
Convertible and Constrained Nucleotides: The 2’-Deoxyribose 5’-C-Functionalization Approach, a French Touch


Abstract
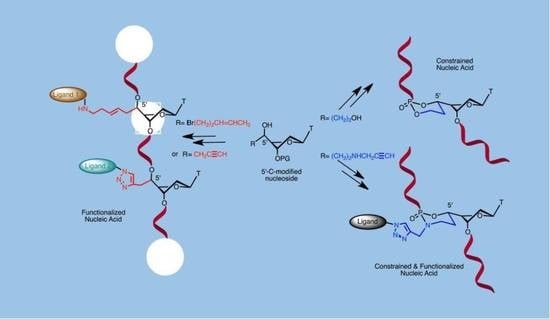
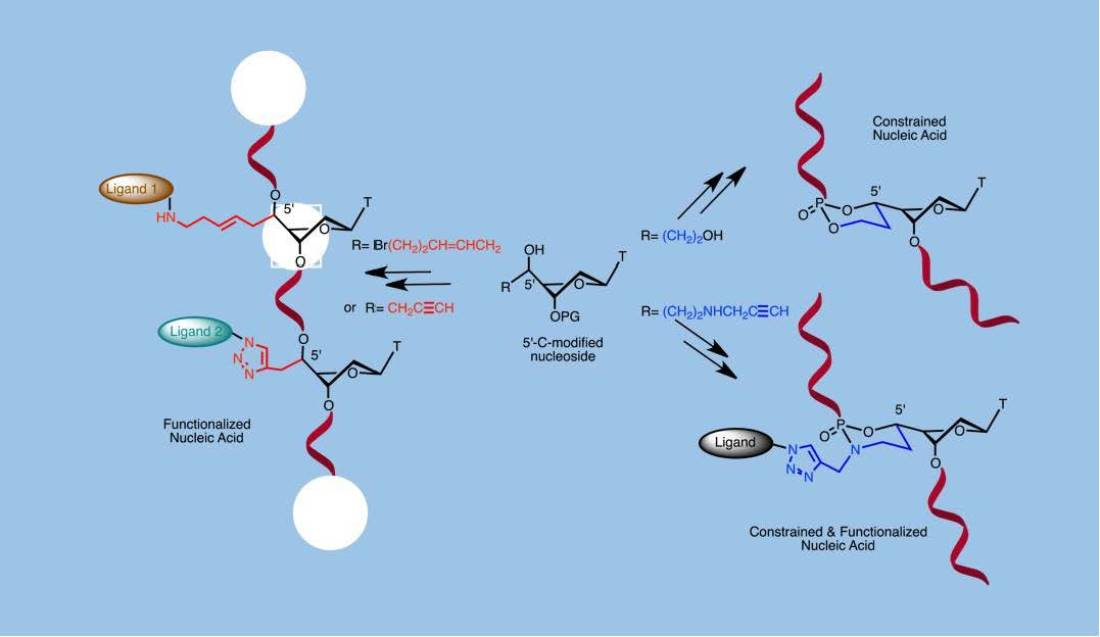
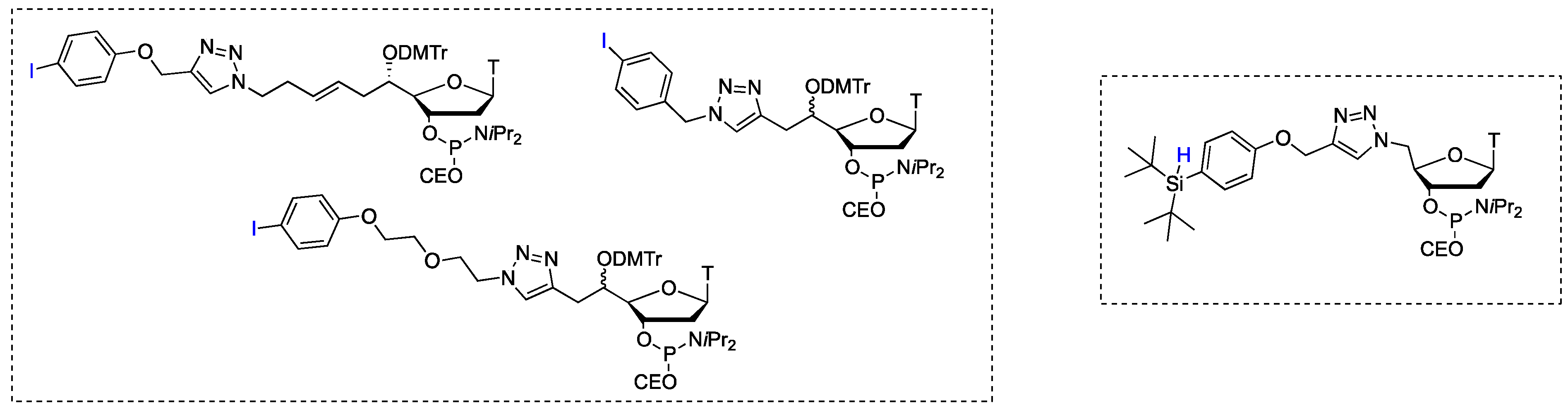
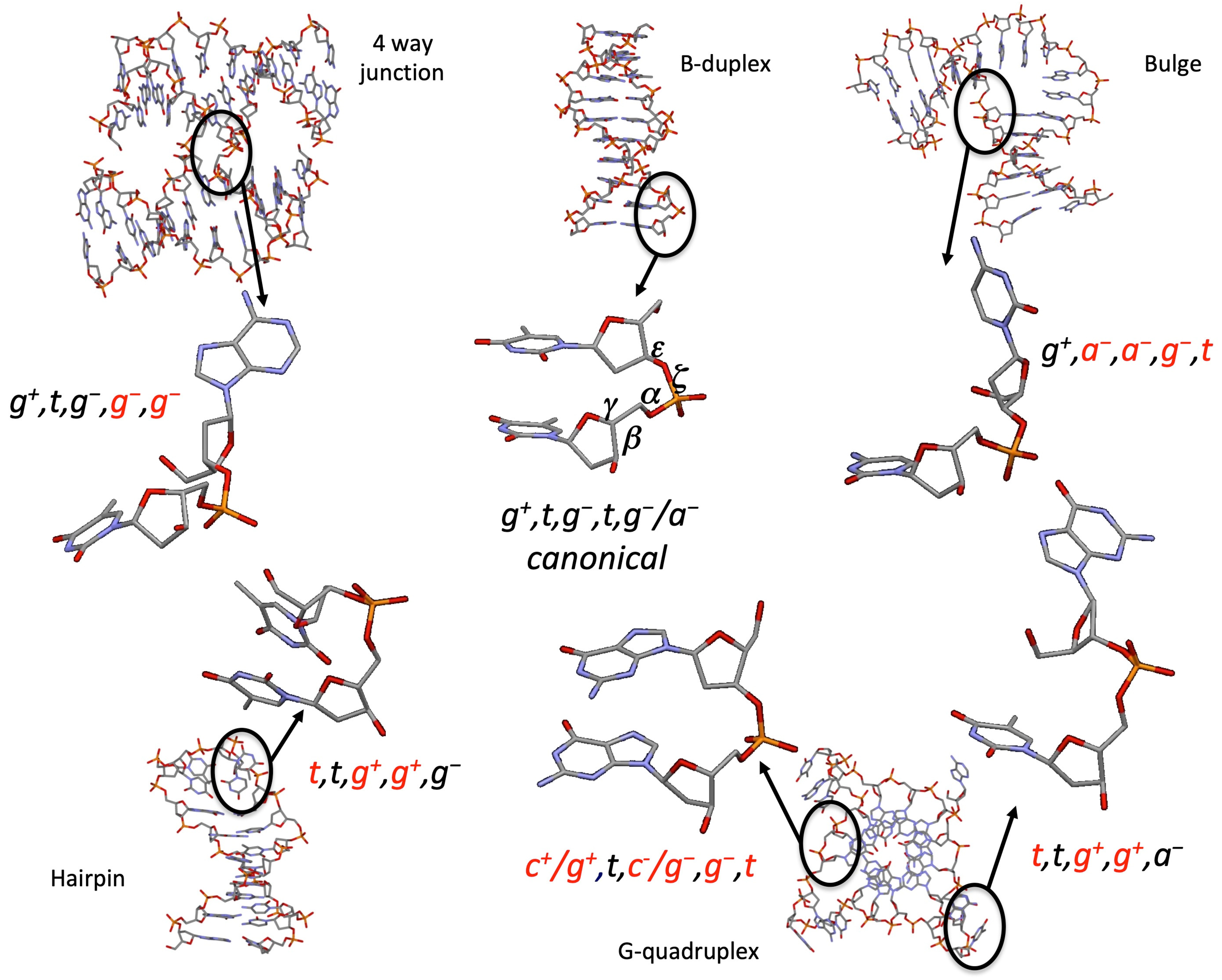
:
{kind=link}
{kind=link}
{kind=link}
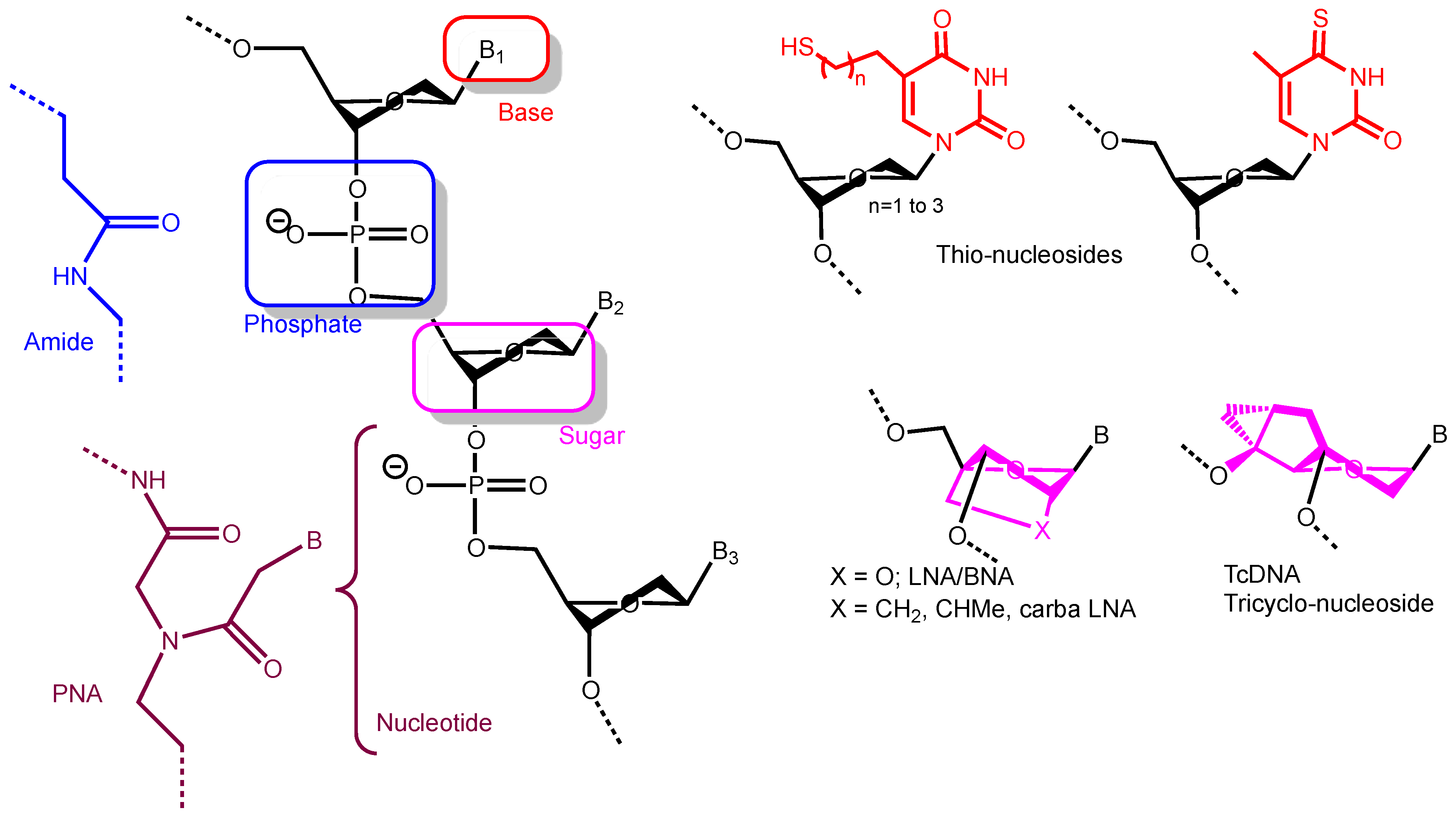{kind=link}
{kind=link}
{kind=link}
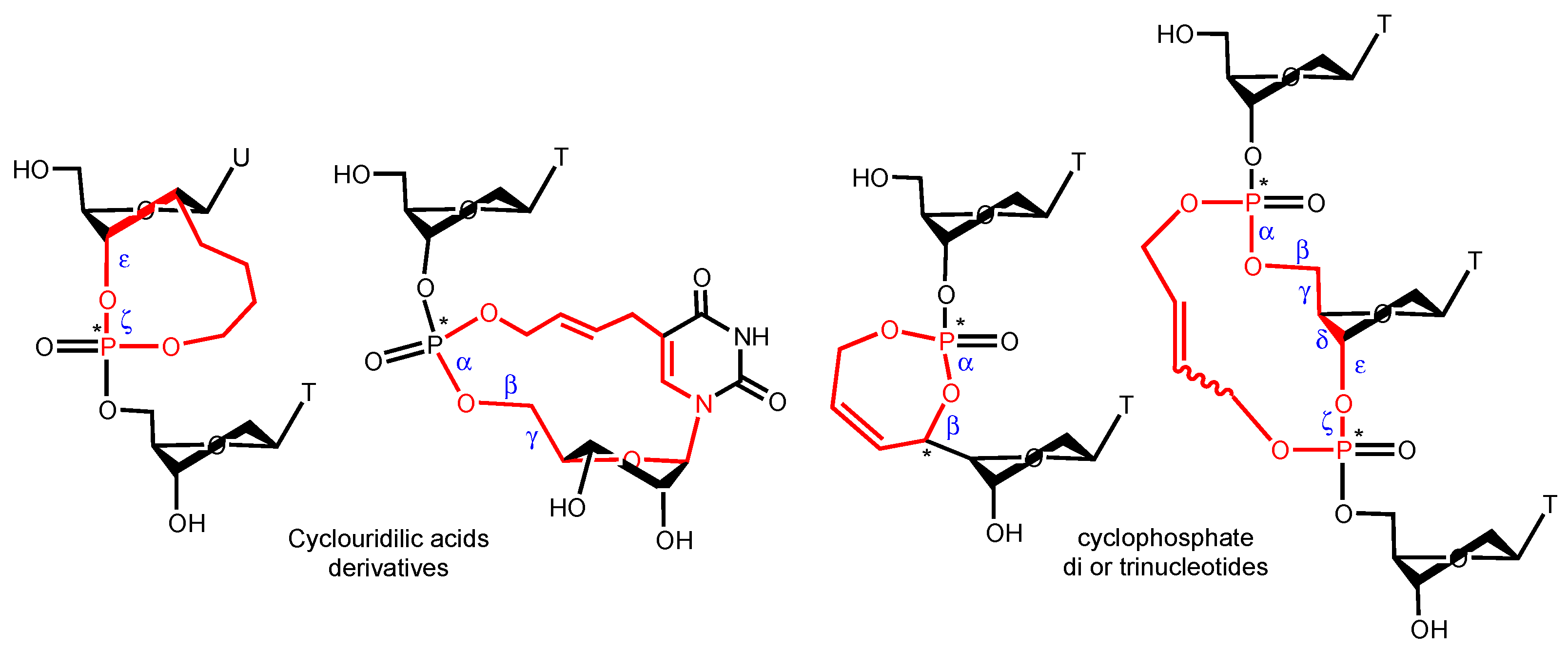
{kind=link}
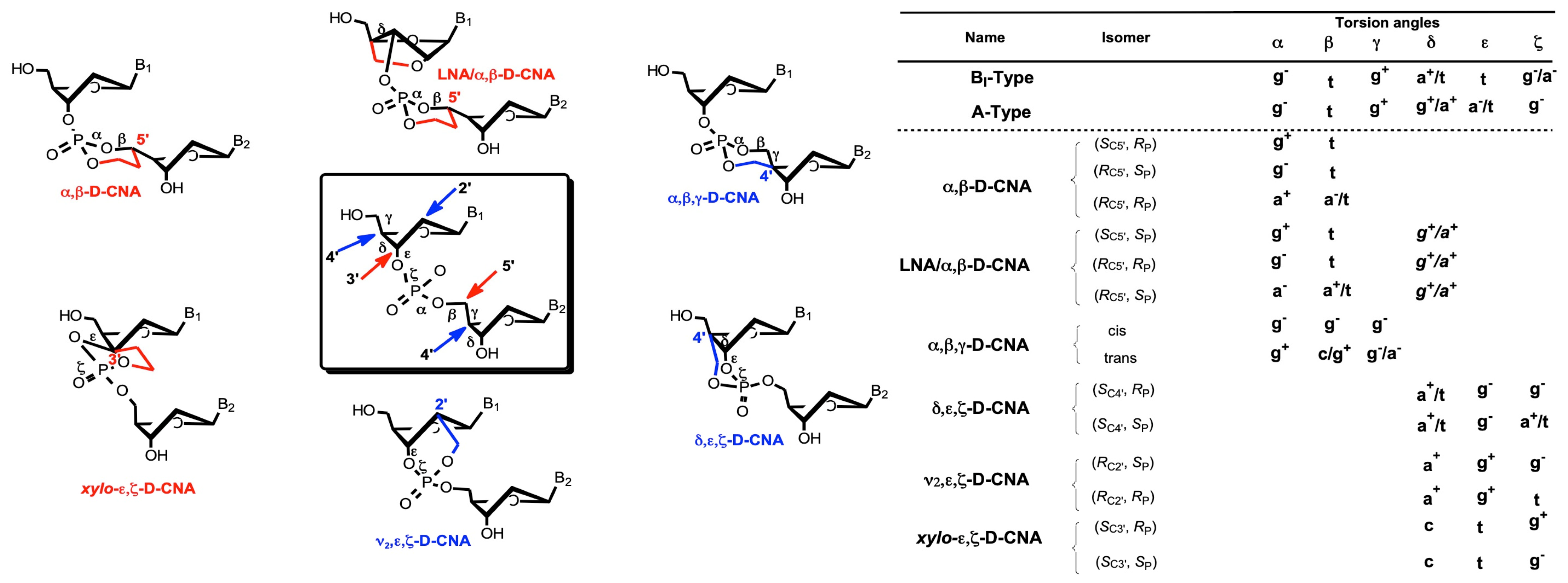
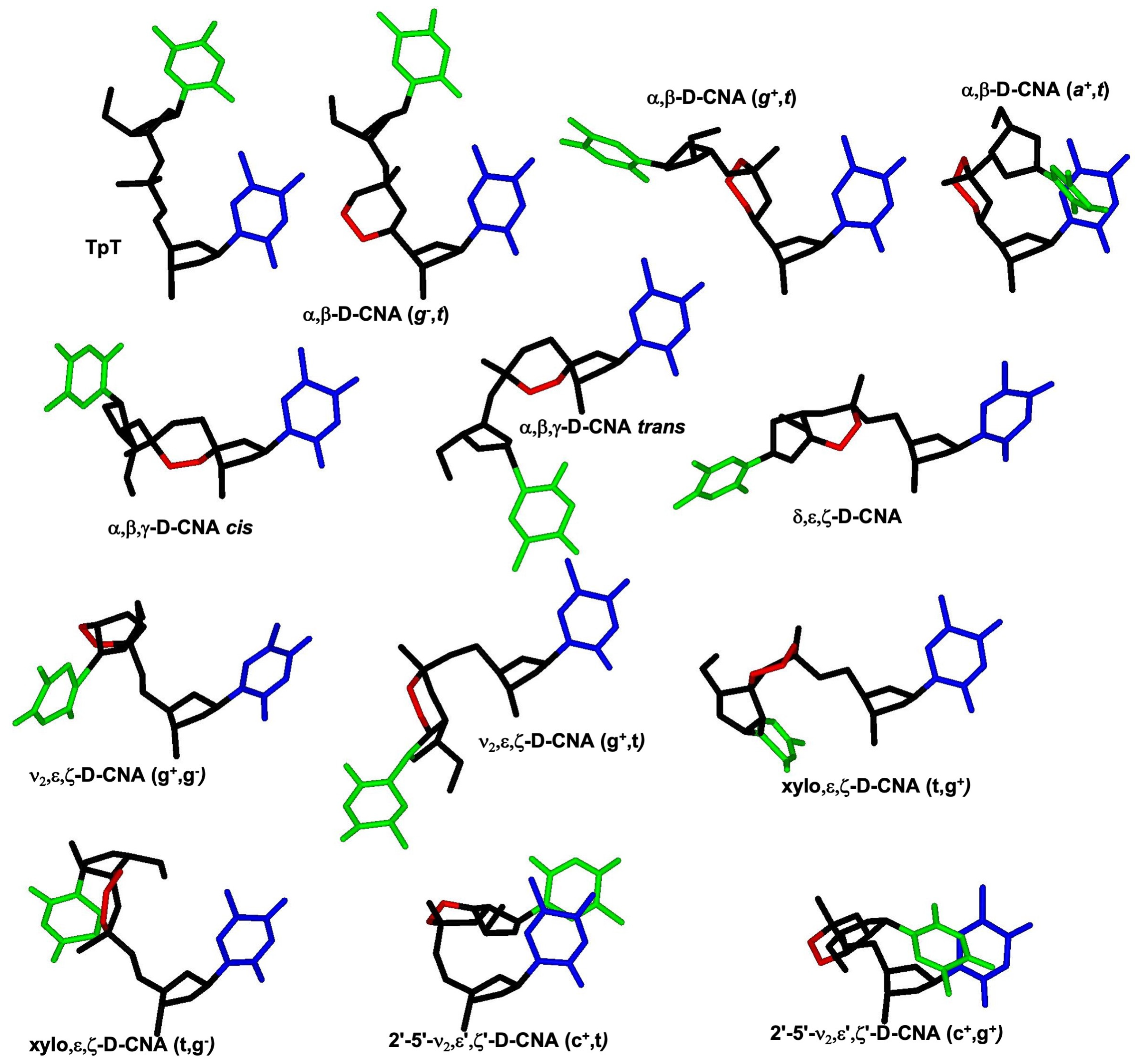{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
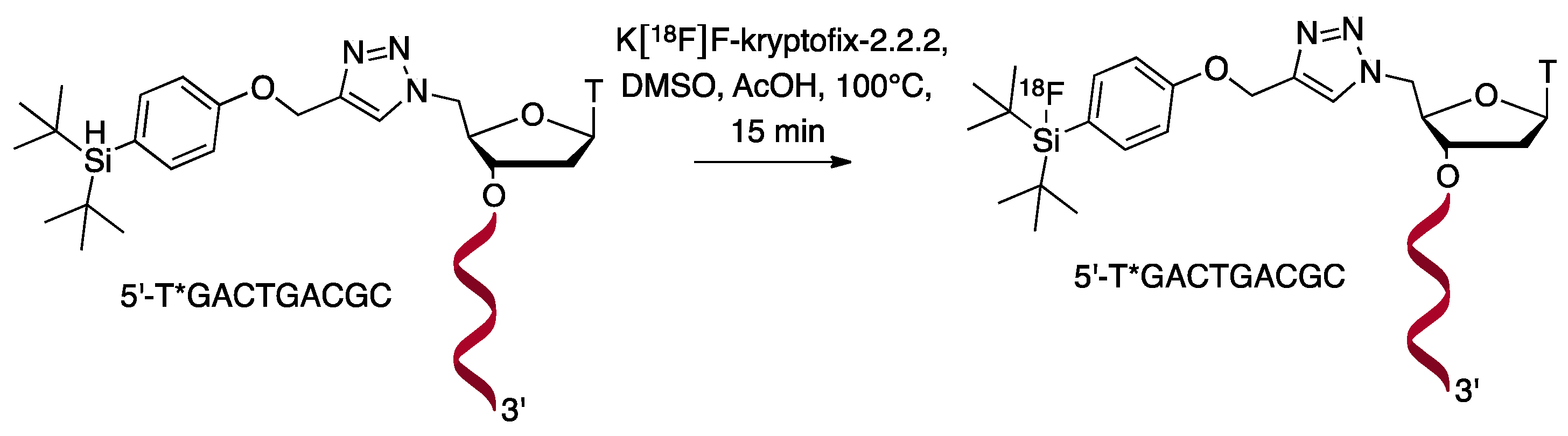{kind=link}
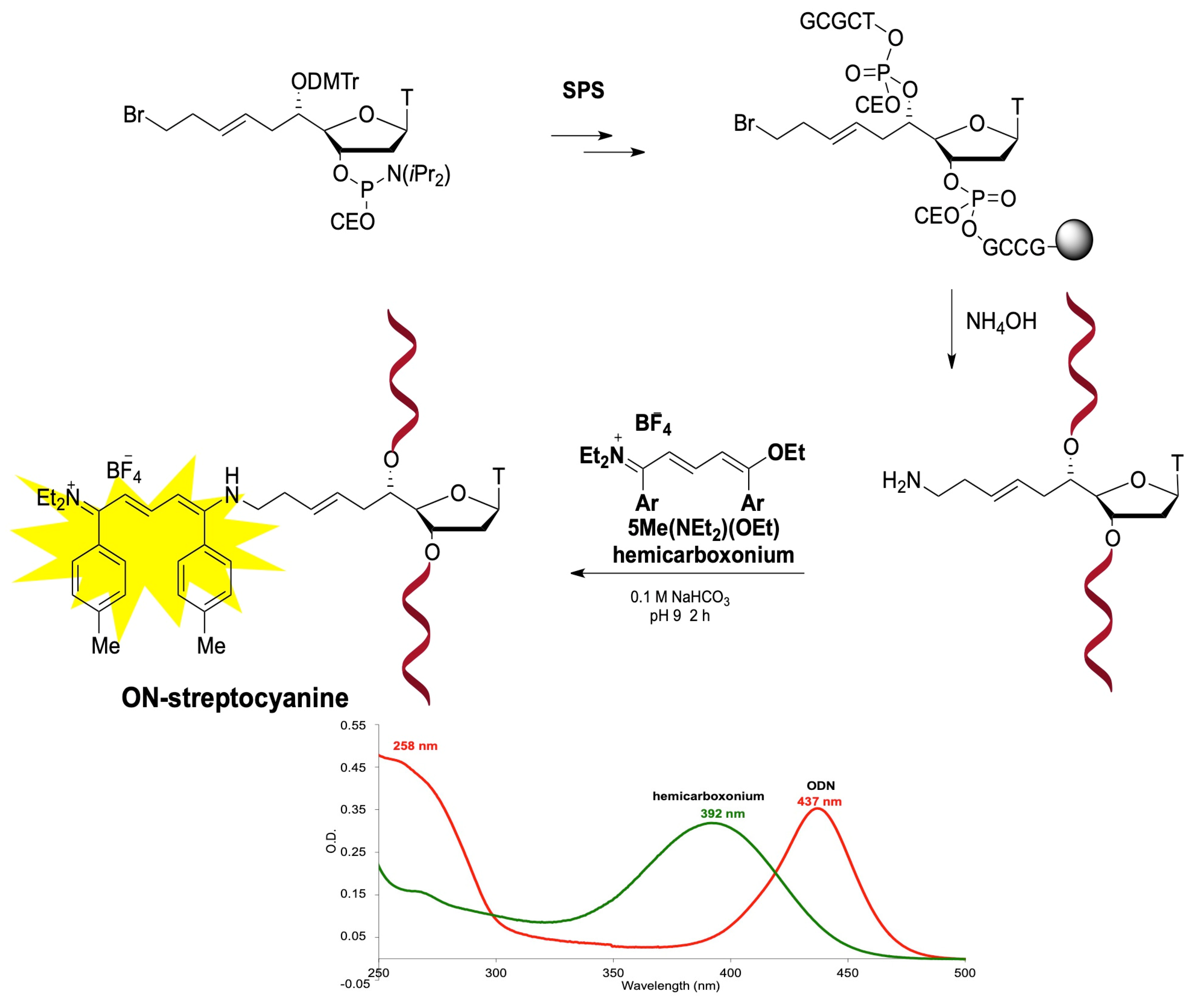{kind=link}
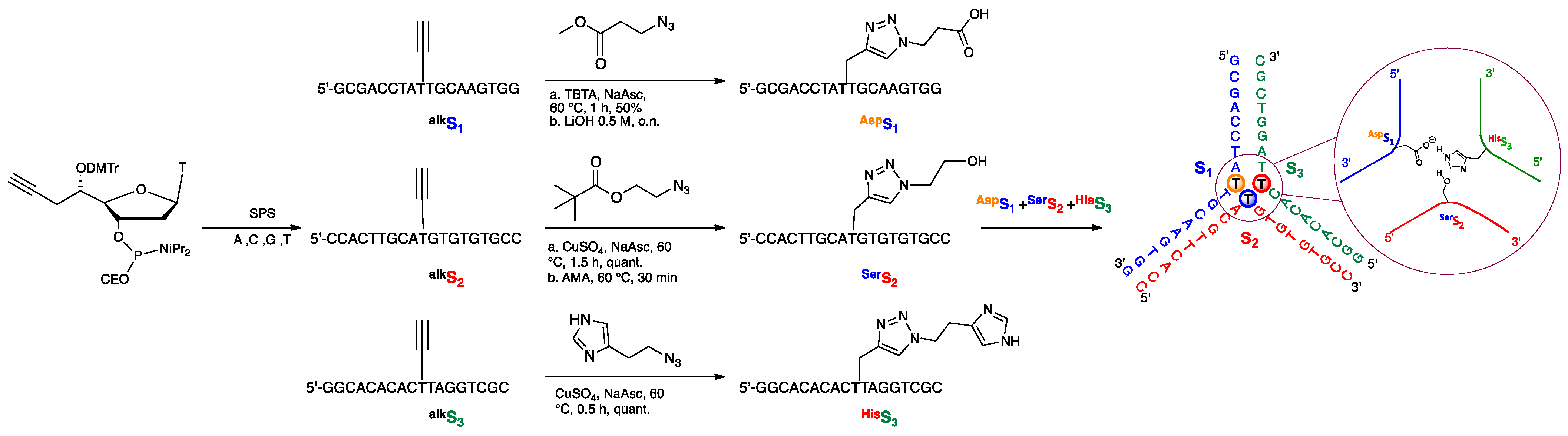{kind=link}
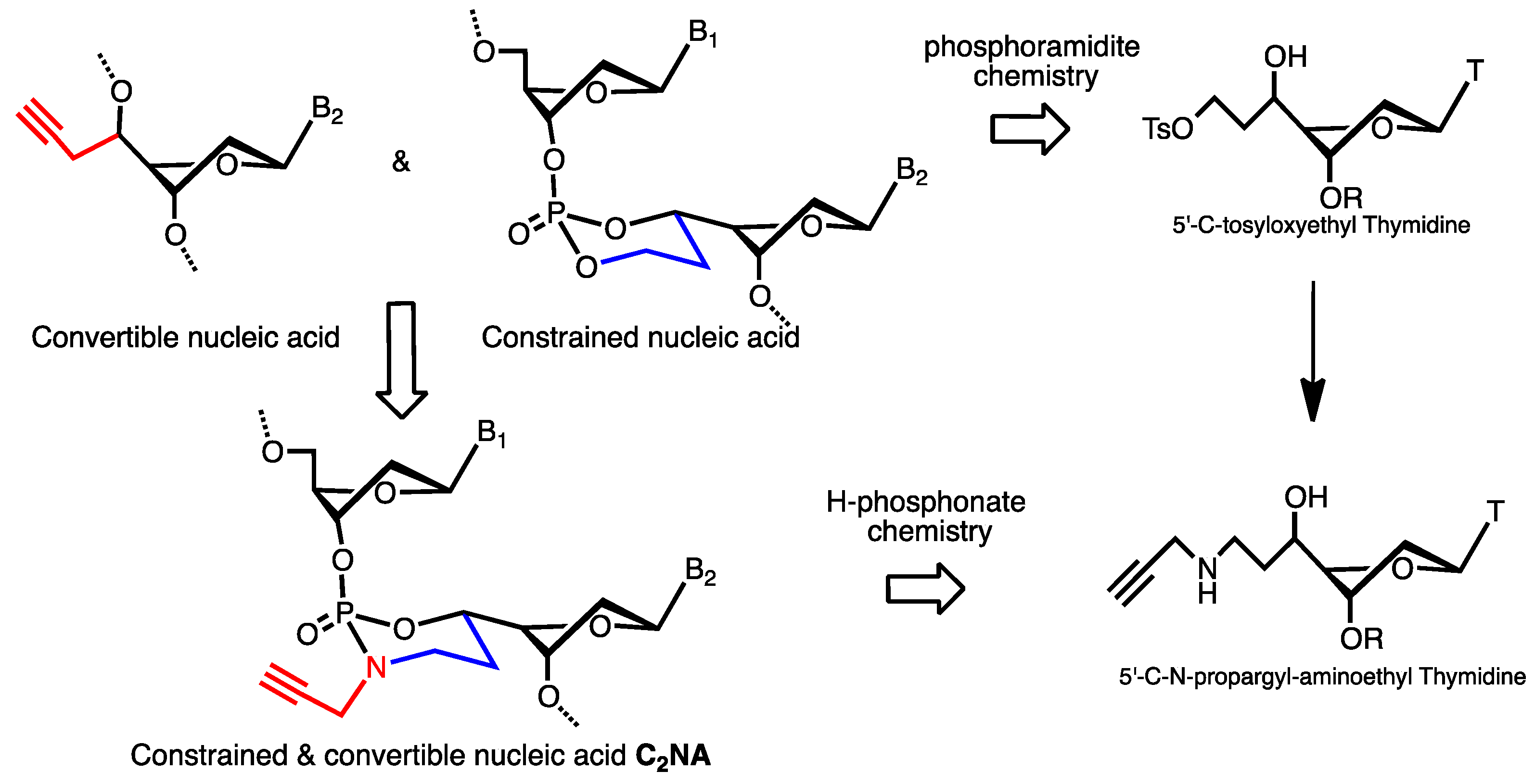{kind=link}
1. Introduction
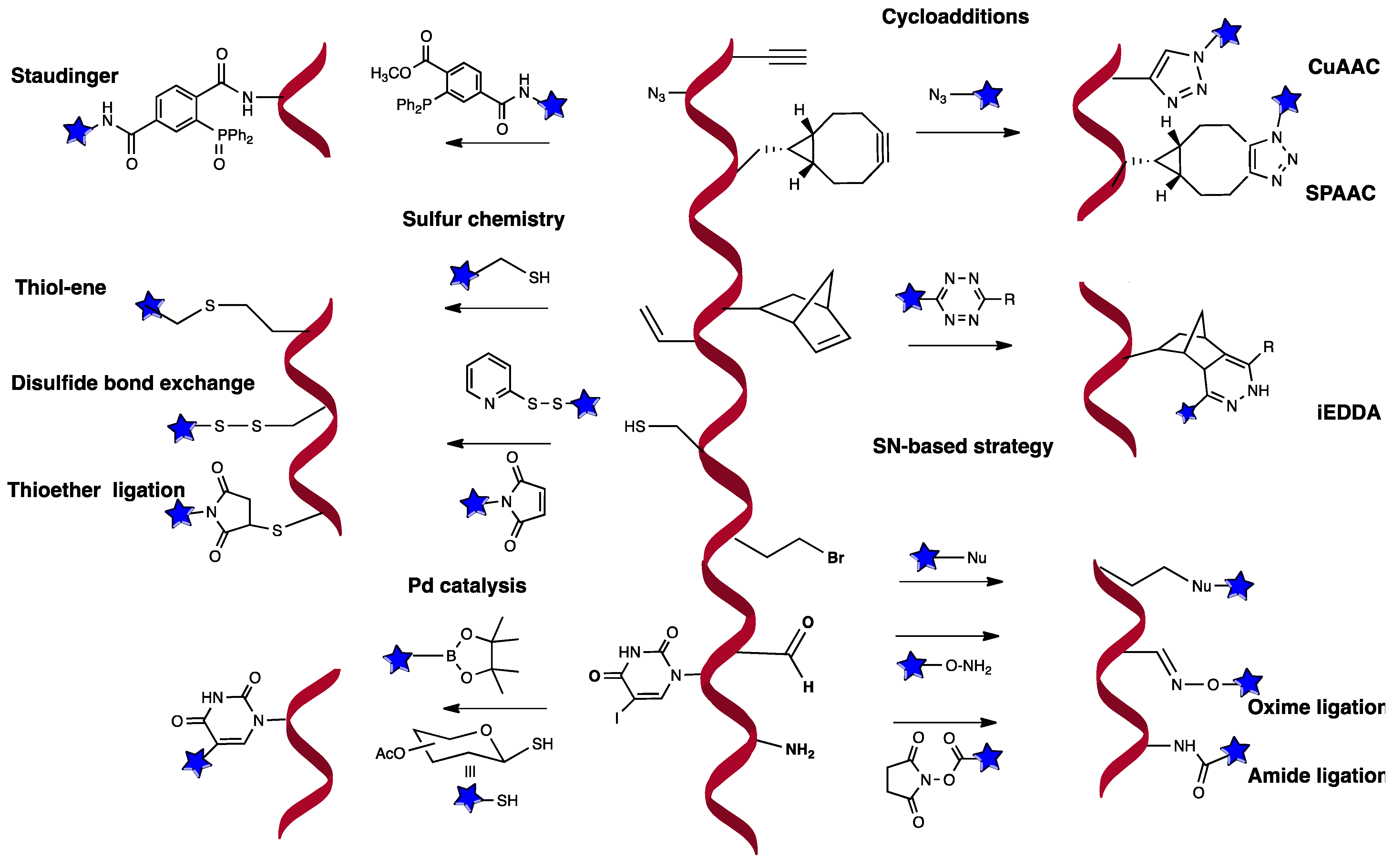
2. Chemical Strategies for Post-Synthetic Functionalization
2.1. Nucleophilic Displacement
2.2. Cycloadditions
2.3. Palladium Catalyzed Conjugation
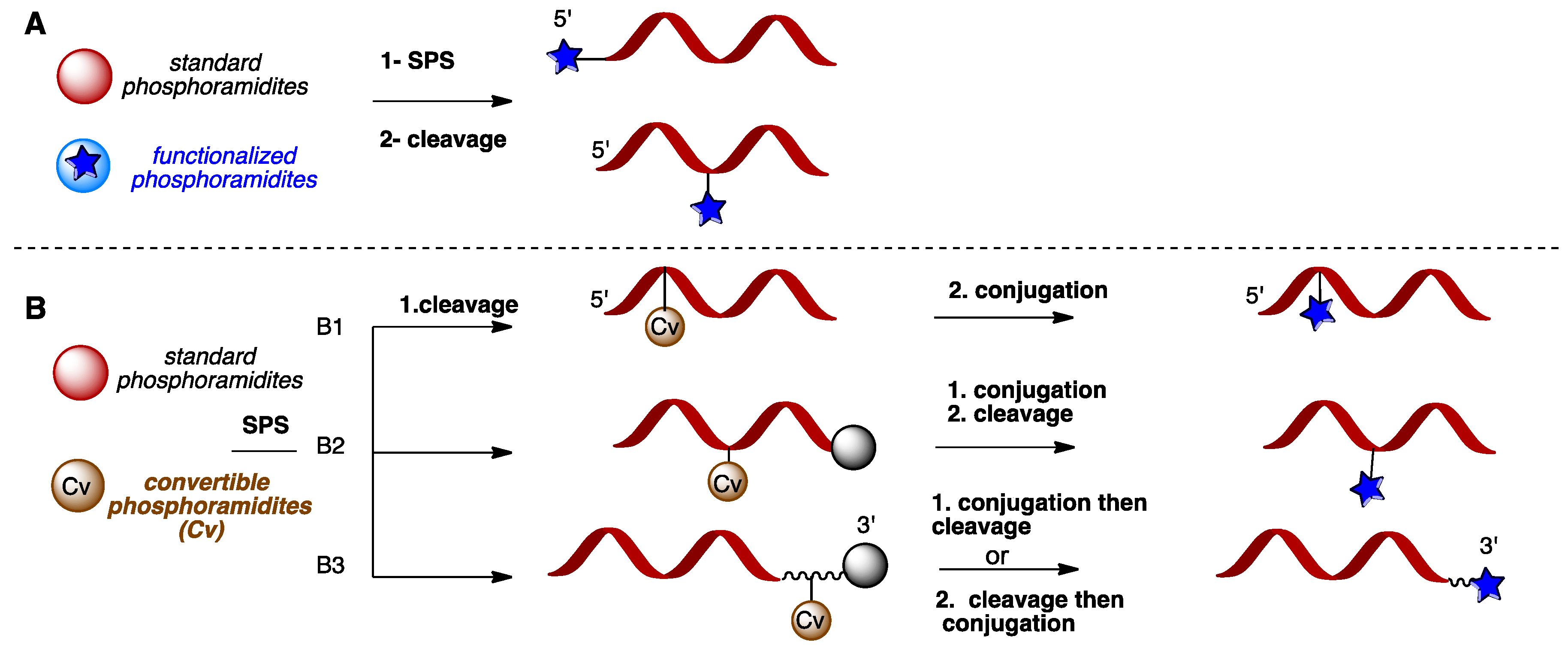
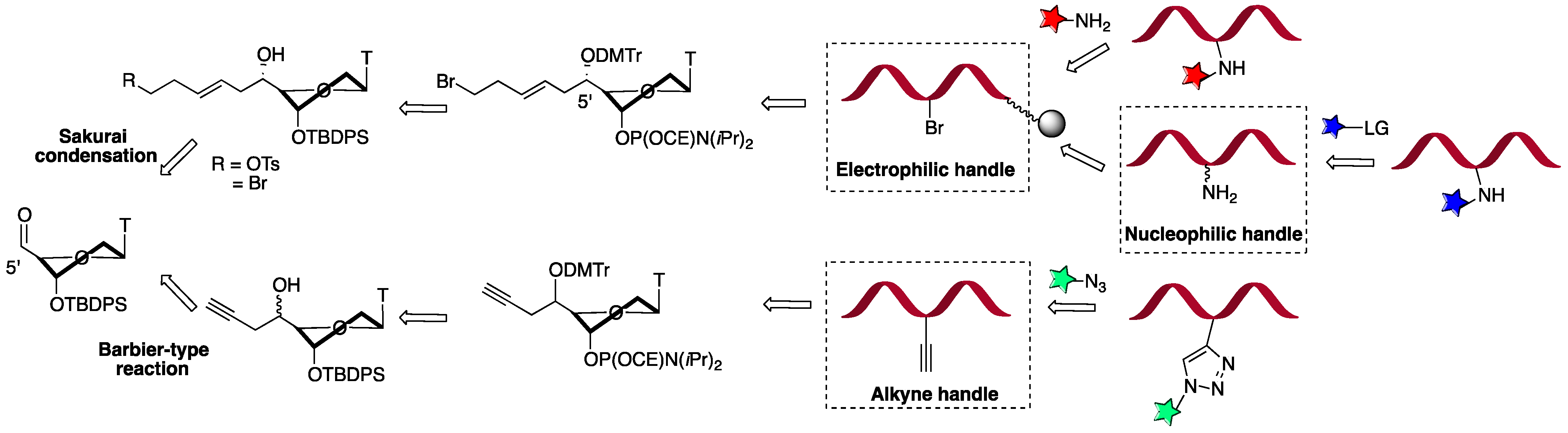
2.4. Narrowing to the 5’-C Functionalization of Nucleic Acids
2.4.1. 5’-C-Bromo or Tosyl Pentenyl Thymidine as Convertible Nucleotides (CvN)
2.4.2. 5’-C-propargyl Thymidine as Convertible Nucleotide for the Construction of Protease Mimics
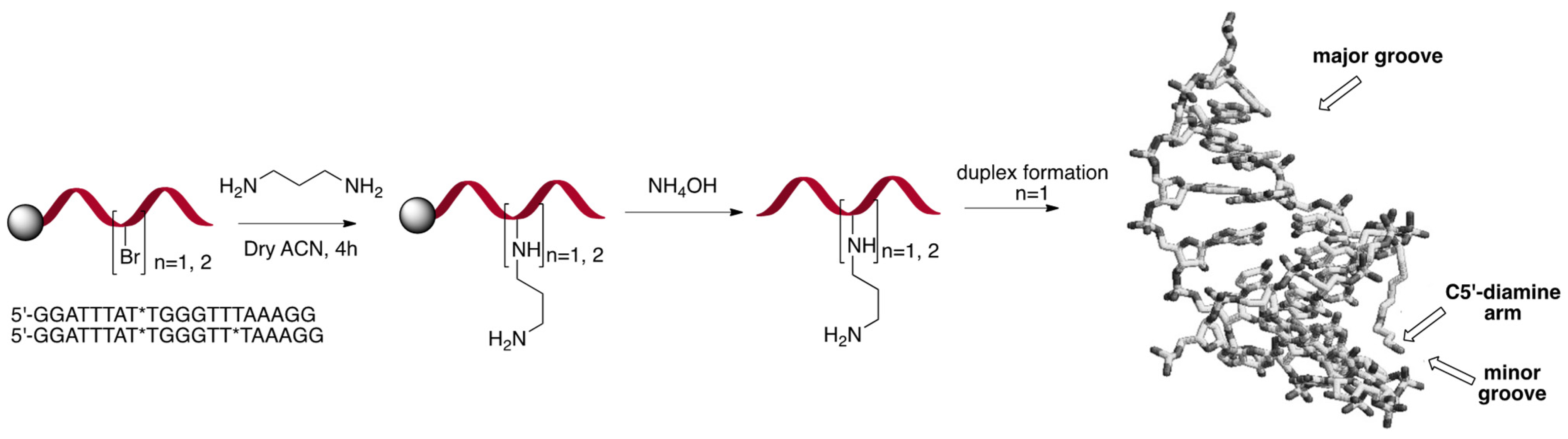
3. Structuration of Nucleic Acids (CNA)
3.1. Dioxaphosphorinane Constrained Nucleic Acids Approach
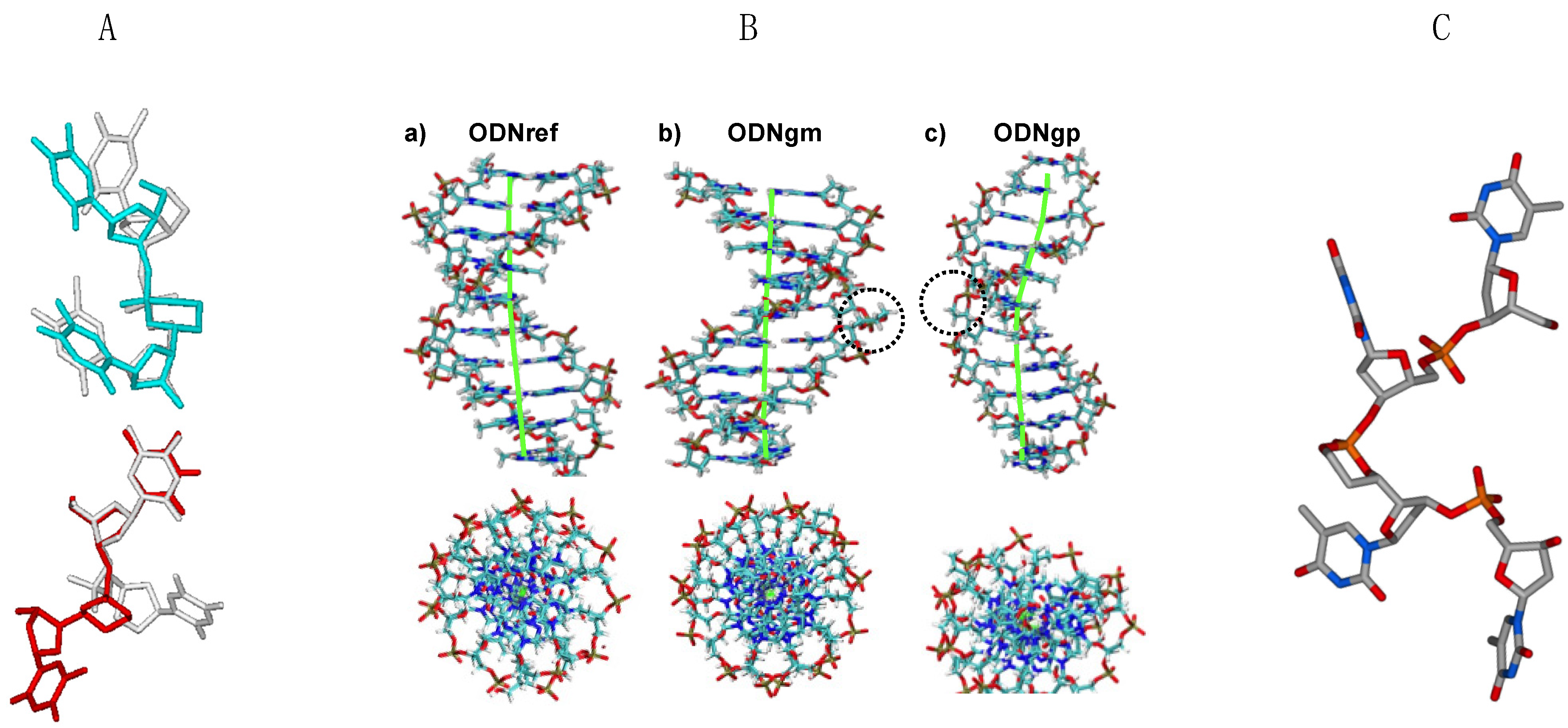
3.2. Behavior of α,β-D-CNA within Oligonucleotides
3.3. In Vitro Properties of α,β-D-CNA
3.3.1. α,β-D-CNA Used as Terminators of Polymerase Chain Reaction
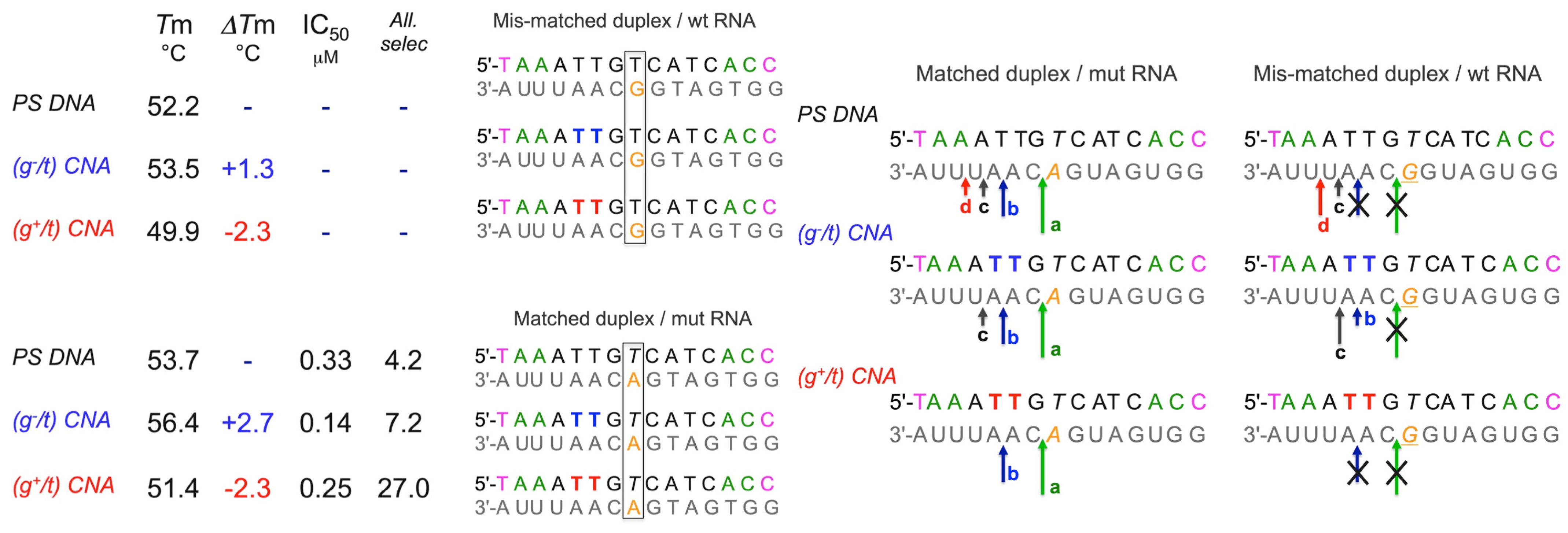
3.3.2. α,β-D-CNA Used for Allele Selective Silencing
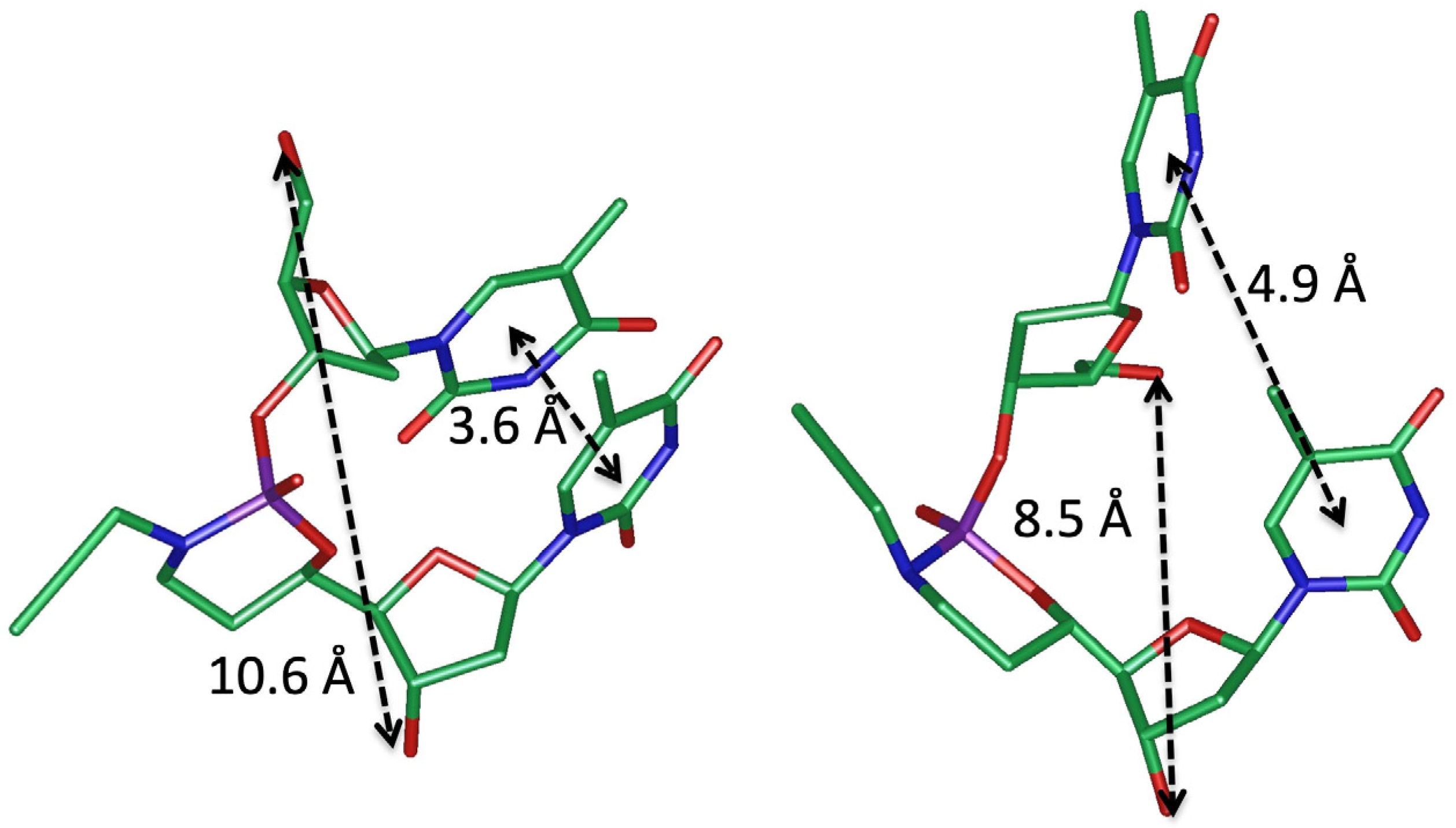
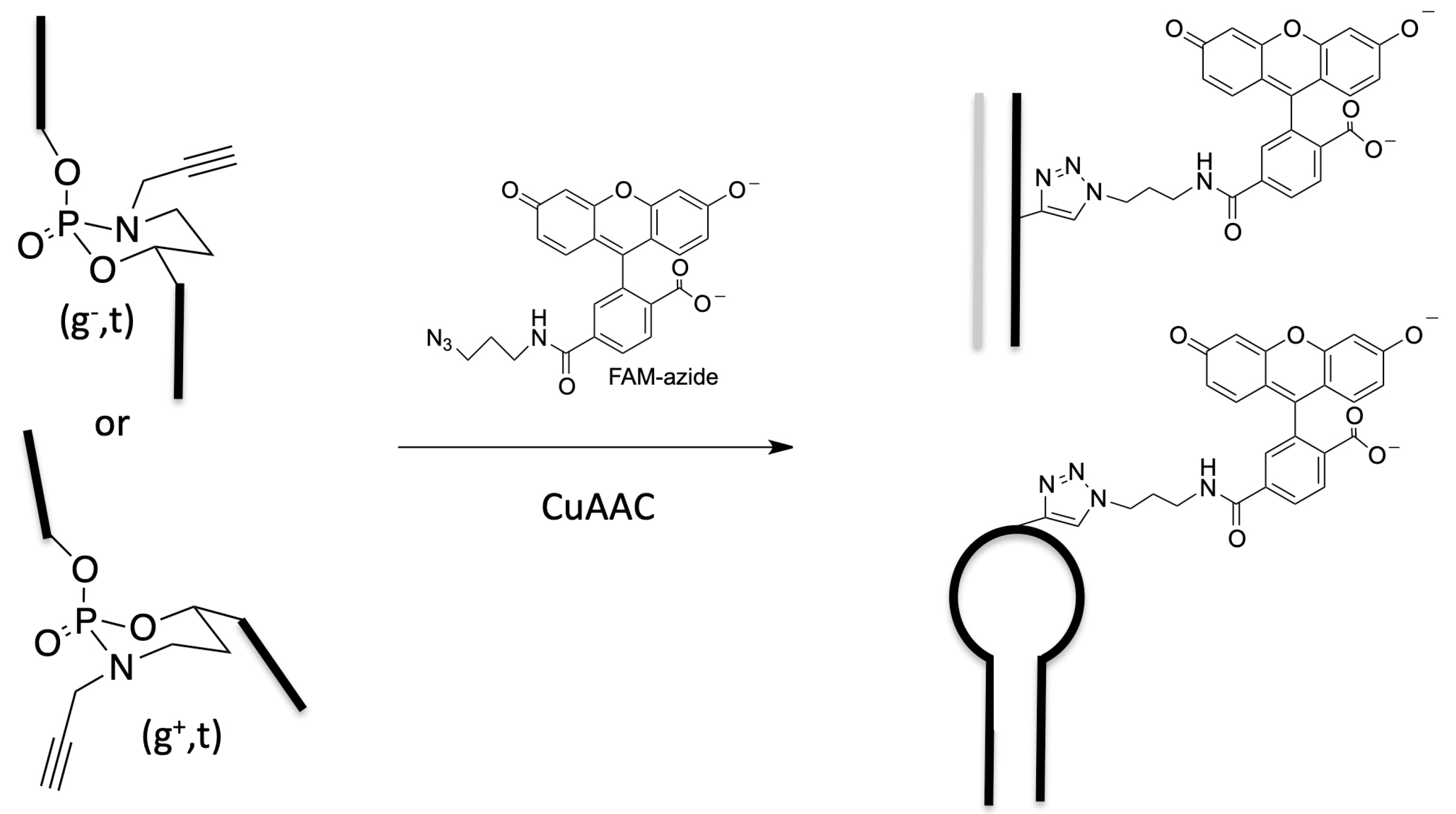
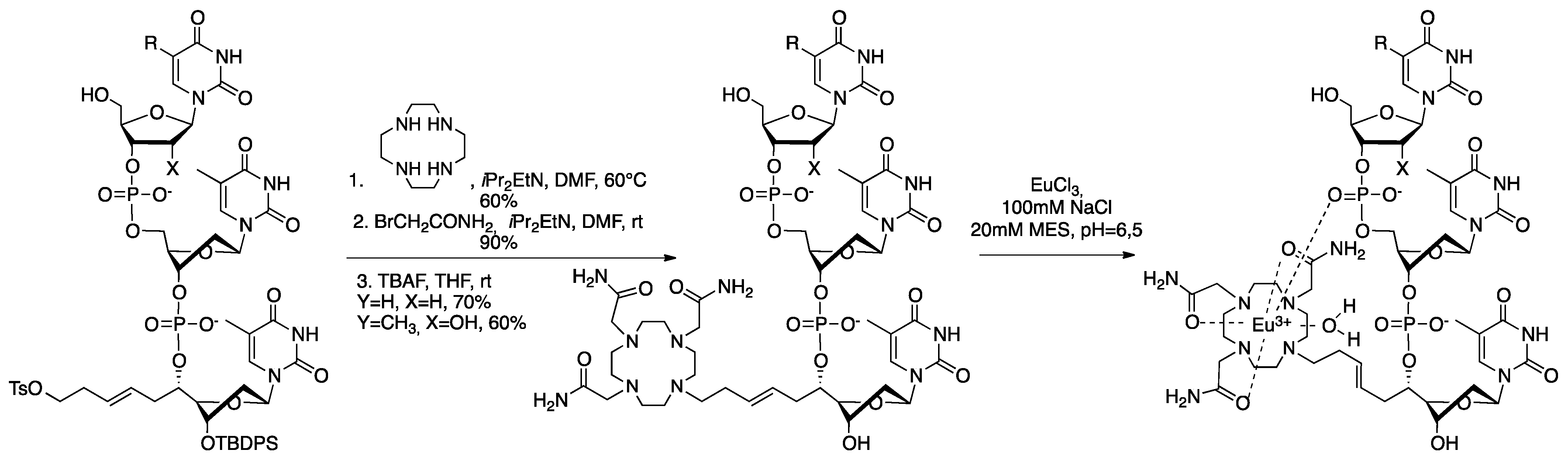
4. Structuration and Functionalization of Nucleic Acids (C2NA)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ochoa, S.; Milam, V.T. Modified nucleic acids: Expanding the capabilities of functional oligonucleotides. Molecules 2020, 25, 4659. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthelemy, P. Bioconjugated oligonucleotides: Recent developments and therapeutic applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Oestergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Taskova, M.; Madsen, C.S.; Jensen, K.J.; Hansen, L.H.; Vester, B.; Astakhova, K. Antisense oligonucleotides internally labeled with peptides show improved target recognition and stability to enzymatic degradation. Bioconjug. Chem. 2017, 28, 768–774. [Google Scholar] [CrossRef]
- Nikan, M.; Tanowitz, M.; Dwyer, C.A.; Jackson, M.; Gaus, H.J.; Swayze, E.E.; Rigo, F.; Seth, P.P.; Prakash, T.P. Targeted delivery of antisense oligonucleotides using neurotensin peptides. J. Med. Chem. 2020, 63, 8471–8484. [Google Scholar] [CrossRef]
- Hawner, M.; Ducho, C. Cellular targeting of oligonucleotides by conjugation with small molecules. Molecules 2020, 25, 5963. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Y.; Tan, J.; Yuan, Q. Chemically modified nucleic acid biopolymers used in biosensing. Mater. Chem. Front. 2020, 4, 1315–1327. [Google Scholar] [CrossRef]
- Li, G.; Moellering, R.E. A concise, modular antibody-oligonucleotide conjugation strategy based on disuccinimidyl ester activation chemistry. ChemBioChem 2019, 20, 1599–1605. [Google Scholar] [CrossRef]
- Peng, T.; Deng, Z.; He, J.; Li, Y.; Tan, Y.; Peng, Y.; Wang, X.-Q.; Tan, W. Functional nucleic acids for cancer theranostics. Coord. Chem. Rev. 2020, 403, 213080. [Google Scholar] [CrossRef]
- Boersma, A.J.; Megens, R.P.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Duchemin, N.; Heath-Apostolopoulos, I.; Smietana, M.; Arseniyadis, S. A decade of DNA-hybrid catalysis: From innovation to comprehension. Org. Biomol. Chem. 2017, 15, 7072–7087. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.; Gothelf, K.V. Chemistries for DNA nanotechnology. Chem. Rev. 2019, 119, 6384–6458. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, J. Conjugates of oligonucleotides and modified oligonucleotides: A review of their synthesis and properties. Bioconjug. Chem. 1990, 1, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Ferentz, A.E.; Verdine, G.L. The convertible nucleoside approach: Structural engineering of nucleic acids by disulfide crosslinking. Nucleic Acids Mol. Biol. 1994, 8, 14–40. [Google Scholar]
- Allerson, C.R.; Chen, S.L.; Verdine, G.L. A chemical method for site-specific modification of RNA: The convertible nucleoside approach. J. Am. Chem. Soc. 1997, 119, 7423–7433. [Google Scholar] [CrossRef]
- Singh, Y.; Murat, P.; Defrancq, E. Recent developments in oligonucleotide conjugation. Chem. Soc. Rev. 2010, 39, 2054–2070. [Google Scholar] [CrossRef]
- Hocek, M. Enzymatic synthesis of base-functionalized nucleic acids for sensing, cross-linking, and modulation of protein–DNA binding and transcription. Acc. Chem. Res. 2019, 52, 1730–1737. [Google Scholar] [CrossRef] [Green Version]
- Hollenstein, M. Nucleoside triphosphates—building blocks for the modification of nucleic acids. Molecules 2012, 17, 13569–13591. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.H.; Park, S.; Hiraga, R.; Okamura, I.; Notsu, S.; Sugiyama, H. Modular DNA-based hybrid catalysts as a toolbox for enantioselective hydration of α,β-unsaturated ketones. Org. Biomol. Chem. 2019, 17, 2548–2553. [Google Scholar] [CrossRef]
- Castro, V.; Rodríguez, H.; Albericio, F. CuAAC: An efficient click chemistry reaction on solid phase. ACS Comb. Sci. 2016, 18, 1–14. [Google Scholar] [CrossRef]
- Pourceau, G.; Meyer, A.; Vasseur, J.-J.; Morvan, F. Azide solid support for 3′-conjugation of oligonucleotides and their circularization by click chemistry. J. Org. Chem. 2009, 74, 6837–6842. [Google Scholar] [CrossRef]
- Gartner, Z.J.; Liu, D.R. The generality of DNA-templated synthesis as a basis for evolving non-natural small molecules. J. Am. Chem. Soc. 2001, 123, 6961–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usanov, D.L.; Chan, A.I.; Maianti, J.P.; Liu, D.R. Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nature Chem. 2018, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Oltra, N.S.; Roelfes, G. Modular assembly of novel DNA-based catalysts. Chem. Commun. 2008, 6039–6041. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, H.; Li, S.; Bare, G.A.L.; Chen, X.; Wang, C.; Moses, J.E.; Wu, P.; Sharpless, K.B. Biocompatible sufex click chemistry: Thionyl tetrafluoride (SOF4)-derived connective hubs for bioconjugation to DNA and proteins. Angew. Chem. Int. Ed. 2019, 58, 8029–8033. [Google Scholar] [CrossRef] [PubMed]
- Banuls, V.; Escudier, J.-M.; Zedde, C.; Claparols, C.; Donnadieu, B.; Plaisancié, H. Stereoselective synthesis of (5′S)-5′-C-(5-bromo-2-penten-1-yl)-2′-deoxyribofuranosyl thymine, a new convertible nucleoside. Eur. J. Org. Chem. 2001, 4693–4700. [Google Scholar] [CrossRef]
- Zewge, D.; Butora, G.; Sherer, E.C.; Tellers, D.M.; Sidler, D.R.; Gouker, J.; Copeland, G.; Jadhav, V.; Li, Z.; Armstrong, J.; et al. Post-synthetic modification of oligonucleotides via orthogonal amidation and copper catalyzed cycloaddition reactions. Bioconjug. Chem. 2018, 29, 1859–1865. [Google Scholar] [CrossRef]
- Fournier, P.; Fiammengo, R.; Jäschke, A. Allylic amination by a DNA–diene–iridium(i) hybrid catalyst. Angew. Chem. Int. Ed. 2009, 48, 4426–4429. [Google Scholar] [CrossRef]
- Trévisiol, E.; Renard, A.; Defrancq, E.; Lhomme, J. The oxyamino-aldehyde coupling reaction: An efficient method for the derivatization of oligonucleotides. Tetrahedron Lett. 1997, 38, 8687–8690. [Google Scholar] [CrossRef]
- Salo, H.; Virta, P.; Hakala, H.; Prakash, T.P.; Kawasaki, A.M.; Manoharan, M.; Lönnberg, H. Aminooxy functionalized oligonucleotides: Preparation, on-support derivatization, and postsynthetic attachment to polymer support. Bioconjug. Chem. 1999, 10, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, J.; Virta, P.; Lönnberg, H. Solid-phase synthesis of multiantennary oligonucleotide glycoconjugates utilizing on-support oximation. Bioconjug. Chem. 2004, 15, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Noel, M.; Clement-Blanc, C.; Meyer, A.; Vasseur, J.-J.; Morvan, F. Solid supports for the synthesis of 3’-aminooxy deoxy- or ribo-oligonucleotides and their 3’-conjugation by oxime ligation. J. Org. Chem. 2019, 84, 14854–14860. [Google Scholar] [CrossRef]
- Sánchez, A.; Pedroso, E.; Grandas, A. Easy introduction of maleimides at different positions of oligonucleotide chains for conjugation purposes. Org. Biomol. Chem. 2012, 10, 8478–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, H.; Diezmann, F.; Seitz, O. DNA as a molecular ruler: Interrogation of a tandem SH2 domain with self-assembled, bivalent DNA–peptide complexes. Angew. Chem. Int. Ed. 2011, 50, 4146–4150. [Google Scholar] [CrossRef]
- Fu, J.; Liu, M.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef] [Green Version]
- Stasińska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide bridge as a linker in nucleic acids’ bioconjugation. Part i: An overview of synthetic strategies. Bioorg. Chem. 2019, 92, 103223. [Google Scholar] [CrossRef]
- Stasińska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide bridge as a linker in nucleic acids’ bioconjugation. Part ii: A summary of practical applications. Bioorg. Chem. 2020, 95, 103518. [Google Scholar] [CrossRef]
- Matyašovský, J.; Pohl, R.; Hocek, M. 2-allyl- and propargylamino-dATPs for site-specific enzymatic introduction of a single modification in the minor groove of DNA. Chem. Eur. J. 2018, 24, 14938–14941. [Google Scholar] [CrossRef] [Green Version]
- Matyašovský, J.; Perlíková, P.; Malnuit, V.; Pohl, R.; Hocek, M. 2-substituted dATP derivatives as building blocks for polymerase-catalyzed synthesis of DNA modified in the minor groove. Angew. Chem. Int. Ed. 2016, 55, 15856–15859. [Google Scholar] [CrossRef]
- Ingale, S.A.; Mei, H.; Leonard, P.; Seela, F. Ethynyl side chain hydration during synthesis and workup of “clickable” oligonucleotides: Bypassing acetyl group formation by triisopropylsilyl protection. J. Org. Chem. 2013, 78, 11271–11282. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Graham, D.; Parkinson, J.A.; Brown, T. DNA duplexes stabilized by modified monomer residues: Synthesis and stability. J. Chem. Soc. Perkin Trans. 1 1998, 1131–1138. [Google Scholar] [CrossRef]
- Gramlich, P.M.E.; Wirges, C.T.; Manetto, A.; Carell, T. Postsynthetic DNA modification through the copper-catalyzed azide–alkyne cycloaddition reaction. Angew. Chem. Int. Ed. 2008, 47, 8350–8358. [Google Scholar] [CrossRef]
- Panattoni, A.; Pohl, R.; Hocek, M. Flexible alkyne-linked thymidine phosphoramidites and triphosphates for chemical or polymerase synthesis and fast postsynthetic DNA functionalization through copper-catalyzed alkyne-azide 1,3-dipolar cycloaddition. Org. Lett. 2018, 20, 3962–3965. [Google Scholar] [CrossRef]
- Qu, Y.; Wen, H.; Ge, R.; Xu, Y.; Gao, H.; Shi, X.; Wang, J.; Cui, W.; Su, W.; Yang, H.; et al. Copper-mediated DNA-compatible one-pot click reactions of alkynes with aryl borates and TMS-N3. Org. Lett. 2020, 22, 4146–4150. [Google Scholar] [CrossRef]
- Farzan, V.M.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Kvach, M.V.; Aparin, I.O.; Brylev, V.A.; Prikazchikova, T.A.; Maklakova, S.Y.; Majouga, A.G.; Ustinov, A.V.; et al. Automated solid-phase click synthesis of oligonucleotide conjugates: From small molecules to diverse N-acetylgalactosamine clusters. Bioconjug. Chem. 2017, 28, 2599–2607. [Google Scholar] [CrossRef]
- Nabo, L.J.; Madsen, C.S.; Jensen, K.J.; Kongsted, J.; Astakhova, K. Ultramild protein-mediated click chemistry creates efficient oligonucleotide probes for targeting and detecting nucleic acids. ChemBioChem 2015, 16, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Gulumkar, V.; Deshpande, P.; Coffey, E.T.; Lonnberg, H.; Virta, P. Synthesis of azide-modified chondroitin sulfate precursors: Substrates for “click”- conjugation with fluorescent labels and oligonucleotides. Bioconjug. Chem. 2018, 29, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Jawalekar, A.M.; Meeuwenoord, N.; Cremers, J.G.O.; Overkleeft, H.S.; van der Marel, G.A.; Rutjes, F.P.J.T.; van Delft, F.L. Conjugation of nucleosides and oligonucleotides by [3 + 2] cycloaddition. J. Org. Chem. 2008, 73, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Gironda-Martínez, A.; Neri, D.; Samain, F.; Donckele, E.J. DNA-compatible diazo-transfer reaction in aqueous media suitable for DNA-encoded chemical library synthesis. Org. Lett. 2019, 21, 9555–9558. [Google Scholar] [CrossRef]
- Wang, C.C.Y.; Seo, T.S.; Li, Z.; Ruparel, H.; Ju, J. Site-specific fluorescent labeling of DNA using Staudinger ligation. Bioconjug. Chem. 2003, 14, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Pourceau, G.; Meyer, A.; Vasseur, J.-J.; Morvan, F. Synthesis of mannose and galactose oligonucleotide conjugates by bi-click chemistry. J. Org. Chem. 2009, 74, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Fomich, M.A.; Kvach, M.V.; Navakouski, M.J.; Weise, C.; Baranovsky, A.V.; Korshun, V.A.; Shmanai, V.V. Azide phosphoramidite in direct synthesis of azide-modified oligonucleotides. Org. Lett. 2014, 16, 4590–4593. [Google Scholar] [CrossRef]
- Hill, K.W.; Taunton-Rigby, J.; Carter, J.D.; Kropp, E.; Vagle, K.; Pieken, W.; McGee, D.P.C.; Husar, G.M.; Leuck, M.; Anziano, D.J.; et al. Diels−Alder bioconjugation of diene-modified oligonucleotides. J. Org. Chem. 2001, 66, 5352–5358. [Google Scholar] [CrossRef]
- Graham, D.; Grondin, A.; McHugh, C.; Fruk, L.; Smith, W.E. Internal labeling of oligonucleotide probes by Diels–Alder cycloaddition. Tetrahedron Lett. 2002, 43, 4785–4788. [Google Scholar] [CrossRef]
- Schoch, J.; Wiessler, M.; Jäschke, A. Post-synthetic modification of DNA by inverse-electron-demand Diels−Alder reaction. J. Am. Chem. Soc. 2010, 132, 8846–8847. [Google Scholar] [CrossRef]
- Schoch, J.; Staudt, M.; Samanta, A.; Wiessler, M.; Jäschke, A. Site-specific one-pot dual labeling of DNA by orthogonal cycloaddition chemistry. Bioconjug. Chem. 2012, 23, 1382–1386. [Google Scholar] [CrossRef]
- Peewasan, K.; Wagenknecht, H.-A. 1,2,4-triazine-modified 2′-deoxyuridine triphosphate for efficient bioorthogonal fluorescent labeling of DNA. ChemBioChem 2017, 18, 1473–1476. [Google Scholar] [CrossRef]
- Lehmann, B.; Wagenknecht, H.-A. Fluorogenic “photoclick” labelling of DNA using a Cy3 dye. Org. Biomol. Chem. 2018, 16, 7579–7582. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Wagenknecht, H.-A. “Photoclick” postsynthetic modification of DNA. Angew. Chem. Int. Ed. 2014, 53, 14580–14582. [Google Scholar] [CrossRef]
- Ganz, D.; Harijan, D.; Wagenknecht, H.-A. Labelling of DNA and RNA in the cellular environment by means of bioorthogonal cycloaddition chemistry. RSC Chem. Biol. 2020, 1, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Omumi, A.; Beach, D.G.; Baker, M.; Gabryelski, W.; Manderville, R.A. Postsynthetic guanine arylation of DNA by Suzuki−Miyaura cross-coupling. J. Am. Chem. Soc. 2011, 133, 42–50. [Google Scholar] [CrossRef]
- Cahová, H.; Jäschke, A. Nucleoside-based diarylethene photoswitches and their facile incorporation into photoswitchable DNA. Angew. Chem. Int. Ed. 2013, 52, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Defrancq, E.; Messaoudi, S. Palladium-mediated labeling of nucleic acids. ChemBioChem 2017, 18, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Jbara, M.; Rodriguez, J.; Dhanjee, H.H.; Loas, A.; Buchwald, S.L.; Pentelute, B.L. Oligonucleotide bioconjugation with bifunctional palladium reagents. Angew. Chem. Int. Ed. 2021, 60, 12109–12115. [Google Scholar] [CrossRef] [PubMed]
- Probst, N.; Lartia, R.; Thery, O.; Alami, M.; Defrancq, E.; Messaoudi, S. Efficient Buchwald-Hartwig-Migita cross-coupling for DNA thio-glycoconjugation. Chem. Eur. J. 2018, 24, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Gierlich, J.; Burley, G.A.; Gramlich, P.M.E.; Hammond, D.M.; Carell, T. Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Org. Lett. 2006, 8, 3639–3642. [Google Scholar] [CrossRef] [PubMed]
- Winz, M.-L.; Linder, E.C.; Becker, J.; Jäschke, A. Site-specific one-pot triple click labeling for DNA and RNA. Chem. Commun. 2018, 54, 11781–11784. [Google Scholar] [CrossRef]
- Meyer, A.; Vasseur, J.-J.; Dumy, P.; Morvan, F. Phthalimide-oxy derivatives for 3’- or 5’-conjugation of oligonucleotides by oxime ligation and circularization of DNA by “bis- or tris-click” oxime ligation. Eur. J. Org. Chem. 2017, 6931–6941. [Google Scholar] [CrossRef]
- Meyer, A.; Vasseur, J.-J.; Morvan, F. Synthesis of monoconjugated and multiply conjugated oligonucleotides by “click thiol” thiol-Michael-type additions and by combination with CuAAC “click Huisgen”. Eur. J. Org. Chem. 2013, 465–473. [Google Scholar] [CrossRef]
- Banuls, V.; Escudier, J.-M. Allylsilanes in the preparation of 5′-C-hydroxy or bromo alkylthymidines. Tetrahedron 1999, 55, 5831–5838. [Google Scholar] [CrossRef]
- James, D.; Escudier, J.-M.; Amigues, E.; Schulz, J.; Vitry, C.; Bordenave, T.; Szlosek-Pinaud, M.; Fouquet, E. A ‘click chemistry’ approach to the efficient synthesis of modified nucleosides and oligonucleotides for pet imaging. Tetrahedron Lett. 2010, 51, 1230–1232. [Google Scholar] [CrossRef]
- Jastrząb, R.; Nowak, M.; Skrobańska, M.; Tolińska, A.; Zabiszak, M.; Gabryel, M.; Marciniak, Ł.; Kaczmarek, M.T. DNA as a target for lanthanide(iii) complexes influence. Coord. Chem. Rev. 2019, 382, 145–159. [Google Scholar] [CrossRef]
- Franklin, S.J. Lanthanide-mediated DNA hydrolysis. Curr. Opin. Chem. Biol. 2001, 5, 201–208. [Google Scholar] [CrossRef]
- Huiban, M.; Huet, A.; Barré, L.; Sobrio, F.; Fouquet, E.; Perrio, C. Methyl transfer reaction from monomethyltin reagent under palladium(0) catalysis: A versatile method for labelling with carbon-11. Chem. Commun. 2006, 97–99. [Google Scholar] [CrossRef] [PubMed]
- James, D.; Escudier, J.-M.; Szlosek-Pinaud, M.; Fouquet, E. Pd-catalyzed methyl transfer on nucleosides and oligonucleotides, envisaged as a PET tracer. Molecules 2013, 18, 13654–13665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, J.; Vimont, D.; Bordenave, T.; James, D.; Escudier, J.-M.; Allard, M.; Szlosek-Pinaud, M.; Fouquet, E. Silicon-based chemistry: An original and efficient one-step approach to [18F]-nucleosides and [18F]-oligonucleotides for PET imaging. Chem. Eur. J. 2011, 17, 3096. [Google Scholar] [CrossRef]
- Li, X.-G.; Roivainen, A.; Bergman, J.; Heinonen, A.; Bengel, F.; Thum, T.; Knuuti, J. Enabling [18F]-bicyclo[6.1.0]nonyne for oligonucleotide conjugation for positron emission tomography applications: [18F]-anti-microRNA-21 as an example. Chem. Commun. 2015, 51, 9821–9824. [Google Scholar] [CrossRef]
- Guo, F.; Li, Q.; Zhou, C. Synthesis and biological applications of fluoro-modified nucleic acids. Org. Biomol. Chem. 2017, 15, 9552–9565. [Google Scholar] [CrossRef]
- Lange, C.W.; VanBrocklin, H.F.; Taylor, S.E. Photoconjugation of 3-azido-5-nitrobenzyl-[18F]fluoride to an oligonucleotide aptamer. J. Label. Compd. Radiopharm. 2002, 45, 257–268. [Google Scholar] [CrossRef]
- Le Droumaguet, C.; Wang, C.; Wang, Q. Fluorogenic click reaction. Chem. Soc. Rev. 2010, 39, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Guo, G.; Zheng, J.; Xing, D.; Zhang, T. Fluorogenic “photoclick” labeling and imaging of DNA with coumarin-fused tetrazole in vivo. ACS Sens. 2019, 4, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Biniuri, Y.; Albada, B.; Wolff, M.; Golub, E.; Gelman, D.; Willner, I. Cu2+ or Fe3+ terpyridine/aptamer conjugates: Nucleoapzymes catalyzing the oxidation of dopamine to aminochrome. ACS Catal. 2018, 8, 1802–1809. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Li, Y.; Liu, X.; Yuan, Q. Self-assembled DNA nanomaterials with highly programmed structures and functions. Mater. Chem. Front. 2018, 2, 423–436. [Google Scholar] [CrossRef]
- Nothling, M.D.; Xiao, Z.; Bhaskaran, A.; Blyth, M.T.; Bennett, C.W.; Coote, M.L.; Connal, L.A. Synthetic catalysts inspired by hydrolytic enzymes. ACS Catal. 2019, 9, 168–187. [Google Scholar] [CrossRef]
- Ameta, S.; Jaschke, A. An RNA catalyst that reacts with a mechanistic inhibitor of serine proteases. Chem. Sci. 2013, 4, 957–964. [Google Scholar] [CrossRef]
- Chandra, M.; Sachdeva, A.; Silverman, S.K. DNA-catalyzed sequence-specific hydrolysis of DNA. Nat. Chem. Biol. 2009, 5, 718–720. [Google Scholar] [CrossRef] [Green Version]
- Hollenstein, M. Deoxynucleoside triphosphates bearing histamine, carboxylic acid, and hydroxyl residues synthesis and biochemical characterization. Org. Biomol. Chem. 2013, 11, 5162–5172. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Avins, J.L.; Klauser, P.C.; Brandsen, B.M.; Lee, Y.; Silverman, S.K. DNA-catalyzed amide hydrolysis. J. Am. Chem. Soc. 2016, 138, 2106–2109. [Google Scholar] [CrossRef] [Green Version]
- Catry, M.; Madder, A. Synthesis of functionalised nucleosides for incorporation into nucleic acid-based serine protease mimics. Molecules 2007, 12, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addamiano, C.; Gerland, B.; Payrastre, C.; Escudier, J.M. DNA three way junction core decorated with amino acids-like residues-synthesis and characterization. Molecules 2016, 21, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Yao, C.; Zhu, Y.; Yang, L.; Luo, D.; Yang, D. DNA functional materials assembled from branched DNA: Design, synthesis, and applications. Chem. Rev. 2020, 120, 9420–9481. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.; Constant, J.-F.; Demeunynck, M. Nucleic acid conformation diversity: From structure to function and regulation. Chem. Soc. Rev. 2001, 30, 70–81. [Google Scholar] [CrossRef]
- Schneider, B.; Neidle, S.; Berman, H.M. Conformations of the sugar-phosphate backbone in helical DNA crystal structures. Biopolymers 1997, 42, 113–124. [Google Scholar] [CrossRef]
- Wengel, J. Synthesis of 3‘-C- and 4‘-C-branched oligodeoxynucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res. 1999, 32, 301–310. [Google Scholar] [CrossRef]
- Leumann, C.J. DNA analogues: From supramolecular principles to biological properties. Bioorg. Med. Chem. 2002, 10, 841–854. [Google Scholar] [CrossRef]
- Cobb, A.J.A. Recent highlights in modified oligonucleotide chemistry. Org. Biomol. Chem. 2007, 5, 3260–3275. [Google Scholar] [CrossRef]
- Petersen, M.; Bondensgaard, K.; Wengel, J.; Jacobsen, J.P. Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc. 2002, 124, 5974–5982. [Google Scholar] [CrossRef]
- McKenzie, L.K.; El-Khoury, R.; Thorpe, J.D.; Damha, M.J.; Hollenstein, M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. [Google Scholar] [CrossRef]
- Bell, N.M.; Micklefield, J. Chemical modification of oligonucleotides for therapeutic, bioanalytical and other applications. ChemBioChem 2009, 10, 2691–2703. [Google Scholar] [CrossRef]
- Zhou, C.; Chattopadhyaya, J. The synthesis of therapeutic locked nucleos(t)ides. Curr. Opin. Drug Discov. Dev. 2009, 12, 876–898. [Google Scholar]
- Stevens, S.Y.; Swanson, P.C.; Voss, E.W.; Glick, G.D. Evidence for induced fit in antibody-DNA complexes. J. Am. Chem. Soc. 1993, 115, 1585–1586. [Google Scholar] [CrossRef]
- Glick, G.D. Design, synthesis, and analysis of conformationally constrained nucleic acids. Biopolymers 1998, 48, 83–96. [Google Scholar] [CrossRef]
- Renneberg, D.; Leumann, C.J. Watson−crick base-pairing properties of tricyclo-DNA. J. Am. Chem. Soc. 2002, 124, 5993–6002. [Google Scholar] [CrossRef]
- Obika, S.; Nanbu, D.; Hari, Y.; Morio, K.-i.; In, Y.; Ishida, T.; Imanishi, T. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3, -endo sugar puckering. Tetrahedron Lett. 1997, 38, 8735–8738. [Google Scholar] [CrossRef]
- Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 4, 455–456. [Google Scholar] [CrossRef]
- Lebreton, J.; Escudier, J.-M.; Arzel, L.; Len, C. Synthesis of bicyclonucleosides having a C-C bridge. Chem. Rev. 2010, 110, 3371–3418. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Babu, B.R.; Maiti, S. Perspectives on chemistry and therapeutic applications of locked nucleic acid (LNA). Chem. Rev. 2007, 107, 4672–4697. [Google Scholar] [CrossRef] [PubMed]
- De Mesmaeker, A.; Lesueur, C.; Bévièrre, M.-O.; Waldner, A.; Fritsch, V.; Wolf, R.M. Amide backbones with conformationally restricted furanose rings: Highly improved affinity of the modified oligonucleotides for their RNA complements. Angew. Chem. Int. Ed. 1996, 35, 2790–2794. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Haaima, G. Peptide nucleic acid (PNA). A DNA mimic with a pseudopeptide backbone. Chem. Soc. Rev. 1997, 26, 73–78. [Google Scholar] [CrossRef]
- Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA: Synthetic polyamide nucleic acids with unusual binding properties. Angew. Chem. Int. Ed. 1998, 37, 2796–2823. [Google Scholar] [CrossRef]
- Sekine, M.; Kurasawa, O.; Shohda, K.-I.; Seio, K.; Wada, T. Synthesis and properties of oligonucleotides having a phosphorus chiral center by incorporation of conformationally rigid 5‘-cyclouridylic acid derivatives. J. Org. Chem. 2000, 65, 6515–6524. [Google Scholar] [CrossRef]
- Seio, K.; Wada, T.; Sekine, M. Synthesis and properties of oligothymidylates incorporating an artificial bend motif. Helv. Chim. Acta 2000, 83, 162–180. [Google Scholar] [CrossRef]
- Seio, K.; Wada, T.; Sakamoto, K.; Yokoyama, S.; Sekine, M. Chemical synthesis and conformational properties of a new cyclouridylic acid having an ethylene bridge between the uracil 5-position and 5‘-phosphate group. J. Org. Chem. 1996, 61, 1500–1504. [Google Scholar] [CrossRef]
- Sekine, M.; Kurasawa, O.; Shohda, K.-i.; Seio, K.; Wada, T. Synthesis and properties of oligodeoxynucleotides incorporating a conformationally rigid uridine unit having a cyclic structure at the 5‘-terminal site. J. Org. Chem. 2000, 65, 3571–3578. [Google Scholar] [CrossRef]
- Sørensen, A.M.; Nielsen, P. Synthesis of conformationally restricted dinucleotides by ring-closing metathesis. Org. Lett. 2000, 2, 4217–4219. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.M.; Nielsen, K.E.; Vogg, B.; Jacobsen, J.P.; Nielsen, P. Synthesis and NMR-studies of dinucleotides with conformationally restricted cyclic phosphotriester linkages. Tetrahedron 2001, 57, 10191–10201. [Google Scholar] [CrossRef]
- Børsting, P.; Nielsen, P. Tandem ring-closing metathesis and hydrogenation towards cyclic dinucleotides. Chem. Commun. 2002, 2140–2141. [Google Scholar] [CrossRef]
- Børsting, P.; Nielsen, K.E.; Nielsen, P. Stabilisation of a nucleic acid three-way junction by an oligonucleotide containing a single 2′-C to 3′-O-phosphate butylene linkage prepared by a tandem RCM-hydrogenation method. Org. Biomol. Chem. 2005, 3, 2183–2190. [Google Scholar] [CrossRef] [PubMed]
- Børsting, P.; Christensen, M.S.; Steffansen, S.I.; Nielsen, P. Synthesis of dinucleotides with 2′-C to phosphate connections by ring-closing metathesis. Tetrahedron 2006, 62, 1139–1149. [Google Scholar] [CrossRef]
- Børsting, P.; Sørensen, A.; Nielsen, P. A ring-closing metathesis strategy towards conformationally restricted di and trinucleotides. Synthesis 2002, 2002, 797–801. [Google Scholar] [CrossRef]
- Catana, D.-A.; Maturano, M.; Payrastre, C.; Lavedan, P.; Tarrat, N.; Escudier, J.-M. Synthesis of phostone-constrained nucleic acid (P-CNA) dinucleotides through intramolecular Arbuzov’s reaction. Eur. J. Org. Chem. 2011, 6857–6863. [Google Scholar] [CrossRef]
- Gerland, B.; Addamiano, C.; Renard, B.-L.; Payrastre, C.; Gopaul, D.; Escudier, J.-M. Thio- and seleno-dioxaphosphorinane-constrained dinucleotides (D-CNA): Synthesis and conformational study. Eur. J. Org. Chem. 2017, 1450–1464. [Google Scholar] [CrossRef]
- Le Clézio, I.; Escudier, J.-M.; Vigroux, A. Diastereoselective synthesis of a conformationally restricted dinucleotide with predefined -α and β torsional angles for the construction of α,β-constrained nucleic acids (α,β-CNA). Org. Lett. 2003, 5, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Le Clézio, I.; Gornitzka, H.; Escudier, J.-M.; Vigroux, A. Constrained nucleic acids (CNA). Part 2. Synthesis of conformationally restricted dinucleotide units featuring noncanonical a,b,g or d,e,z torsion angle combinations. J. Org. Chem. 2005, 70, 1620–1629. [Google Scholar] [CrossRef]
- Dupouy, C.; Le Clézio, I.; Lavedan, P.; Gornitzka, H.; Escudier, J.-M.; Vigroux, A. Diastereoselective synthesis of conformationally restricted dinucleotides featuring canonical and noncanonical α/β torsion angle combinations (α,β-D-CNA). Eur. J. Org. Chem. 2006, 5515–5525. [Google Scholar] [CrossRef]
- Le Clézio, I.; Dupouy, C.; Lavedan, P.; Escudier, J.-M. Synthesis and structure of an α,β-D-CNA featuring a noncanonical α/β torsion angle combination within a tetranucleotide. Eur. J. Org. Chem. 2007, 3894–3900. [Google Scholar] [CrossRef]
- Le Clézio, I.; Vigroux, A.; Escudier, J.-M. Diastereoselective and regioselective synthesis of conformationally restricted thio-dioxa- and oxo-oxathiaphosphorinane dinucleotides featuring noncanonical α/β torsion angle combinations (α,β-CNAs). Eur. J. Org. Chem. 2007, 1935–1941. [Google Scholar] [CrossRef]
- Dupouy, C.; Lavedan, P.; Escudier, J.-M. Synthesis and structure of dinucleotides featuring canonical and non-canonical A-type duplex α, β and δ torsion angle combinations (LNA/α,β-D-CNA). Eur. J. Org. Chem. 2007, 5256–5264. [Google Scholar] [CrossRef]
- Dupouy, C.; Lavedan, P.; Escudier, J.-M. Synthesis of spiro e,z-D-CNA in xylo configuration featuring noncanonical d/e/z torsion angle combination. Tetrahedron 2007, 63, 11235–11243. [Google Scholar] [CrossRef]
- Dupouy, C.; Lavedan, P.; Escudier, J.-M. Synthesis and structure of dinucleotides with S-type sugar puckering and noncanonical ϵ and ζ torsion angle combination (ν2,ϵ,ζ-D-CNA). Eur. J. Org. Chem. 2008, 1285–1294. [Google Scholar] [CrossRef]
- Dupouy, C.; Iché-Tarrat, N.; Durrieu, M.-P.; Rodriguez, F.; Escudier, J.-M.; Vigroux, A. Watson–crick base-pairing properties of nucleic acid analogues with stereocontrolled α and β torsion angles (α,β-D-CNAs). Angew. Chem. Int. Ed. 2006, 45, 3623–3627. [Google Scholar] [CrossRef]
- Dupouy, C.; Iche-Tarrat, N.; Durrieu, M.-P.; Vigroux, A.; Escudier, J.-M. a,β-D-CNA induced rigidity within oligonucleotides. Org. Biomol. Chem. 2008, 6, 2849–2851. [Google Scholar] [CrossRef]
- Boissonnet, A.; Dupouy, C.; Millard, P.; Durrieu, M.-P.; Tarrat, N.; Escudier, J.-M. a,β-D-CNA featuring canonical and noncanonical a/b torsional angles behaviours within oligonucleotides. New, J. Chem. 2011, 35, 1528–1533. [Google Scholar] [CrossRef]
- Maturano, M.; Catana, D.-A.; Lavedan, P.; Tarrat, N.; Saffon, N.; Payrastre, C.; Escudier, J.-M. Synthesis and structural study of ribo-dioxaphosphorinane-constrained nucleic acid dinucleotides (ribo-α,β-D-CNA). Eur. J. Org. Chem. 2012, 721–730. [Google Scholar] [CrossRef]
- Dupouy, C.; Payrastre, C.; Escudier, J.-M. Cyclic phospho di- and tri-ester as structural elements of nucleic acids. Targets Heterocycl. Syst. 2008, 12, 185–211. [Google Scholar]
- Dupouy, C.; Millard, P.; Boissonnet, A.; Escudier, J.-M. a,β-D-CNA preorganization of unpaired loop moiety stabilizes DNA hairpin. Chem. Commun. 2010, 46, 5142–5144. [Google Scholar] [CrossRef] [PubMed]
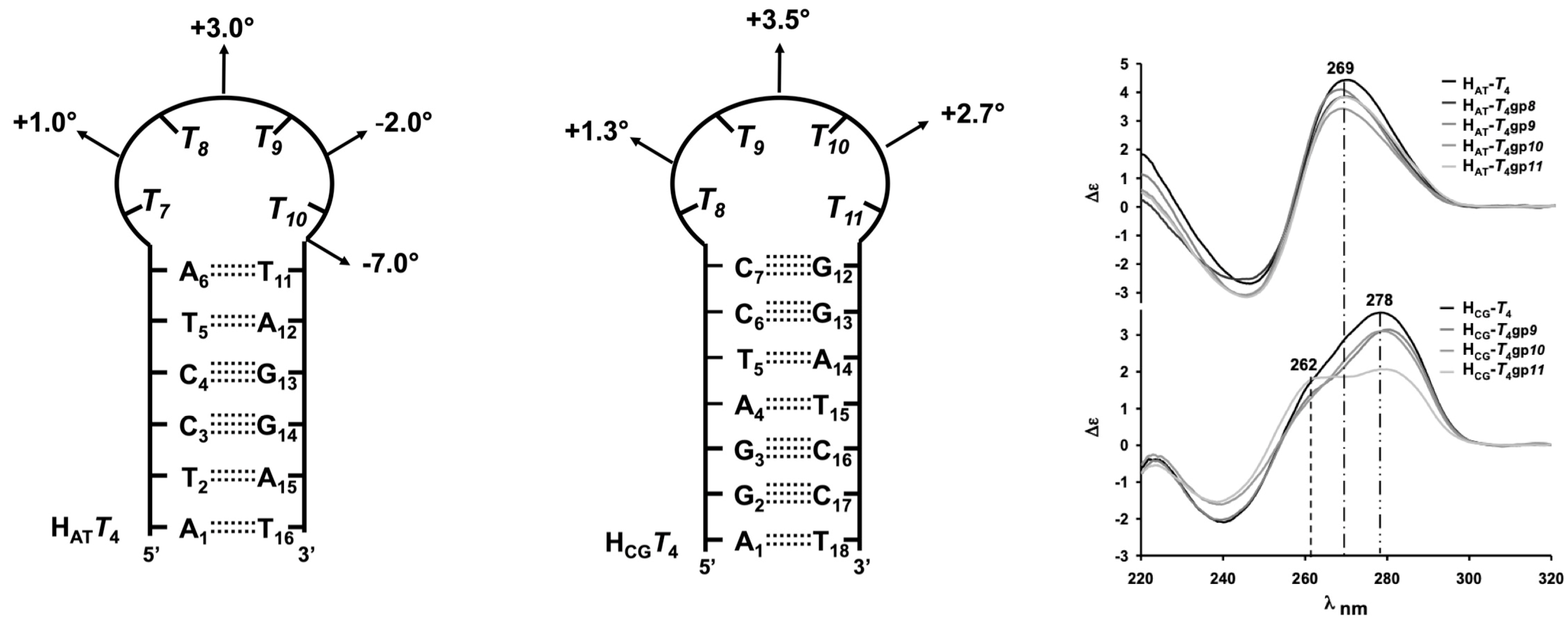
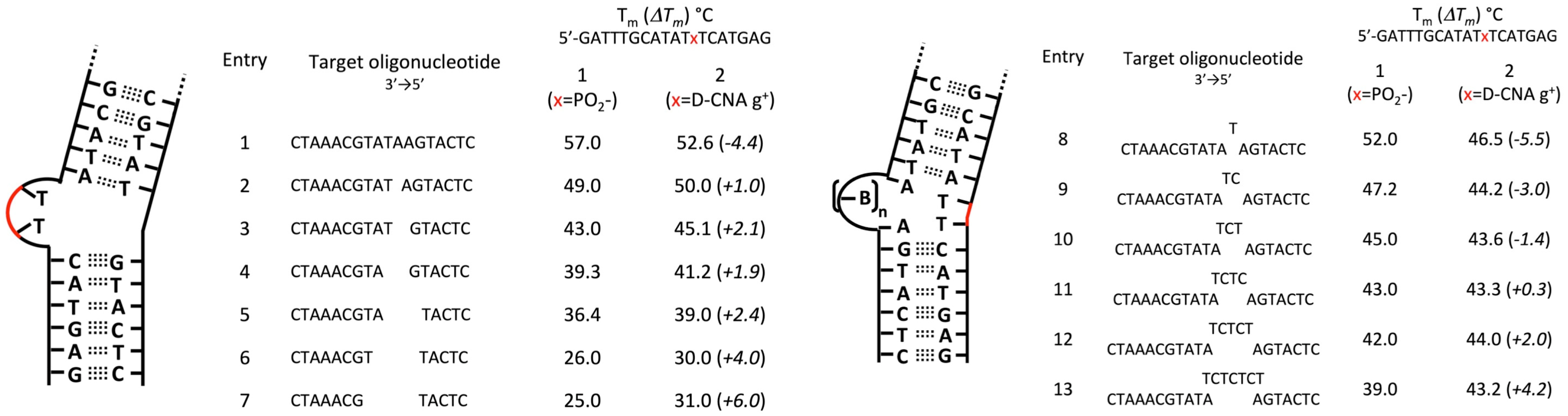
- Gerland, B.; Millard, P.; Dupouy, C.; Renard, B.-L.; Escudier, J.-M. Stabilization of hairpins and bulged secondary structures of nucleic acids by single incorporation of α,β-D-CNA featuring a gauche(+) alpha torsional angle. RSC Adv. 2014, 4, 48821–48826. [Google Scholar] [CrossRef]
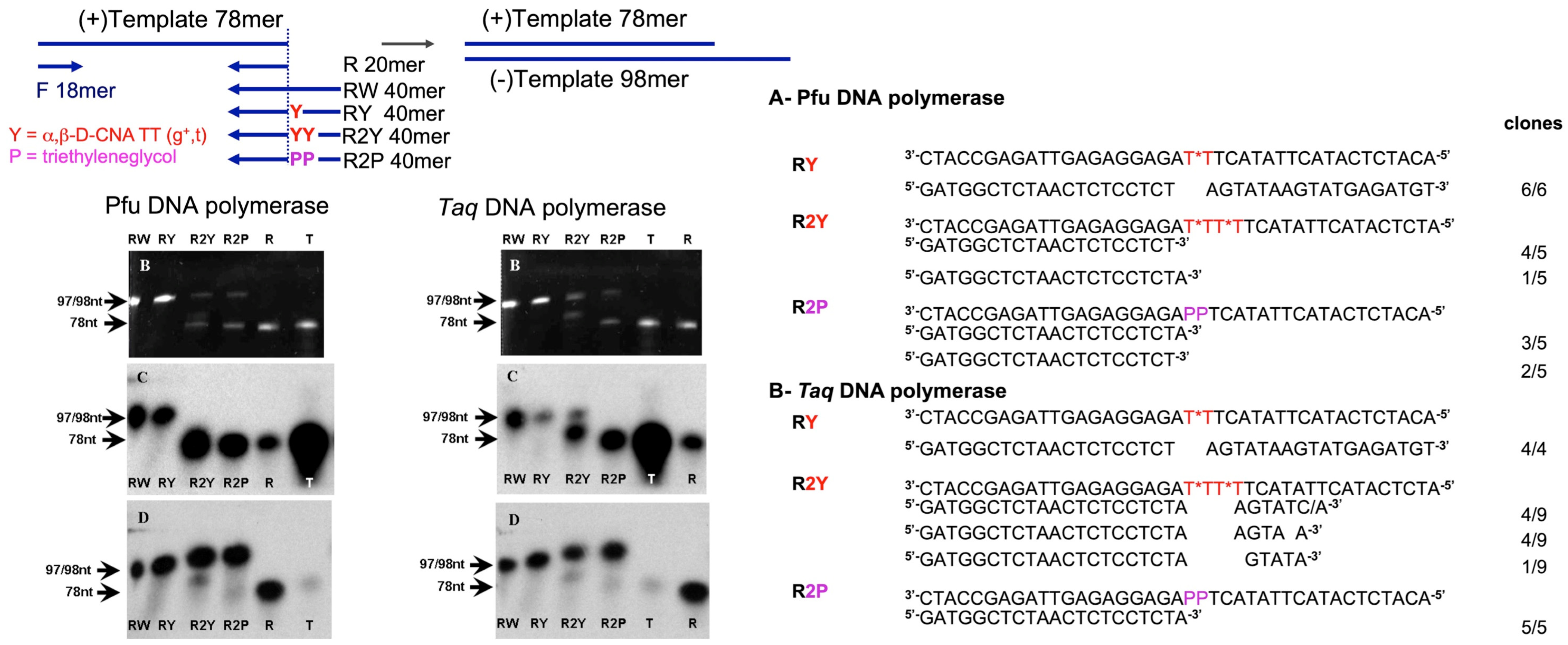
- Martinez, O.; Ecochard, V.; Mahéo, S.; Gross, G.; Bodin, P.; Teissié, J.; Escudier, J.-M.; Paquereau, L. a,β-D-constrained nucleic acids are strong terminators of thermostable DNA polymerases in polymerase chain reaction. PLoS ONE 2011, 6, e25510. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Gerland, B.; Escudier, J.-M.; Swayze, E.E.; Seth, P.P. Differential effects on allele selective silencing of mutant huntingtin by two stereoisomers of α,β-constrained nucleic acid. ACS Chem. Biol. 2014, 9, 1975–1979. [Google Scholar] [CrossRef]
- Højland, T.; Kumar, S.; Babu, B.R.; Umemoto, T.; Albæk, N.; Sharma, P.K.; Nielsen, P.; Wengel, J. LNA (Locked Nucleic Acid) and analogs as triplex-forming oligonucleotides. Org. Biomol. Chem. 2007, 5, 2375–2379. [Google Scholar] [CrossRef]
- Lou, C.; Vester, B.; Wengel, J. Oligonucleotides containing a piperazino-modified 2′-amino-LNA monomer exhibit very high duplex stability and remarkable nuclease resistance. Chem. Commun. 2015, 51, 4024–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, C.; Samuelsen, S.V.; Christensen, N.J.; Vester, B.; Wengel, J. Oligonucleotides containing aminated 2’-amino-LNA nucleotides: Synthesis and strong binding to complementary DNA and RNA. Bioconjug. Chem. 2017, 28, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, M.B.; Christensen, N.J.; Joergensen, P.T.; Jensen, K.J.; Wengel, J.; Lou, C. Polyamine-functionalized 2’-amino-LNA in oligonucleotides: Facile synthesis of new monomers and high-affinity binding towards ssDNA and dsDNA. Chem. Eur. J. 2021, 27, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Escudier, J.-M.; Payrastre, C.; Gerland, B.; Tarrat, N. Convertible and conformationally constrained nucleic acids (C2NAs). Org. Biomol. Chem. 2019, 17, 6386–6397. [Google Scholar] [CrossRef]
- Cadoni, E.; De Paepe, L.; Manicardi, A.; Madder, A. Behyond small molecules: Targeting G-Quadruplex structures with oligonucleotides and their analogues. Nucleic Acids Res. 2021, 49, 6638–6659. [Google Scholar] [CrossRef]
- Tateishi-Karimata, H.; Sugimito, N. Roles of non-canonical structres of nucleic acids in cancer and neurodegenerative diseases. Nucleic Acids Res. 2021, 49, 7839–7855. [Google Scholar] [CrossRef]























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chardet, C.; Payrastre, C.; Gerland, B.; Escudier, J.-M. Convertible and Constrained Nucleotides: The 2’-Deoxyribose 5’-C-Functionalization Approach, a French Touch. Molecules 2021, 26, 5925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195925
Chardet C, Payrastre C, Gerland B, Escudier J-M. Convertible and Constrained Nucleotides: The 2’-Deoxyribose 5’-C-Functionalization Approach, a French Touch. Molecules. 2021; 26(19):5925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195925
Chicago/Turabian StyleChardet, Crystalle, Corinne Payrastre, Béatrice Gerland, and Jean-Marc Escudier. 2021. "Convertible and Constrained Nucleotides: The 2’-Deoxyribose 5’-C-Functionalization Approach, a French Touch" Molecules 26, no. 19: 5925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26195925