
Computational Analysis of Chemical Space of Natural Compounds Interacting with Sulfotransferases

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Natural SULT Ligands
2.1.1. Compounds Collection for the In-House Library of Natural SULT Ligands
- SMILES and InChi keys structure notations;
- the trivial name of the ligand as provided in the literature;
- PubChem CID;
- ligand type (substrate/inhibitor);
- binding affinity data (IC50, Km), where available;
- the SULT isoform the ligand interacts to;
- physico-chemical parameters important for ADME properties: molecular weight, n-octanol/water partition coefficient, aqueous solubility, number of hydrogen atom donors/acceptors, calculated in MOE;
- literature sources.
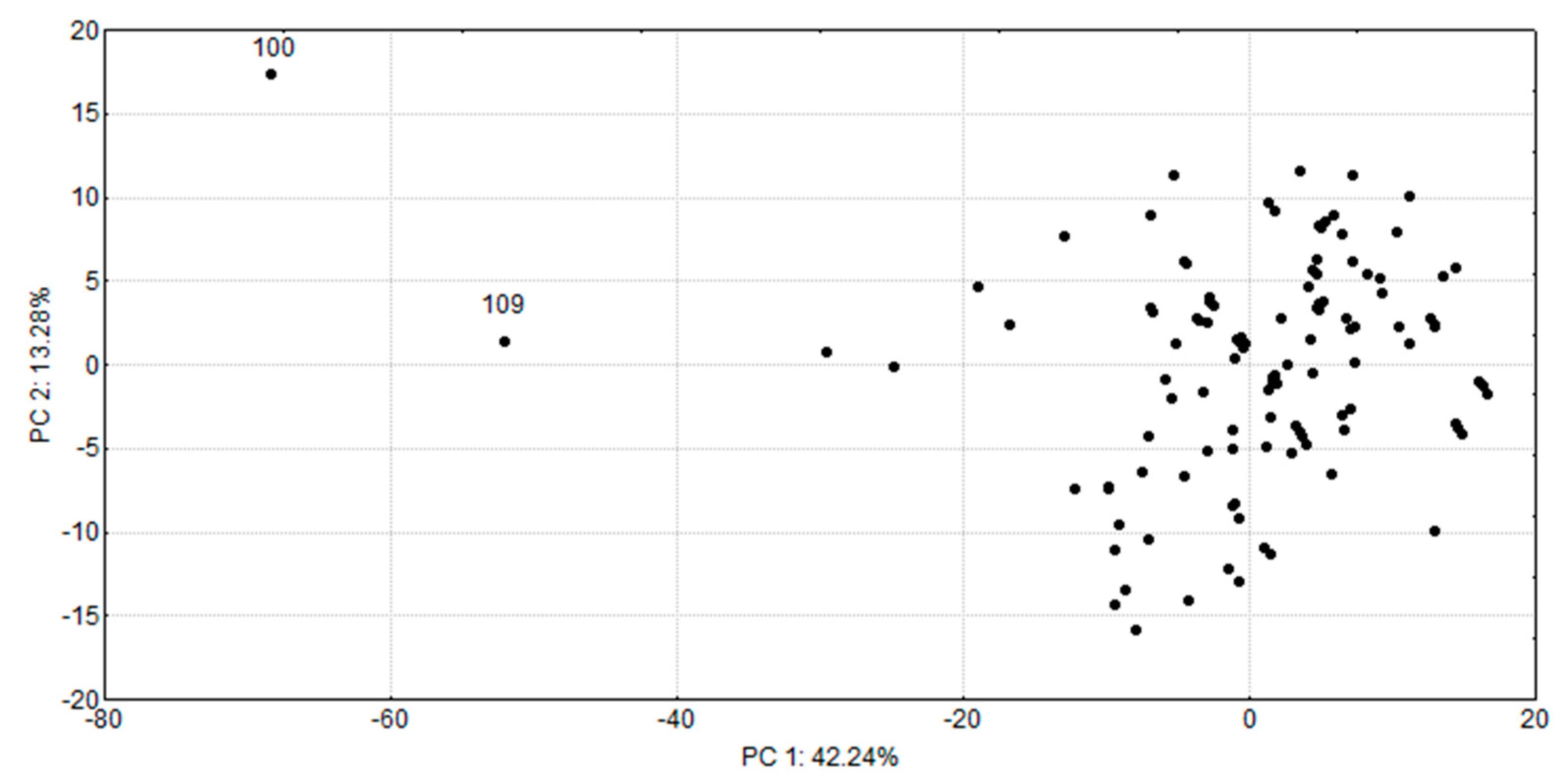
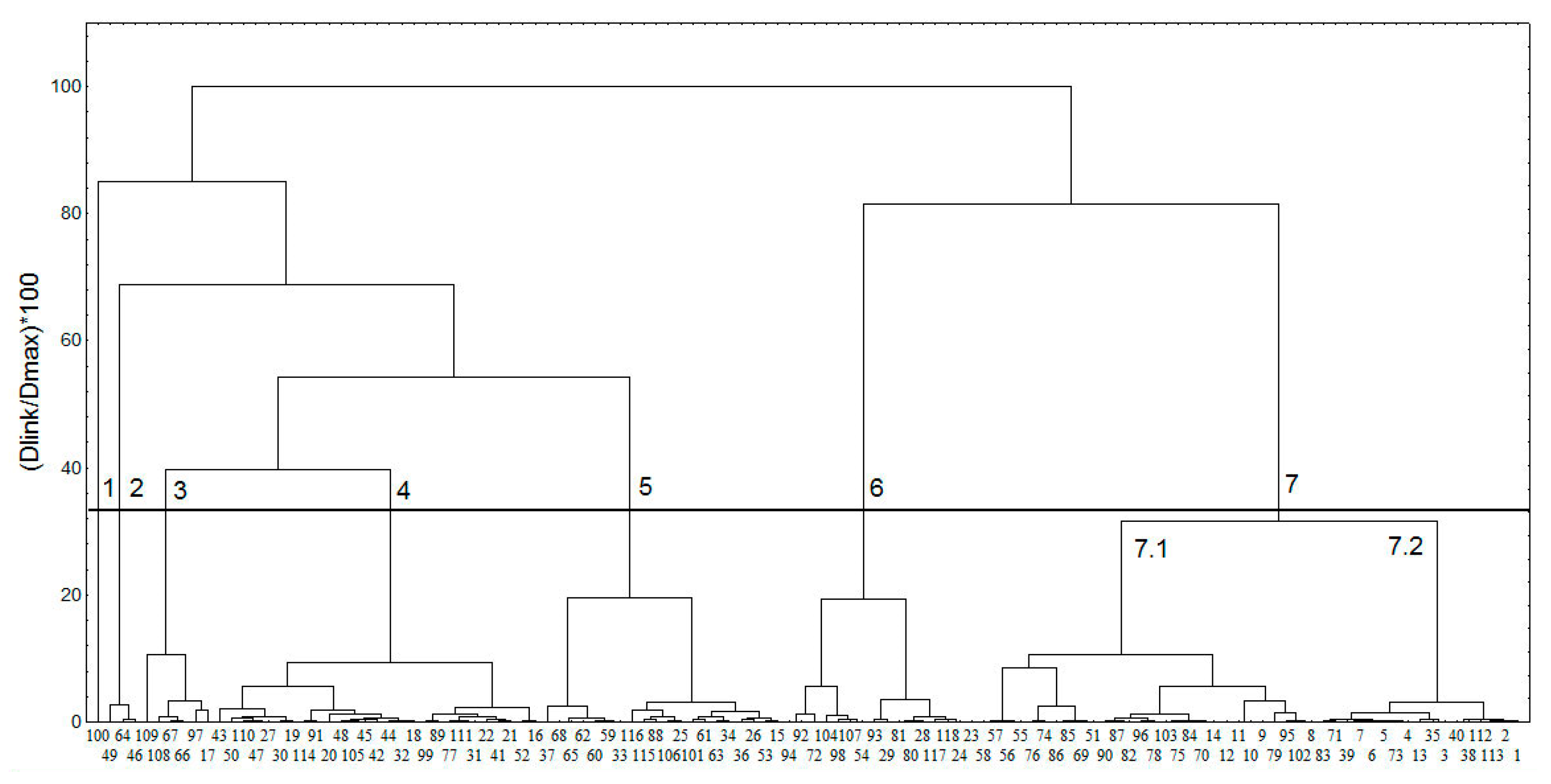
2.1.2. Characterization of the Chemical Space of the Natural SULT Ligands
- ASA+—water accessible surface area of all atoms with positive partial charge;
- ASA−—water accessible surface area of all atoms with negative partial charge;
- ASA_H—water accessible surface area of all hydrophobic atoms;
- ASA_P—water accessible surface area of all polar atoms;
- n_acc—number of H-bond acceptor atoms;
- n_don—number of H-bond donor atoms;
- rings—number of rings;
- volume—van der Waals volume;
- surface—van der Waals surface area;
- weight—molecular weight;
- logP(o/w)—octanol-water partition coefficient.
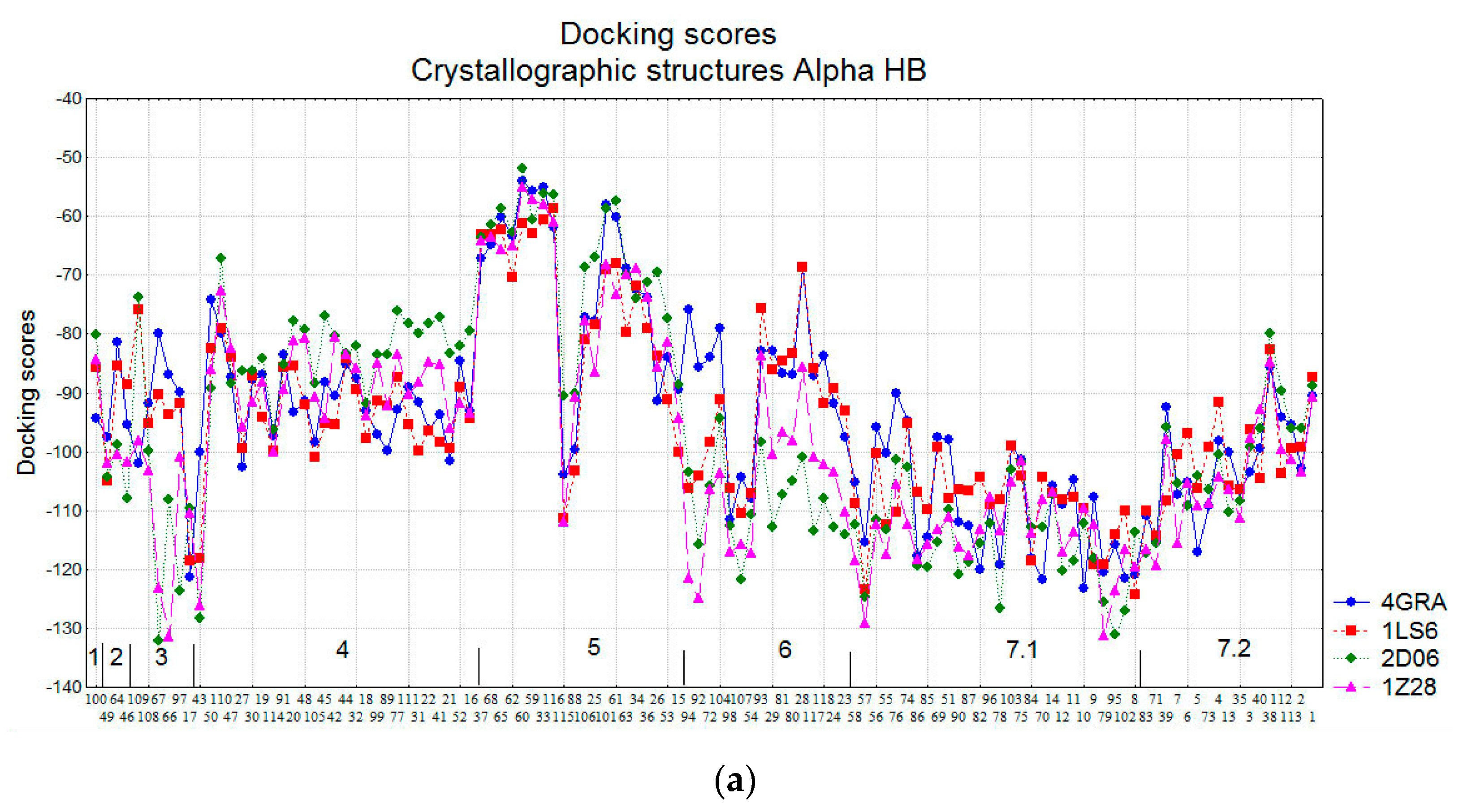
2.1.3. Docking Results
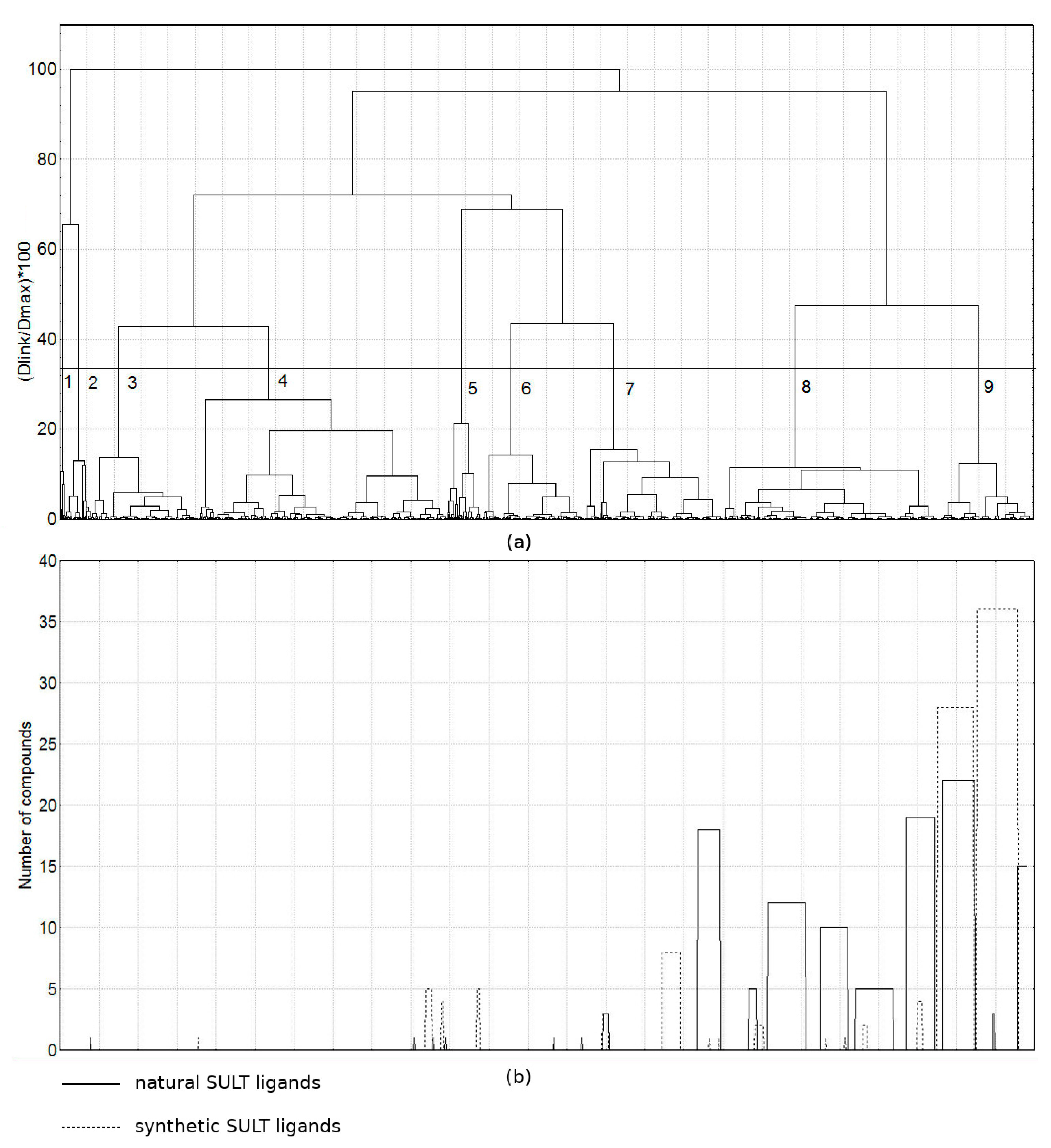
2.2. Comparison of the Groups of Natural SULT Ligands (118 Compounds), Synthetic SULT Ligands (102 Compounds) and Other Natural Products (1220 Compounds)
2.2.1. Chemical Space Analysis
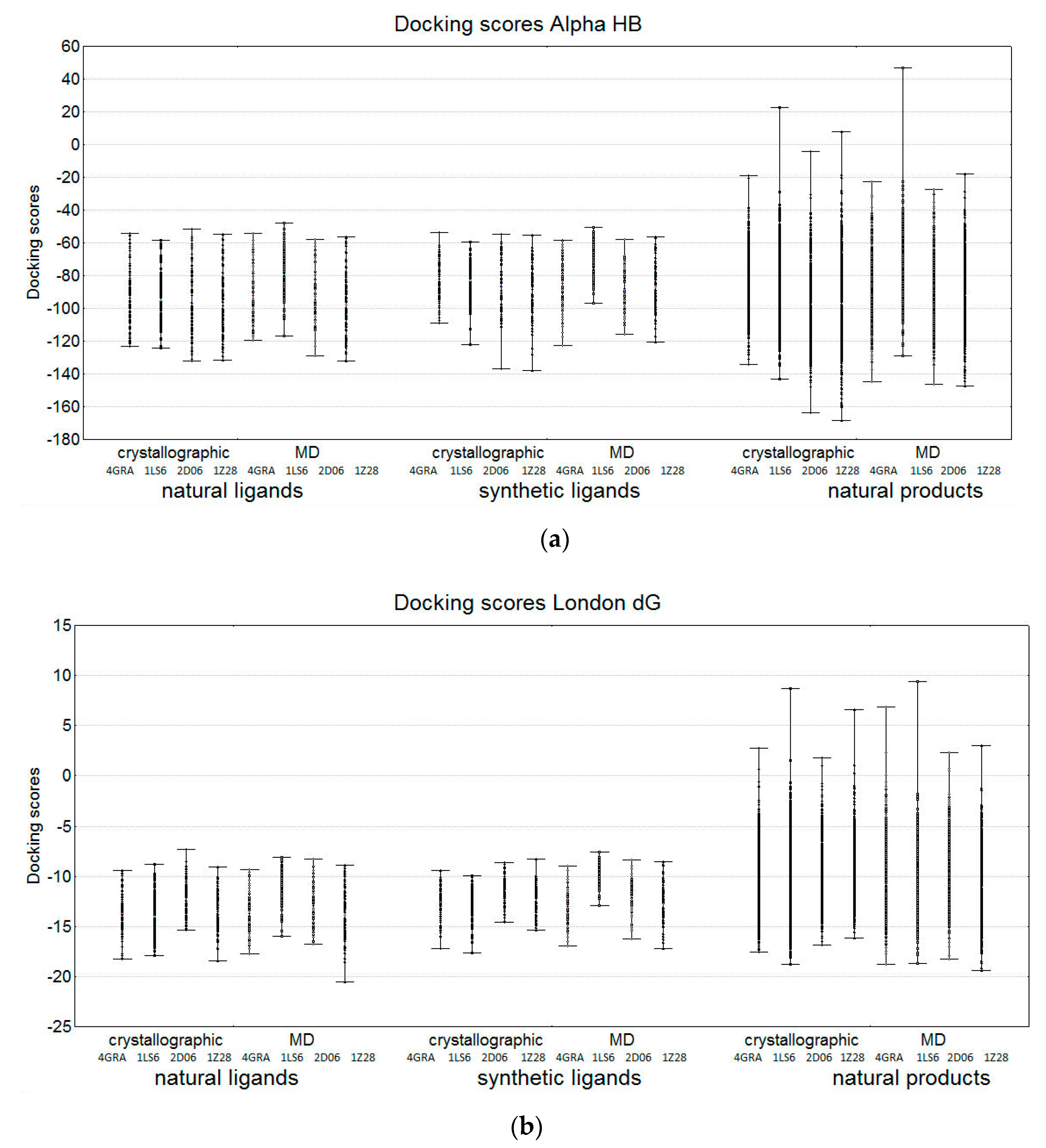
2.2.2. Comparison of the Docking Results in the Three Groups of Compounds
2.2.3. Search for Potential SULT Ligands among the Group of other Natural Products
3. Materials and Methods
3.1. Protein Targets
3.2. Datasets Preparation
3.3. Chemical Structures Preparation and Calculation of Molecular Descriptors
3.4. Molecular Dynamics
3.5. Docking
3.6. Statistical Analysis
3.7. ROCS Software
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sun, H.; Scott, D.O. Structure-Based Drug Metabolism Predictions for Drug Design. Chem. Biol. Drug Des. 2010, 75, 3–17. [Google Scholar] [CrossRef]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and Enzymes in the Metabolism of Drugs and Other Xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef]
- Shimada, T. Xenobiotic-Metabolizing Enzymes Involved in Activation and Detoxification of Carcinogenic Polycyclic Aromatic Hydrocarbons. Drug Metab. Pharmacokinet. 2006, 21, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in Silico Structure-Based ADMET Prediction in Drug Discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef]
- Pratt, W.B.; Taylor, P. (Eds.) Principles of Drug Action: The Basis of Pharmacology, 3rd ed.; Churchill Livingstone: New York, NY, USA, 1990; ISBN 978-0-443-08676-2. [Google Scholar]
- Tibbs, Z.E.; Rohn-Glowacki, K.J.; Crittenden, F.; Guidry, A.L.; Falany, C.N. Structural Plasticity in the Human Cytosolic Sulfotransferase Dimer and Its Role in Substrate Selectivity and Catalysis. Drug Metab. Pharmacokinet. 2015, 30, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ako, R.; Wu, B. Crystal Structures of Human Sulfotransferases: Insights into the Mechanisms of Action and Substrate Selectivity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Bojarová, P.; Williams, S.J. Sulfotransferases, Sulfatases and Formylglycine-Generating Enzymes: A Sulfation Fascination. Curr. Opin. Chem. Biol. 2008, 12, 573–581. [Google Scholar] [CrossRef]
- Chapman, E.; Best, M.D.; Hanson, S.R.; Wong, C.-H. Sulfotransferases: Structure, Mechanism, Biological Activity, Inhibition, and Synthetic Utility. Angew. Chem. Int. Ed. 2004, 43, 3526–3548. [Google Scholar] [CrossRef]
- Coughtrie, M.W.H. Function and Organization of the Human Cytosolic Sulfotransferase (SULT) Family. Chem. Biol. Interact. 2016, 259, 2–7. [Google Scholar] [CrossRef]
- Vrba, J.; Papoušková, B.; Kosina, P.; Lněničková, K.; Valentová, K.; Ulrichová, J. Identification of Human Sulfotransferases Active towards Silymarin Flavonolignans and Taxifolin. Metabolites 2020, 10, 329. [Google Scholar] [CrossRef]
- Brand, W.; Boersma, M.G.; Bik, H.; den Hil, E.F.H.; Vervoort, J.; Barron, D.; Meinl, W.; Glatt, H.; Williamson, G.; van Bladeren, P.J.; et al. Phase II Metabolism of Hesperetin by Individual UDP-Glucuronosyltransferases and Sulfotransferases and Rat and Human Tissue Samples. Drug Metab. Dispos. 2010, 38, 617–625. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Carbonell, P.; Lagorce, D.; Villoutreix, B.O.; Moroy, G.; Miteva, M.A. In Silico Mechanistic Profiling to Probe Small Molecule Binding to Sulfotransferases. PLoS ONE 2013, 8, e73587. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Pan, P.W.; Dombrovski, L.; Najmanovich, R.; Tempel, W.; Dong, A.; Loppnau, P.; Martin, F.; Thonton, J.; Edwards, A.M.; et al. Structural and Chemical Profiling of the Human Cytosolic Sulfotransferases. PLoS Biol. 2007, 5, e97. [Google Scholar] [CrossRef]
- Dajani, R.; Hood, A.M.; Coughtrie, M.W.H. A Single Amino Acid, Glu146, Governs the Substrate Specificity of a Human Dopamine Sulfotransferase, SULT1A3. Mol. Pharmacol. 1998, 54, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Fuda, H.; Lee, Y.C.; Negishi, M.; Strott, C.A.; Pedersen, L.C. Crystal Structure of Human Cholesterol Sulfotransferase (SULT2B1b) in the Presence of Pregnenolone and 3′-Phosphoadenosine 5′-Phosphate: Rationale for Specificity Differences between Prototypical SULT2A1 and the SULT2B1 Isoforms. J. Biol. Chem. 2003, 278, 44593–44599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Cook, I.; Leyh, T.S. The NSAID Allosteric Site of Human Cytosolic Sulfotransferases. J. Biol. Chem. 2017, 292, 20305–20312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, I.; Wang, T.; Girvin, M.; Leyh, T.S. The Structure of the Catechin-Binding Site of Human Sulfotransferase 1A1. Proc. Natl. Acad. Sci. USA 2016, 113, 14312–14317. [Google Scholar] [CrossRef] [Green Version]
- Cook, I.; Wang, T.; Falany, C.N.; Leyh, T.S. The Allosteric Binding Sites of Sulfotransferase 1A1. Drug Metab. Dispos. 2015, 43, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Cook, I.; Wang, T.; Falany, C.N.; Leyh, T.S. High Accuracy in Silico Sulfotransferase Models. J. Biol. Chem. 2013, 288, 34494–34501. [Google Scholar] [CrossRef] [Green Version]
- Cook, I.; Wang, T.; Almo, S.C.; Kim, J.; Falany, C.N.; Leyh, T.S. The Gate That Governs Sulfotransferase Selectivity. Biochemistry 2013, 52, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Qi, R.; Liu, Y.; Zhao, L.; Han, W. Mechanistic Insights into the Effect of Ligands on Structural Stability and Selectivity of Sulfotransferase 2A1 (SULT2A1). ACS Omega 2019, 4, 22021–22034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakers, C.; Schumacher, F.; Meinl, W.; Glatt, H.; Kleuser, B.; Wolber, G. In Silico Prediction of Human Sulfotransferase 1E1 Activity Guided by Pharmacophores from Molecular Dynamics Simulations. J. Biol. Chem. 2016, 291, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Dudas, B.; Toth, D.; Perahia, D.; Nicot, A.B.; Balog, E.; Miteva, M.A. Insights into the Substrate Binding Mechanism of SULT1A1 through Molecular Dynamics with Excited Normal Modes Simulations. Sci. Rep. 2021, 11, 13129. [Google Scholar] [CrossRef]
- Gamage, N.U.; Duggleby, R.G.; Barnett, A.C.; Tresillian, M.; Latham, C.F.; Liyou, N.E.; McManus, M.E.; Martin, J.L. Structure of a Human Carcinogen-Converting Enzyme, SULT1A1: Structural and Kinetic Implications of Substrate Inhibition. J. Biol. Chem. 2003, 278, 7655–7662. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Gamage, N.; Martin, J.L.; McManus, M.E. Human Cytosolic Sulfotransferase SULT1A1. Int. J. Biochem. Cell Biol. 2007, 39, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, K.V.; Agarwal, V.; James, M.O. Sulfonation and Glucuronidation of Hydroxylated Bromodiphenyl Ethers in Human Liver. Chemosphere 2019, 226, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Yang, L.; He, D.; Li, Y.; Shi, M.; Zhang, J. Metabolic Pathways and Pharmacokinetics of Natural Medicines with Low Permeability. Drug Metab. Rev. 2017, 49, 464–476. [Google Scholar] [CrossRef]
- Wu, B.; Basu, S.; Meng, S.; Wang, X.; Zhang, S. Regioselective Sulfation and Glucuronidation of Phenolics: Insights into the Structural Basis of Conjugation. Curr. Drug Metab. 2011, 12, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Glatt, H. Sulfotransferases in the Bioactivation of Xenobiotics. Chem. Biol. Interact. 2000, 129, 141–170. [Google Scholar] [CrossRef]
- Vrba, J.; Papoušková, B.; Lněničková, K.; Kosina, P.; Křen, V.; Ulrichová, J. Identification of UDP-Glucuronosyltransferases Involved in the Metabolism of Silymarin Flavonolignans. J. Pharm. Biomed. Anal. 2020, 178, 112972. [Google Scholar] [CrossRef]
- Vrba, J.; Papoušková, B.; Roubalová, L.; Zatloukalová, M.; Biedermann, D.; Křen, V.; Valentová, K.; Ulrichová, J.; Vacek, J. Metabolism of Flavonolignans in Human Hepatocytes. J. Pharm. Biomed. Anal. 2018, 152, 94–101. [Google Scholar] [CrossRef]
- Vacek, J.; Papoušková, B.; Kosina, P.; Vrba, J.; Křen, V.; Ulrichová, J. Biotransformation of Flavonols and Taxifolin in Hepatocyte in Vitro Systems as Determined by Liquid Chromatography with Various Stationary Phases and Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry. J. Chromatogr. B 2012, 899, 109–115. [Google Scholar] [CrossRef]
- Vrba, J.; Kren, V.; Vacek, J.; Papouskova, B.; Ulrichova, J. Quercetin, Quercetin Glycosides and Taxifolin Differ in Their Ability to Induce AhR Activation and CYP1A1 Expression in HepG2 Cells. Phytother. Res. 2012, 26, 1746–1752. [Google Scholar] [CrossRef]
- Falany, C.N.; Wheeler, J.; Oh, T.S.; Falany, J.L. Steroid Sulfation by Expressed Human Cytosolic Sulfotransferases. J. Steroid Biochem. Mol. Biol. 1994, 48, 369–375. [Google Scholar] [CrossRef]
- Eaton, E.A.; Walle, U.K.; Lewis, A.J.; Hudson, T.; Wilson, A.A.; Walle, T. Flavonoids, Potent Inhibitors of the Human P-Form Phenolsulfotransferase. Potential Role in Drug Metabolism and Chemoprevention. Drug Metab. Dispos. 1996, 24, 232–237. [Google Scholar]
- Harris, R.M.; Hawker, R.J.; Langman, M.J.S.; Singh, S.; Waring, R.H. Inhibition of Phenolsulphotransferase by Salicylic Acid: A Possible Mechanism by Which Aspirin May Reduce Carcinogenesis. Gut 1998, 42, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Dajani, R.; Cleasby, A.; Neu, M.; Wonacott, A.J.; Jhoti, H.; Hood, A.M.; Modi, S.; Hersey, A.; Taskinen, J.; Cooke, R.M.; et al. X-Ray Crystal Structure of Human Dopamine Sulfotransferase, SULT1A3: Molecular Modeling and Quantitative Structure-Activity Relationship Analysis Demonstrate a Molecu-Lar Basis for Sulfotransferase Substrate Specificity. J. Biol. Chem. 1999, 274, 37862–37868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.M.; Waring, R.H.; Kirk, C.J.; Hughes, P.J. Sulfation of “Estrogenic” Alkylphenols and 17β-Estradiol by Human Platelet Phenol Sulfotransferases. J. Biol. Chem. 2000, 275, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spink, B.C.; Katz, B.H.; Hussain, M.M.; Pang, S.; Connor, S.P.; Aldous, K.M.; Gierthy, J.F.; Spink, D.C. SULT1A1 Catalyzes 2-Methoxyestradiol Sulfonation in MCF-7 Breast Cancer Cells. Carcinogenesis 2000, 21, 1947–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatt, H.; Boeing, H.; Engelke, C.E.H.; Ma, L.; Kuhlow, A.; Pabel, U.; Pomplun, D.; Teubner, W.; Meinl, W. Human Cytosolic Sulphotransferases: Genetics, Characteristics, Toxicological Aspects. Mutat. Res. Mol. Mech. Mutagen. 2001, 482, 27–40. [Google Scholar] [CrossRef]
- Honma, W.; Kamiyama, Y.; Yoshinari, K.; Sasano, H.; Shimada, M.; Nagata, K.; Yamazoe, Y. Enzymatic Characterization and Interspecies Difference of Phenol Sulfotransferases, ST1A Forms. Drug Metab. Dispos. 2001, 29, 274–281. [Google Scholar] [PubMed]
- Li, X.; Clemens, D.L.; Cole, J.R.; Anderson, R.J. Characterization of Human Liver Thermostable Phenol Sulfotransferase (SULT1A1) Allozymes with 3,3′,5-Triiodothyronine as the Substrate. J. Endocrinol. 2001, 171, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Mesía-Vela, S.; Sańchez, R.I.; Estrada-Muñiz, E.; Alavez-Solano, D.; Torres-Sosa, C.; Jiménez-Estrada, M.; Reyes-Chilpa, R.; Kauffman, F.C. Natural Products Isolated from Mexican Medicinal Plants: Novel Inhibitors of Sulfotransferases, SULT1A1 and SULT2A1. Phytomedicine 2001, 8, 481–488. [Google Scholar] [CrossRef]
- Mesía-Vela, S.; Kauffman, F.C. Inhibition of Rat Liver Sulfotransferases SULT1A1 and SULT2A1 and Glucuronosyltransferase by Dietary Flavonoids. Xenobiotica 2003, 33, 1211–1220. [Google Scholar] [CrossRef]
- Taskinen, J.; Ethell, B.T.; Pihlavisto, P.; Hood, A.M.; Burchell, B.; Coughtrie, M.W.H. Conjugation of Catechols by Recombinant Human Sulfotransferases, Udp-Glucuronosyltransferases, and Soluble Catechol O-Methyltransferase: Structure-Conjugation Relationships and Predictive Models. Drug Metab. Dispos. 2003, 31, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-T.; Yen, G.-C. Effects of Phenolic Acids on Human Phenolsulfotransferases in Relation to Their Antioxidant Activity. J. Agric. Food Chem. 2003, 51, 1474–1479. [Google Scholar] [CrossRef]
- Harris, R.M.; Wood, D.M.; Bottomley, L.; Blagg, S.; Owen, K.; Hughes, P.J.; Waring, R.H.; Kirk, C.J. Phytoestrogens Are Potent Inhibitors of Estrogen Sulfation: Implications for Breast Cancer Risk and Treatment. J. Clin. Endocrinol. Metab. 2004, 89, 1779–1787. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Ogura, K.; Takahashi, E.; Harada, T.; Nishiyama, T.; Muro, K.; Hiratsuka, A.; Kadota, S.; Watabe, T. Regioselective Monosulfation and Disulfation of the Phytoestrogens Daidzein and Genistein by Human Liver Sulfotransferases. Drug Metab. Pharmacokinet. 2004, 19, 216–226. [Google Scholar] [CrossRef]
- Aust, S.; Jaeger, W.; Klimpfinger, M.; Mayer, K.; Baravalle, G.; Ekmekcioglu, C.; Thalhammer, T. Biotransformation of Melatonin in Human Breast Cancer Cell Lines: Role of Sulfotransferase 1A1. J. Pineal Res. 2005, 39, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Meinl, W.; Pabel, U.; Osterloh-Quiroz, M.; Hengstler, J.G.; Glatt, H. Human Sulphotransferases Are Involved in the Activation of Aristolochic Acids and Are Expressed in Renal Target Tissue. Int. J. Cancer 2006, 118, 1090–1097. [Google Scholar] [CrossRef]
- Nishimuta, H.; Ohtani, H.; Tsujimoto, M.; Ogura, K.; Hiratsuka, A.; Sawada, Y. Inhibitory Effects of Various Beverages on Human Recombinant Sulfotransferase Isoforms SULT1A1 and SULT1A3. Biopharm. Drug Dispos. 2007, 28, 491–500. [Google Scholar] [CrossRef]
- Riches, Z.; Bloomer, J.C.; Coughtrie, M.W.H. Comparison of 2-Aminophenol and 4-Nitrophenol as in Vitro Probe Substrates for the Major Human Hepatic Sulfotransferase, SULT1A1, Demonstrates Improved Selectivity with 2-Aminophenol. Biochem. Pharmacol. 2007, 74, 352–358. [Google Scholar] [CrossRef]
- Ung, D.; Nagar, S. Variable Sulfation of Dietary Polyphenols by Recombinant Human Sulfotransferase (SULT) 1A1 Genetic Variants and SULT1E1. Drug Metab. Dispos. 2007, 35, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, S.; Idell, S.; Fu, J.; Carter, G.; Snow, R.; Liu, M.-C. Cigarette Smoke Toxicants as Substrates and Inhibitors for Human Cytosolic SULTs. Toxicol. Appl. Pharmacol. 2007, 221, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Liu, M.-Y.; Suiko, M.; Sakakibara, Y.; Liu, M.-C. Hydroxylated Serotonin and Dopamine as Substrates and Inhibitors for Human Cytosolic SULT1A3. J. Neurochem. 2007, 103, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Saruwatari, A.; Okamura, S.; Nakajima, Y.; Narukawa, Y.; Takeda, T.; Tamura, H. Pomegranate Juice Inhibits Sulfoconjugation in Caco-2 Human Colon Carcinoma Cells. J. Med. Food 2008, 11, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Waring, R.H.; Ayers, S.; Gescher, A.J.; Glatt, H.-R.; Meinl, W.; Jarratt, P.; Kirk, C.J.; Pettitt, T.; Rea, D.; Harris, R.M. Phytoestrogens and Xenoestrogens: The Contribution of Diet and Environment to Endocrine Disruption. J. Steroid Biochem. Mol. Biol. 2008, 108, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Chen, Y.; Zhou, T.; Chen, G. Sulfation of Dietary Flavonoids by Human Sulfotransferases. Xenobiotica 2009, 39, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senggunprai, L.; Yoshinari, K.; Yamazoe, Y. Inhibitory Effects of Kynurenic Acid, a Tryptophan Metabolite, and Its Derivatives on Cytosolic Sulfotransferases. Biochem. J. 2009, 422, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Meinl, W.; Glatt, H.-R.; Barron, D.; Stalmach, A.; Steiling, H.; Crozier, A.; Williamson, G. In Vitro and in Vivo Conjugation of Dietary Hydroxycinnamic Acids by UDP-Glucuronosyltransferases and Sulfotransferases in Humans. J. Nutr. Biochem. 2010, 21, 1060–1068. [Google Scholar] [CrossRef]
- Kurogi, K.; Chepak, A.; Hanrahan, M.T.; Liu, M.-Y.; Sakakibara, Y.; Suiko, M.; Liu, M.-C. Sulfation of Opioid Drugs by Human Cytosolic Sulfotransferases: Metabolic Labeling Study and Enzymatic Analysis. Eur. J. Pharm. Sci. 2014, 62, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Xi, Y.; Luo, L.; Zhou, C.; Kurogi, K.; Sakakibara, Y.; Suiko, M.; Liu, M.-C. Sulfate Conjugation of Daphnetin by the Human Cytosolic Sulfotransferases. J. Ethnopharmacol. 2016, 189, 250–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy: The Principles and Practice of Numerical Classification; WF Freeman & Co.: San Francisco, CA, USA, 1973. [Google Scholar]
- Glatt, H.; Meinl, W. Pharmacogenetics of Soluble Sulfotransferases (SULTs). Naunyn. Schmiedebergs Arch. Pharmacol. 2004, 369, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, R.M.; Pearce, L.B.; Roth, J.A. Purification and Kinetic Characterization of a Phenol-Sulfating Form of Phenol Sulfotransferase from Human Brain. Arch. Biochem. Biophys. 1986, 249, 464–471. [Google Scholar] [CrossRef]
- Costa, M.G.S.; Batista, P.R.; Bisch, P.M.; Perahia, D. Exploring Free Energy Landscapes of Large Conformational Changes: Molecular Dynamics with Excited Normal Modes. J. Chem. Theory Comput. 2015, 11, 2755–2767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, J.; Shi, X.; Miao, S.; Li, Y.; Wen, A. Absorption and Metabolism Characteristics of Rutin in Caco-2 Cells. Sci. World, J. 2013, 2013, e382350. [Google Scholar] [CrossRef]
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. A Fragment Library of Natural Products and Its Comparative Chemoinformatic Characterization. Mol. Inform. 2020, 39, 2000050. [Google Scholar] [CrossRef]
- Squirewell, E.J.; Duffel, M.W. The Effects of Endoxifen and Other Major Metabolites of Tamoxifen on the Sulfation of Estradiol Catalyzed by Human Cytosolic Sulfotransferases HSULT1E1 and HSULT1A1*1. Drug Metab. Dispos. 2015, 43, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Lagarde, N.; Rey, J.; Gyulkhandanyan, A.; Tufféry, P.; Miteva, M.A.; Villoutreix, B.O. Online Structure-Based Screening of Purchasable Approved Drugs and Natural Compounds: Retrospective Examples of Drug Repositioning on Cancer Targets. Oncotarget 2018, 9, 32346–32361. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain Χ1 and Χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.O.; Ambadapadi, S. Interactions of Cytosolic Sulfotransferases with Xenobiotics. Drug Metab. Rev. 2013, 45, 401–414. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Group | Designation | Number of Compounds | Compound Number Range in the Paper |
|---|---|---|---|
| Natural SULT ligands (hormones, neurotransmitters, plant-derived compounds and their metabolites produced by different enzymes) | natural SULT ligands | 118 | 1–118 |
| SULT ligands of non-natural (synthetic) origin and their metabolites produced by different enzymes | synthetic SULT ligands | 102 | 119–220 |
| Other natural products for which interactions with SULTs were not reported | other natural products | 1220 | 221–1440 |
| Descriptor | Mean | Minimum | Maximum | Standard Deviation |
|---|---|---|---|---|
| ASA+ | 210 | 99 | 514 | 66 |
| ASA− | 172 | 64 | 455 | 73 |
| ASA_H | 294 | 127 | 551 | 95 |
| ASA_P | 166 | 38 | 761 | 95 |
| n_acc | 4 | 1 | 24 | 3 |
| n_don | 3 | 0 | 17 | 2 |
| rings | 3 | 1 | 11 | 1 |
| volume | 240 | 107 | 805 | 106 |
| surface | 268 | 128 | 809 | 108 |
| weight | 272 | 108 | 1085 | 147 |
| logP(o/w) | 2.2 | −2.1 | 5.3 | 1.2 |
| Comparison of the Descriptors between the Groups of Natural and Synthetic SULT Ligands | Comparison of the Descriptors between the Groups of Natural SULT Ligands and other Natural Products | |||
|---|---|---|---|---|
| Descriptor | F | p | F | p |
| ASA+ | 30.6 | 0.0000 | 7.0 | 0.0084 |
| ASA− | 0.0 | 0.8895 | 13.5 | 0.0002 |
| ASA_H | 13.1 | 0.0004 | 91.9 | 0.0000 |
| ASA_P | 37.7 | 0.0000 | 7.8 | 0.0052 |
| n_acc | 33.8 | 0.0000 | 18.4 | 0.0000 |
| n_don | 22.1 | 0.0000 | 2.1 | 0.1501 |
| rings | 6.9 | 0.0093 | 28.6 | 0.0000 |
| volume | 1.1 | 0.2860 | 81.0 | 0.0000 |
| surface | 1.1 | 0.2949 | 77.6 | 0.0000 |
| weight | 3.2 | 0.0746 | 57.1 | 0.0000 |
| logP(o/w) | 31.8 | 0.0000 | 0.7 | 0.4101 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lessigiarska, I.; Peng, Y.; Tsakovska, I.; Alov, P.; Lagarde, N.; Jereva, D.; Villoutreix, B.O.; Nicot, A.B.; Pajeva, I.; Pencheva, T.; et al. Computational Analysis of Chemical Space of Natural Compounds Interacting with Sulfotransferases. Molecules 2021, 26, 6360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216360
Lessigiarska I, Peng Y, Tsakovska I, Alov P, Lagarde N, Jereva D, Villoutreix BO, Nicot AB, Pajeva I, Pencheva T, et al. Computational Analysis of Chemical Space of Natural Compounds Interacting with Sulfotransferases. Molecules. 2021; 26(21):6360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216360
Chicago/Turabian StyleLessigiarska, Iglika, Yunhui Peng, Ivanka Tsakovska, Petko Alov, Nathalie Lagarde, Dessislava Jereva, Bruno O. Villoutreix, Arnaud B. Nicot, Ilza Pajeva, Tania Pencheva, and et al. 2021. "Computational Analysis of Chemical Space of Natural Compounds Interacting with Sulfotransferases" Molecules 26, no. 21: 6360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216360