragment-based lead identification and optimization. J. Med. Chem. 2014, 57, 3484–3493. [Google Scholar] [CrossRef] [PubMed]
- Strang, B.L.; Asquith, C.R.M.; Moshrif, H.F.; Ho, C.M.K.; Zuercher, W.J.; Al-Ali, H. Identification of lead anti-human cytomegalovirus compounds targeting MAP4K4 via machine learning analysis of kinase inhibitor screening data. PLoS ONE 2018, 13, e0201321. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Godoi, P.H.; East, M.P.; Tizzard, G.H.; Graves, L.M.; Johnson, G.L.; Dornsife, R.E.; Wells, C.I.; et al. Identification and optimization of 4-anilinoquinolines as inhibitors of cyclin G associated kinase. ChemMedChem 2018, 13, 48–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Tung, R.M.; Blenis, J. A novel human SPS1/STE20 homologue, KHS, activates Jun N-terminal kinase. Oncogene 1997, 14, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Diener, K.; Wang, X.S.; Chen, C.; Meyer, C.F.; Keesler, G.; Zukowski, M.; Tan, T.H.; Yao, Z. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 9687–9692. [Google Scholar] [CrossRef] [Green Version]
- Duquesnes, N.; Lezoualc’h, F.; Crozatier, B. PKC-delta and PKC-epsilon: Foes of the same family or strangers? J. Mol. Cell Cardiol. 2011, 51, 665–673. [Google Scholar] [CrossRef]
- Watanabe, G.; Saito, Y.; Madaule, P.; Ishizaki, T.; Fujisawa, K.; Morii, N.; Mukai, H.; Ono, Y.; Kakizuka, A.; Narumiya, S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 1996, 271, 645–648. [Google Scholar] [CrossRef]
- Small-Molecule Drug Discovery Suite 2020-4; Schrödinger, LLC: New York, NY, USA, 2020.
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [Green Version]
- Goyal, P.; Behring, A.; Kumar, A.; Siess, W. Identifying and characterizing a novel protein kinase STK35L1 and deciphering its orthologs and close-homologs in vertebrates. PLoS ONE. 2009, 4, e6981. [Google Scholar] [CrossRef] [Green Version]
- Cárcamo, J.; Weis, F.M.; Ventura, F.; Wieser, R.; Wrana, J.L.; Attisano, L.; Massagué, J. Type I receptors specify growth-inhibitory and transcriptional responses to transforming growth factor beta and activin. Mol. Cell Biol. 1994, 14, 3810–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogirou, A.S.; Koutentis, P.A. A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride. Molecules 2015, 20, 14576–14594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, in press. [Google Scholar] [CrossRef]
- Cohen, P.; Alessi, D.R. Kinase drug discovery-what’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorov, O.; Müller, S.; Knapp, S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010, 6, 166–169. [Google Scholar] [CrossRef]
- Rudolf, A.F.; Skovgaard, T.; Knapp, S.; Jensen, L.J.; Berthelsen, J. A comparison of protein kinases inhibitor screening methods using both enzymatic activity and binding affinity determination. PLoS ONE 2014, 9, e98800. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Deacon, S.; Horiuchi, K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin. Drug Discov. 2008, 3, 607–621. [Google Scholar] [CrossRef]
- Elwaie, T.A.; Abbas, S.E.; Aly, E.I.; George, R.F.; Ali, H.; Kraiouchkine, N.; Abdelwahed, K.S.; Fandy, T.E.; El Sayed, K.A.; Elmageed, Z.Y.A.; et al. HER2 kinase-targeted breast cancer therapy: Design, synthesis, and in vitro and in vivo evaluation of novel lapatinib congeners as selective and potent HER2 inhibitors with favorable metabolic stability. J. Med. Chem. 2020, 63, 15906–15945. [Google Scholar] [CrossRef]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Mehdi, O.; Françoise, S.; Sofia, C.L.; Urs, G.; Kevin, Z.; Bernard, S.; Igor, S.; Anabela, C.; Dominique, L.; Eric, M.; et al. HDAC gene expression in pancreatic tumor cell lines following treatment with the HDAC inhibitors panobinostat (LBH589) and trichostatine (TSA). Pancreatology 2012, 12, 146–155. [Google Scholar] [CrossRef]
- Mabuchi, M.; Ueda, M.; Yoshida, Y.; Horiike, K.; Yamaoka, K.; Nakao, S.; Shimizu, T.; Ueda, Y.; Tsujikawa, K.; Tanaka, A. Systematic trial for evaluating docetaxel in a human prostate cancer cell DU145 xenograft model. Anticancer Res. 2017, 37, 1665–1676. [Google Scholar] [CrossRef]
- Cha, M.Y.; Lee, K.; Kim, J.W.; Lee, C.G.; Song, J.Y.; Kim, Y.H.; Lee, G.S.; Park, S.B.; Kim, M.S. Discovery of a novel Her-1/Her-2 dual tyrosine kinase inhibitor for the treatment of Her-1 selective inhibitor-resistant non-small cell lung cancer. J. Med. Chem. 2009, 52, 6880–6888. [Google Scholar] [CrossRef]
- Collins, K.A.L.; Stuhlmiller, T.; Zawistowski, J.J.S.; East, M.P.; Pham, T.T.; Hall, C.R.; Goulet, D.R.; Bevill, S.M.; Angus, S.P.; Velarde, S.H.; et al. Proteomic analysis defines kinase taxonomies specific for subtypes of breast cancer. Oncotarget 2018, 9, 15480–15497. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, D.; Rushe, M.M.; Arduini, R.; Lukacs, C.; Atkins, K.; Sun, X.; Little, K.; Cullivan, M.; Paramasivam, M.; Patterson, T.A.; et al. Germinal-center kinase-like kinase co-crystal structure reveals a swapped activation loop and C-terminal extension. Protein Sci. 2017, 26, 152–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, P.; Delker, S.; Pagarigan, B.; Mahmoudi, A.; Jackson, P.; Abbasian, M.; Muir, J.; Raheja, N.; Cathers, B. Crystal structures of PRK1 in complex with the clinical compounds lestaurtinib and tofacitinib reveal ligand induced conformational changes. PLoS ONE 2014, 9, e103638. [Google Scholar] [CrossRef] [PubMed]
- McHardy, T.; Caldwell, J.J.; Cheung, K.M.; Hunter, L.J.; Taylor, K.; Rowlands, M.; Ruddle, R.; Henley, A.; de Haven, B.A.; Valenti, M.; et al. Discovery of 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as selective, orally active inhibitors of protein kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Tebben, A.J.; Ruzanov, M.; Gao, M.; Xie, D.; Kiefer, S.E.; Yan, C.; Newitt, J.A.; Zhang, L.; Kim, K.; Lu, H.; et al. Crystal structures of apo and inhibitor-bound TGFβR2 kinase domain: Insights into TGFβR isoform selectivity. Acta Crystallogr. D Struct. Biol. 2016, 72, 658–674. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Harwood, L.M. “Dry-Column” Flash Chromatography. Aldrichimica Acta 1985, 18, 25. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Ar1 | Ar2 | 5637 | DU145 | PANC1 | MCF7 | UCH1 | UCH2 | A431 | WS-1 |
| IC50 (μM) a | ||||||||||
| 14 |  |  | 15 | >100 | >100 | >100 | 58 | >100 | >100 | 92 |
| 15 |  | 15 | >100 | >100 | >100 | 67 | >100 | >100 | >100 | |
| 16 |  | 1.6 | 41 | 33 | 18 | 46 | >100 | >100 | >100 | |
| 17 |  | 2.1 | 23 | >100 | 14 | >100 | >100 | >100 | >100 | |
| 18 |  | 55 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | |
| 19 |  | >100 | 14 | 11 | 46 | >100 | >100 | >100 | >100 | |
| 20 |  | >100 | >100 | >100 | >100 | nt | nt | nt | nt | |
| 21 |  | >100 | 51 | >100 | >100 | 97 | >100 | >100 | >100 | |
| 22 |  | >100 | 50 | >100 | 25 | >100 | >100 | >100 | 24 | |
| 23 |  | >100 | 84 | >100 | 92 | 71 | >100 | >100 | >100 | |
| 24 |  | >100 | 15 | 26 | 13 | 67 | >100 | >100 | >100 | |
| 25 |  | 13 | >100 | >100 | 38 | 52 | 99 | >100 | 71 | |
| 26 |  | 8.4 | 5.7 | >100 | 50 | 84 | >100 | >100 | >100 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalogirou, A.S.; East, M.P.; Laitinen, T.; Torrice, C.D.; Maffuid, K.A.; Drewry, D.H.; Koutentis, P.A.; Johnson, G.L.; Crona, D.J.; Asquith, C.R.M. Synthesis and Evaluation of Novel 1,2,6-Thiadiazinone Kinase Inhibitors as Potent Inhibitors of Solid Tumors. Molecules 2021, 26, 5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195911
Kalogirou AS, East MP, Laitinen T, Torrice CD, Maffuid KA, Drewry DH, Koutentis PA, Johnson GL, Crona DJ, Asquith CRM. Synthesis and Evaluation of Novel 1,2,6-Thiadiazinone Kinase Inhibitors as Potent Inhibitors of Solid Tumors. Molecules. 2021; 26(19):5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195911
Chicago/Turabian StyleKalogirou, Andreas S., Michael P. East, Tuomo Laitinen, Chad D. Torrice, Kaitlyn A. Maffuid, David H. Drewry, Panayiotis A. Koutentis, Gary L. Johnson, Daniel J. Crona, and Christopher R. M. Asquith. 2021. "Synthesis and Evaluation of Novel 1,2,6-Thiadiazinone Kinase Inhibitors as Potent Inhibitors of Solid Tumors" Molecules 26, no. 19: 5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195911