
Chiral Nano-Liquid Chromatography and Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Antifungal Drugs in Milk



Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of the Chromatographic Conditions
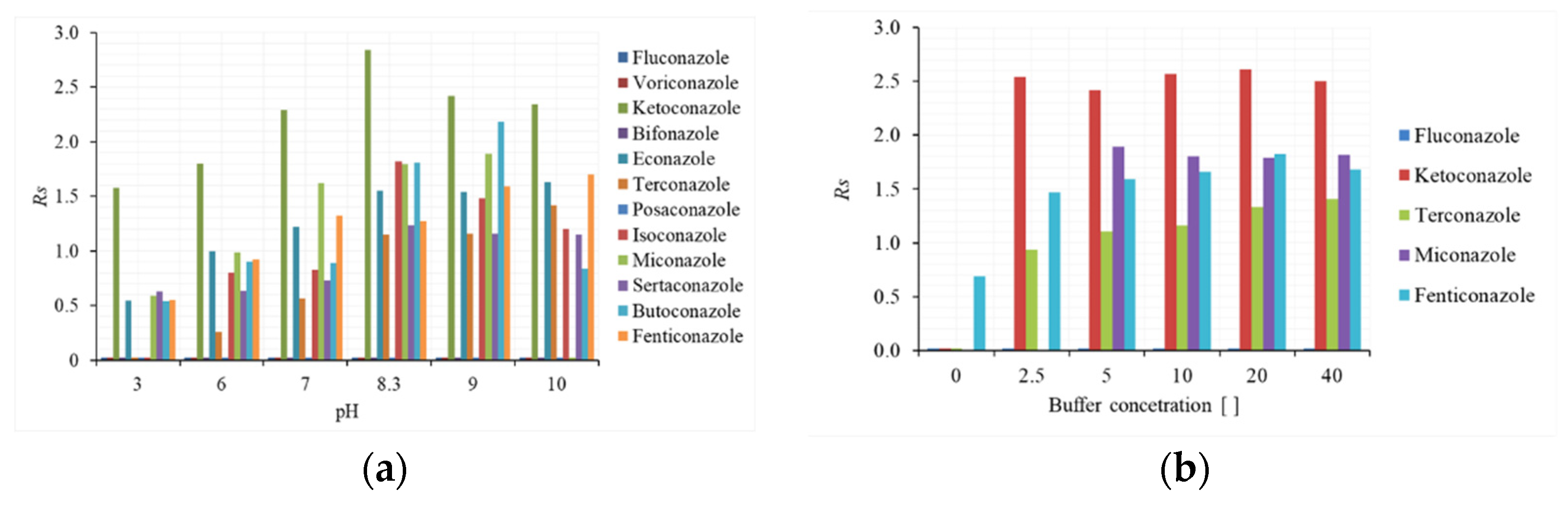
2.1.1. Effect of pH and Buffer Solution on Enantioseparation
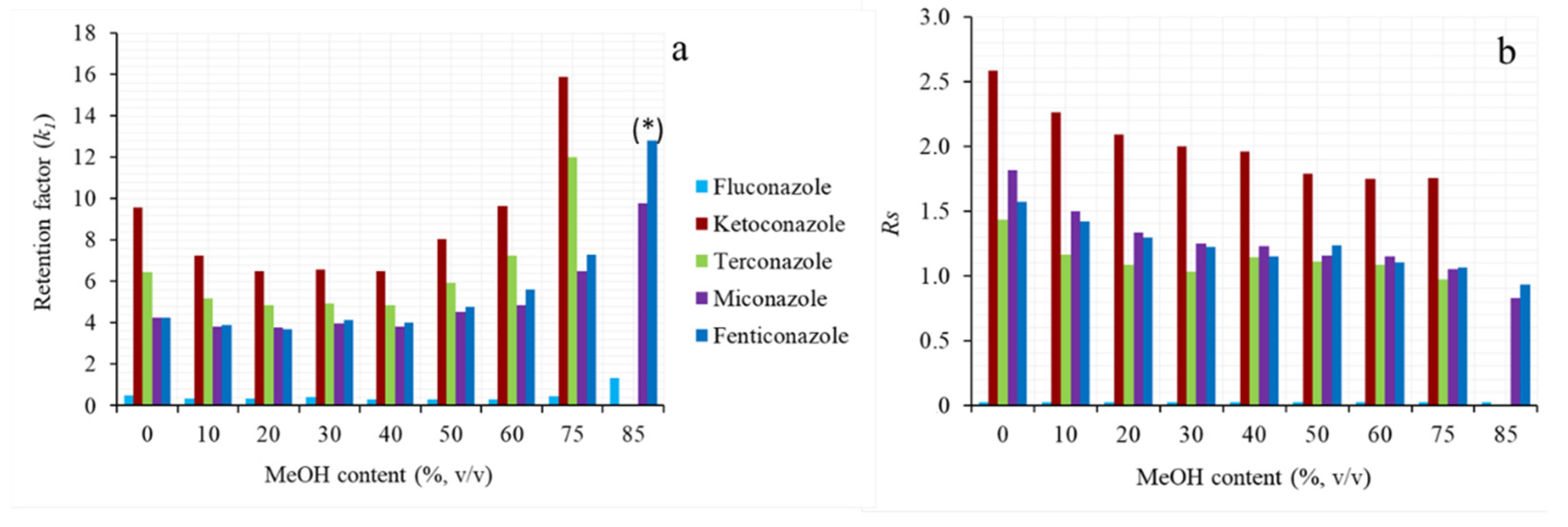
2.1.2. Effect of the Mobile Phase Composition: Water Content and Organic Modifier
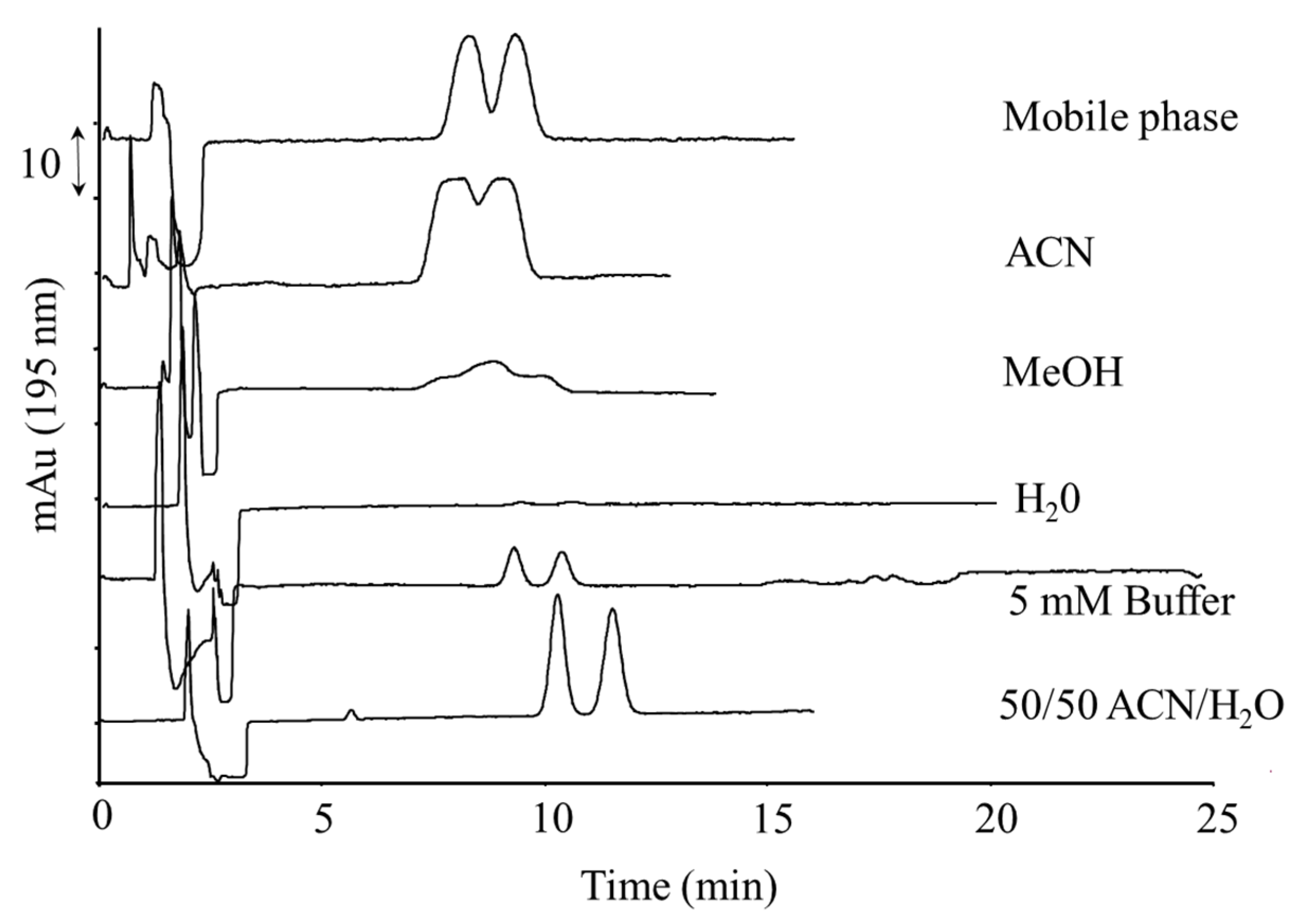
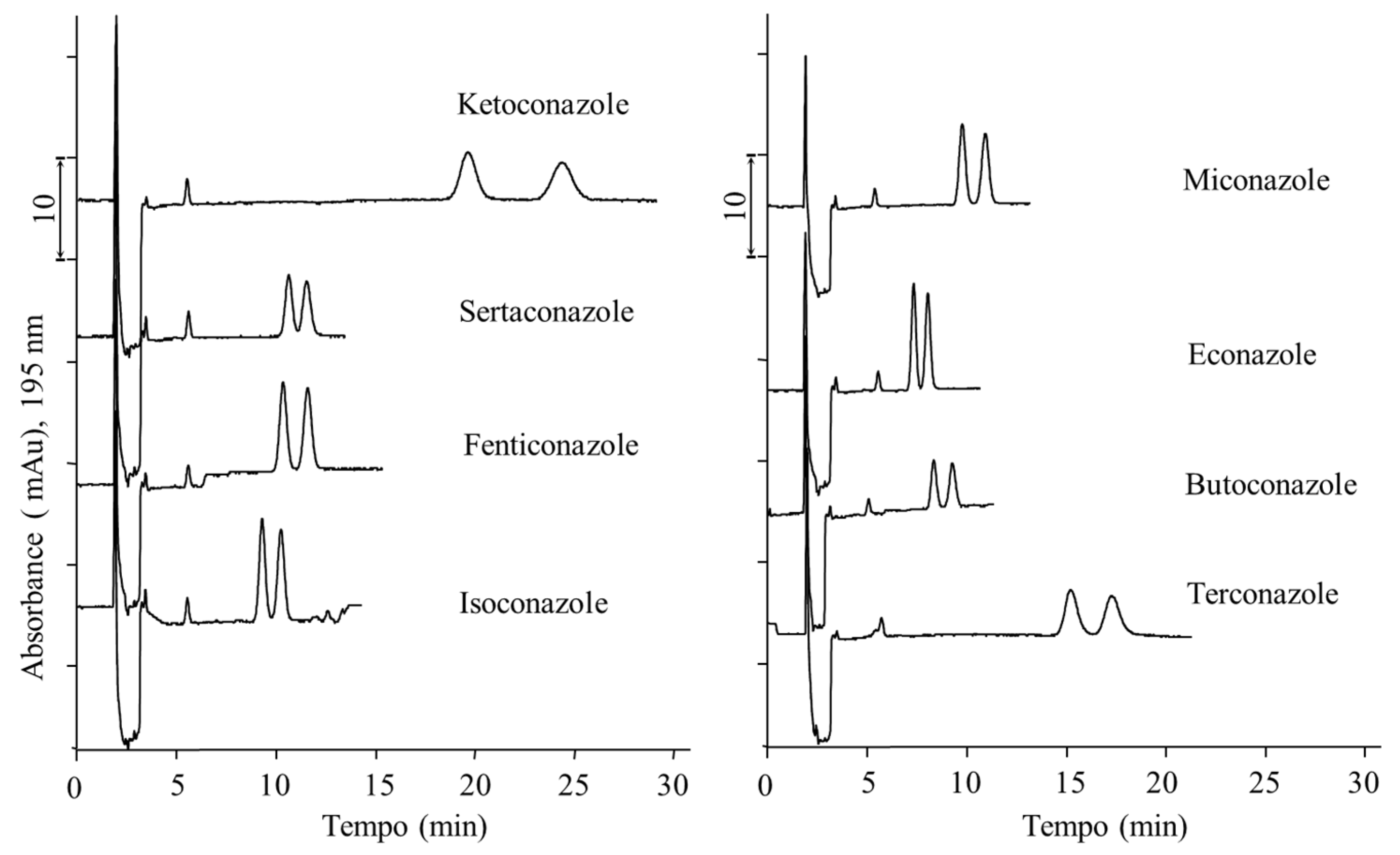
2.1.3. On-Column Pre-Concentration for Sensitivity Improvement
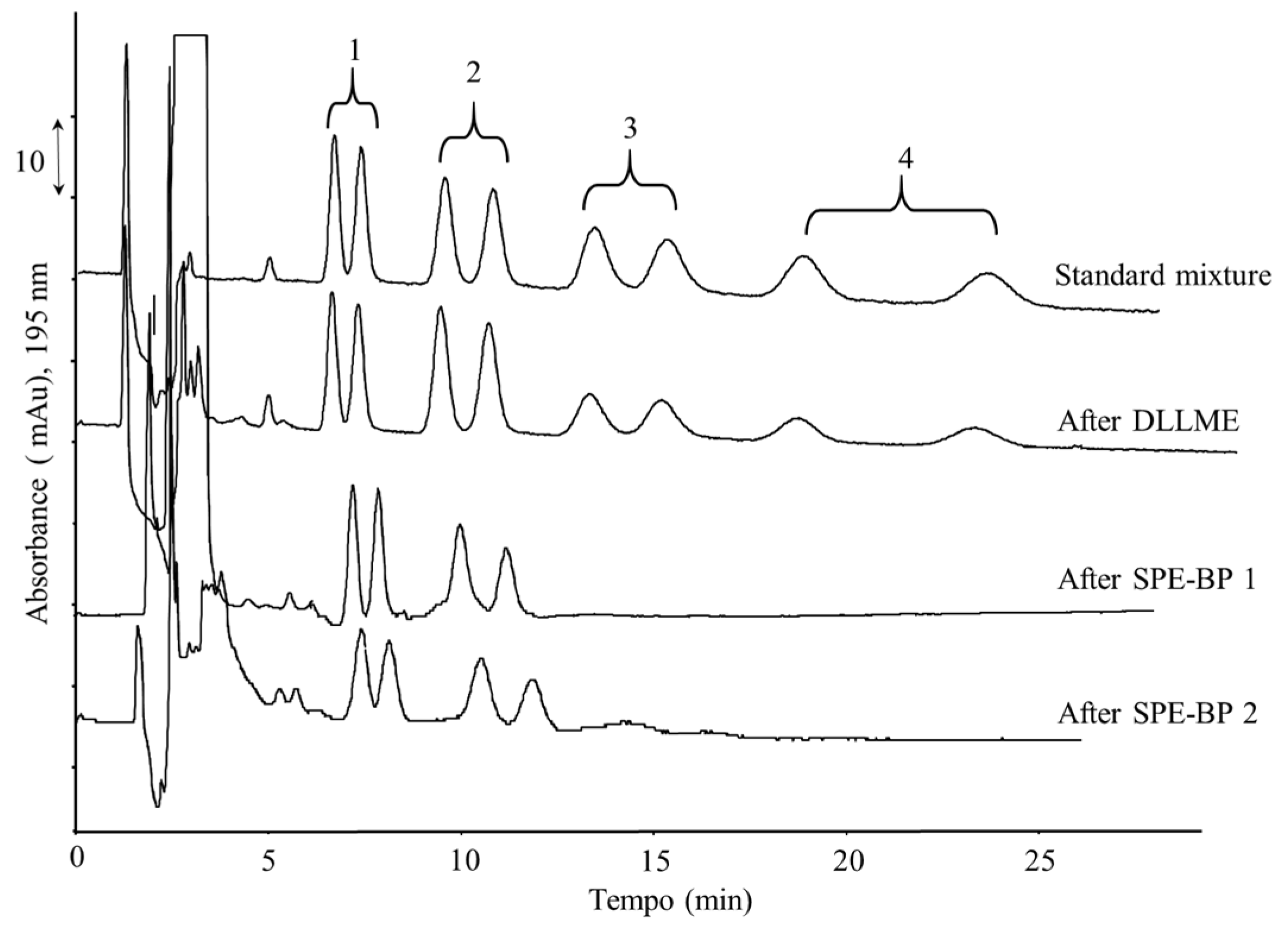
2.2. Optimization of the Extraction Procedure
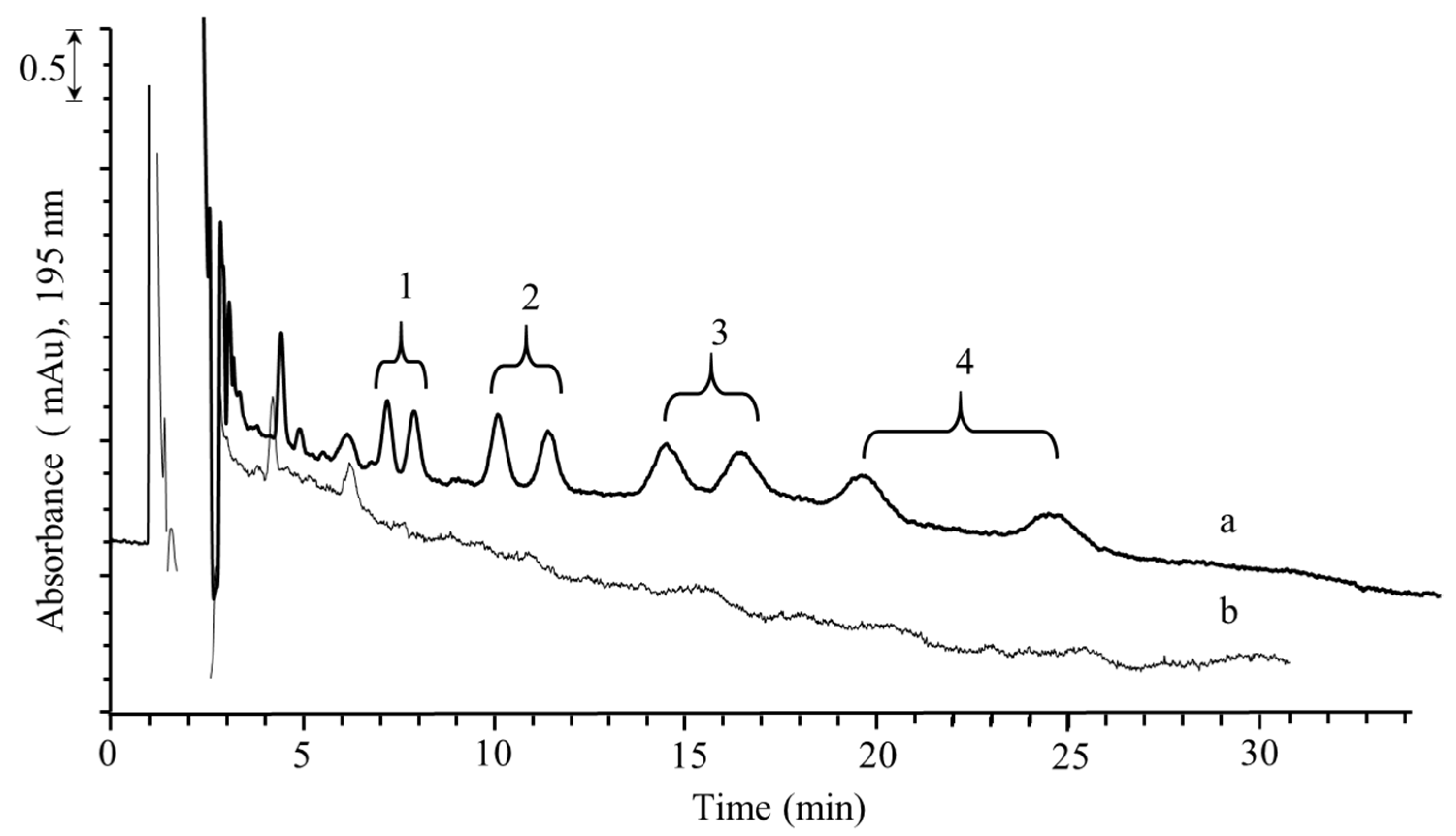
2.3. Validation of the DLLME–Nano-LC–UV Method
2.4. Comparison with Other Methods
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. The Nano-LC System
3.3. Chiral Stationary Phase and Packing of Capillary Column
3.4. Milk Samples
3.5. Sample Preparation
3.6. Method Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hector, R.F. An overview of antifungal drugs and their use for treatment of deep and superficial mycoses in animals. Clin. Tech. Small Anim. Pract. 2005, 20, 240–249. [Google Scholar] [CrossRef]
- Van Den Bossche, H.; Engelen, M.; Rochette, R. Antifungal agents of use in animal health-chemical, biochemical and pharmacological aspects. J. Vet. Pharmacol. Ther. 2003, 26, 5–29. [Google Scholar] [CrossRef]
- Rochette, F.; Engelen, M.; Vanden Bossche, H. Antifungal agents of use in animal health–practical applications. J. Vet. Pharmacol. Ther. 2003, 26, 31–53. [Google Scholar] [CrossRef]
- Farajzadeh, A.; Mogaddam, M.R.; Ghorbanpour, H. Development of a new microextraction method based on elevated temperature dispersive liquid-liquid microextraction for determination of triazole pesticides residues in honey by gas chromatography-nitrogen phosphorus detection. J. Chromatogr. A. 2014, 1347, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour, B.; Yamini, Y.; Esrafili, A. Extraction of azole antifungal drugs from milk and biological fluids using a new hollow fiber liquid-phase microextraction and analysis by GC-FID. Chromatographia 2011, 74, 281–289. [Google Scholar] [CrossRef]
- Toribio, L.; del Nozal, M.J.; Bernal, J.L.; Alonso, C.; Jiménez, J.J. Enantiomeric separation of several antimycotic azole drugs using supercritical fluid chromatography. J. Chromatogr. A 2007, 1144, 255–261. [Google Scholar] [CrossRef]
- Liu, C.; Liao, Y.; Huang, X. Extraction of triazole fungicides in environmental waters utilizing poly (ionic liquid)-functionalized magnetic adsorbent. J. Chromatogr. A 2017, 1524, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Othman, N.; Lim, V.; Ramachandran, M.R.; Sanagi, M.M.; Kamaruzaman, S.; Hirota, Y.; Norikazu, N.; Noorfatimah, Y.; Yahaya, N. Rapid ultrasound-assisted emulsification microextraction combined with COU-2 dispersive micro-solid phase extraction for the determination of azole antifungals in milk samples by HPLC-DAD. Chromatographia 2017, 80, 1553–1562. [Google Scholar] [CrossRef]
- Wang, X.; Qi, P.; Zhang, H.; Xu, H.; Wang, X.; Li, Z.; Wang, Z.; Wang, Q. Enantioselective Analysis and Dissipation of Triazole Fungicide Penconazole in Vegetables by Liquid Chromatography-Tandem Mass Spectrometry. J. Agric. Food Chem. 2014, 62, 11047–11053. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Gao, B.; Gou, G.; Li, L.; Shi, H.; Wang, M. Simultaneous enantioselective determination of the chiral fungicide prothioconazole and its major chiral metabolite prothioconazole-desthio in food and environmental samples by ultra performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2017, 65, 8241–8247. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Dai, S.; Zheng, C.; Yang, S.; Cai, T.; Bie, M. Enantiomeric separation of triazole fungicides with 3μm and 5μm particle chiral columns by reverse-phase high performance liquid chromatography. Chirality 2011, 23, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, F.; Liu, X.; Xu, J.; Li, J.; Kong, Z.; Chen, X.; Liang, X.; Zheng, Y. Simultaneous enantioselective determination of triazole fungicides in soil and water by chiral liquid chromatography/tandem mass spectrometry. J. Chromatogr. A. 2012, 1224, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ma, S.; Zhang, L.; Zhao, P.; Hou, X.; Zhao, L.; Liang, N. Trace enantioselective determination of triazole fungicides in honey by a sensitive and efficient method. J. Food Compos. Anal. 2018, 74, 62–70. [Google Scholar] [CrossRef]
- Ahmed, M.; Ghanem, A. Chiral β-cyclodextrin functionalized polymer monolith for the direct enantioselective reversed phase nano liquid chromatographic separation of racemic pharmaceuticals. J. Chromatogr. A 2014, 1345, 115–127. [Google Scholar] [CrossRef]
- Ghanem, A.; Ahmed, M.; Ishii, H.; Ikegami, T. Immobilized β-cyclodextrin-based silica vs polymer monoliths for chiral nano liquid chromatographic separation of racemates. Talanta 2015, 132, 301–314. [Google Scholar] [CrossRef]
- Fouad, A.; Shaykoon, M.S.A.; Ibrahim, S.M.; El-Adl, S.M.; Ghanem, A. Colistin Sulfate Chiral Stationary Phase for the Enantioselective Separation of Pharmaceuticals Using Organic Polymer Monolithic Capillary Chromatography. Molecules 2019, 24, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, A.; Marzouk, A.A.; Ibrahim, S.M.; El-Adl, S.M.; Ghanem, A. Functionalized polymer monoliths with carbamylated amylose for the enantioselective reversed phase nano-liquid chromatographic separation of a set of racemic pharmaceuticals. J. Chromatogr. A 2017, 1515, 91–99. [Google Scholar] [CrossRef]
- Ahmed, M.; Yajadda, M.M.; Han, Z.J.; Su, D.; Wang, G.; Ostrikov, K.K.; Ghanem, A. Single-walled carbon nanotube-based polymer monoliths for the enantioselective nano-liquid chromatographic separation of racemic pharmaceuticals. J. Chromatogr. A 2014, 1360, 100–109. [Google Scholar] [CrossRef]
- Jesús Valimaña-Traverso, J.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; García, M.Á.; Sierra, I.; Marina, L.M. Periodic mesoporous organosilica materials as sorbents for solid-phase extraction of drugs prior to simultaneous enantiomeric separation by capillary electrophoresis. J. Chromatogr. A 2018, 1566, 135–145. [Google Scholar] [CrossRef]
- Abdel-Megied, A.M.; Hanafi, R.S.; Aboul-Enein, H.Y. A chiral enantioseparation generic strategy for anti-Alzheimer and antifungal drugs by short end injection capillary electrophoresis using an experimental design approach. Chirality 2018, 30, 165–176. [Google Scholar] [CrossRef]
- Rousseau, A.; Gillotin, F.; Chiap, P.; Bodoki, E.; Crommen, J. Generic systems for the enantioseparation of basic drugs in NACE using single-isomer anionic CDs. J. Pharm. Biomed. Anal. 2011, 54, 154–159. [Google Scholar] [CrossRef]
- Gentili, A.; Caretti, F.; Bellante, S.; Rocca, L.M.; Curini, R.; Venditti, A. Development and validation of two multiresidue liquid chromatography tandem mass spectrometry methods based on a versatile extraction procedure for isolating non-steroidal anti-inflammatory drugs from bovine milk and muscle tissue. Anal. Bioanal. Chem. 2012, 404, 1375–1388. [Google Scholar] [CrossRef]
- Caretti, F.; Gentili, A.; Ambrosi, A.; Rocca, L.M.; Delfini, M.; Di Cocco, M.E.; D’Ascenzo, G. Residue analysis of glucocorticoids in bovine milk by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 397, 2477–2490. [Google Scholar] [CrossRef]
- Pérez-Fernández, V.; García, M.Á.; Marina, M.L. Chiral separation of agricultural fungicides. J. Chromatogr. A 2011, 1218, 6561–6582. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B.; Yasima, E.; Okamoto, Y. Dimethyl-, dichloroand chloromethyl-phenylcarbamate derivatives of amylose as chiral stationary phases for high performance liquid chromatography. J. Chromatogr. A 1995, 694, 101–109. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Yashima, E.; Okamoto, Y. Chloromethylphenylcarbamate derivatives of cellulose as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 1994, 670, 39–49. [Google Scholar] [CrossRef]
- Zhu, B.; Zhao, F.; Yu, J.; Wang, Z.; Song, Y.; Li, Q. Chiral separation and a molecular modeling study of eight azole antifungals on the cellulose tris(3,5-dichlorophenylcarbamate) chiral stationary phase. New J. Chem. 2018, 42, 13421–13429. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, J.; Liu, Y.; Yu, J.; Guo, X. Immobilized Cellulose-Based Chiralpak IC Chiral Stationary Phase for Enantioseparation of Eight Imidazole Antifungal Drugs in Normal-Phase, Polar Organic Phase and Reversed-Phase Conditions Using High-Performance Liquid Chromatography. Chromatographia 2019, 82, 649–660. [Google Scholar] [CrossRef]
- Tomai, P.; Martinelli, A.; Gasperi, T.; Bianchi, M.; Purcaro, V.; Teofili, L.; Papacci, P.; Cori, M.S.; Vento, G.; Curini, R.; et al. Rotating-disc micro-solid phase extraction of F2-isoprostanes from maternal and cord plasma by using oxidized buckypaper as sorbent membrane. J. Chromatogr. A 2019, 1586, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Tomai, P.; Gentili, A.; Fanali, S.; Picó, Y. Multi-residue determination of organic micro-pollutants in river sediment by stir-disc solid phase extraction based on oxidized buckypaper. J. Chromatogr. A 2010, 1621, 461080. [Google Scholar] [CrossRef]
- D’Orazio, G.; Fanali, C.; Gentili, A.; Tagliaro, F.; Fanali, S. Nano-liquid chromatography for enantiomers separation of baclofen by using vancomycin silica stationary phase. J. Chromatogr. A 2019, 1605, 360358. [Google Scholar] [CrossRef]
- Tomai, P.; Gentili, A.; Curini, R.; Gottardo, R.; Tagliaro, F.; Fanali, S. Dispersive liquid-liquid microextraction, an effective tool for the determination of synthetic cannabinoids in oral fluid by liquid chromatography–tandem mass spectrometry. J. Pharm. Anal. 2020, 11, 292–298. [Google Scholar] [CrossRef]
- Tomai, P.; Martinelli, A.; Morosetti, S.; Curini, R.; Fanali, S.; Gentili, A. Oxidized buckypaper for stir-disc solid phase extraction: Evaluation of several classes of environmental pollutants recovered from surface water samples. Anal. Chem. 2018, 90, 6827–6834. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Fernández, V.; Gentili, A.; Martinelli, A.; Caretti, F.; Curini, R. Evaluation of oxidized buckypaper as material for the solid phase extraction of cobalamins from milk: Its efficacy as individual and support sorbent of a hydrophilic–lipophilic balance copolymer. J. Chromatogr. A 2016, 1428, 255–266. [Google Scholar] [CrossRef]
- D’Orazio, G.; Rocchi, S.; Fanali, S. Nano-liquid chromatography coupled with mass spectrometry: Separation of sulfonamides employing non-porous core-shell particles. J. Chromatogr. A 2012, 1255, 277–285. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, G.; Hernández-Borges, J.; Vicente Herrera-Herrera, A.; Fanali, S.; Rodríguez-Delgado, M.Á. Determination of estrogenic compounds in milk and yogurt samples by hollow-fibre liquid-phase microextraction-gas chromatography-triple quadrupole mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 7447–7459. [Google Scholar] [CrossRef]
- D’Orazio, G.; Asensio-Ramos, M.; Hernández-Borges, J.; Rodríguez-Delgado, M.A.; Fanali, S. Evaluation of the combination of a dispersive liquid-liquid microextraction method with micellar electrokinetic chromatography coupled to mass-spectrometry for the determination of estrogenic compounds in milk and yogurt. Electrophoresis 2015, 36, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Gentili, A.; Miccheli, A.; Tomai, P.; Baldassarre, M.E.; Curini, R.; Pérez-Fernández, V. Liquid chromatography–tandem mass spectrometry method for the determination of vitamin K homologues in human milk after overnight cold saponification. J. Food Compos. Anal. 2016, 47, 21–30. [Google Scholar] [CrossRef]
- Manz, A.; Simon, W. Injectors for open-tubular column liquid chromatography with 106 theoretical plates at retention times in the minute range. J. Chromatogr. A 1987, 387, 187–196. [Google Scholar] [CrossRef]
- D’Orazio, G.; Fanali, C.; Karchkhadze, M.; Chankvetadze, B.; Fanali, S. Enantiomeric separation of some chiral analytes using amylose 3,5-dimethylphenylcarbamate covalently immobilized on silica by nano-liquid chromatography and capillary electrochromatography. J. Chromatogr. A 2017, 1520, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.M. Bioanalytical methods validation: A critique of the proposed FDA guidance. Chromatographia 2000, 52, S65–S69. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | pKa | LogP (*) | HLB (*)(**) |
|---|---|---|---|
| Fluconazole | 12.68 | 0.56 | 9.56 |
| Voriconazole | 12.7 | 1.82 | 8.24 |
| Ketoconazole | 6.42 | 4.19 | 10.39 |
| Bifonazole | 6.36 | 5.23 | 2.66 |
| Econazole | 6.48 | 5.35 | 5.77 |
| Terconazole | 8.45 | 5.37 | 14.26 |
| Posaconazole | 14.85 | 5.41 | 8.15 |
| Isoconazole | 6.48 | 5.96 | 6.01 |
| Miconazole | 6.48 | 5.96 | 6.01 |
| Sertaconazole | 6.48 | 6.23 | 5.93 |
| Butoconazole | 6.51 | 6.55 | 5.52 |
| Fenticonazole | 6.48 | 6.94 | 4.16 |
| Peak Area (RSD, %) | |||||
|---|---|---|---|---|---|
| Level I (3.0–7.0 µg mL−1) | Level II (6.0–15.0 µg mL−1) | ||||
| Peak | Intraday (n = 6) | Interday (n = 15) | Intraday (n = 6) | Interday (n = 15) | |
| #1 | Econazole | 6.4 | 9.2 | 6.5 | 9.0 |
| #2 | Econazole | 6.7 | 8.8 | 6.8 | 8.8 |
| #1 | Miconazole | 5.2 | 7.3 | 6.4 | 7.5 |
| #2 | Miconazole | 4.7 | 7.8 | 6.1 | 8.4 |
| #1 | Terconazole | 4.9 | 6.9 | 4.7 | 7.3 |
| #2 | Terconazole | 5.2 | 7.2 | 5.5 | 7.5 |
| #1 | Ketoconazole | 6.0 | 8.3 | 5.9 | 8.4 |
| #2 | Ketoconazole | 5.7 | 8.8 | 6.7 | 9.1 |
| Linear Dynamic Range (µg mL−1) | Regression Equation (n = 6) | ||||||
|---|---|---|---|---|---|---|---|
| y = mx + n | |||||||
| Peak # | Analyte | M ± t·Sm (104) | N ± t·Sn (104) | R2 | LODamethod (µg mL−1) | LOQbmethod (µg mL−1) | |
| 1 | Econazole | 0.50–10.0 | 1.9 ± 0.8 | 1.5 ± 1.6 | 0.990 | 0.05 | 0.14 |
| 2 | 0.50–10.0 | 2.0 ± 1.0 | 1.2 ± 1.4 | 0.992 | 0.06 | 0.16 | |
| 1 | Miconazole | 1.5–10.0 | 2.3 ± 1.1 | 1.3 ± 1.4 | 0.991 | 0.20 | 0.80 |
| 2 | 1.5–10.0 | 2.2 ± 1.2 | 1.1 ± 1.2 | 0.990 | 0.30 | 1.10 | |
| 1 | Terconazole | 4.0–20.0 | 1.8 ± 0.8 | 1.5 ± 1.7 | 0.990 | 1.10 | 3.20 |
| 2 | 4.0–20.0 | 1.7 ± 1.1 | 1.7 ± 1.8 | 0.991 | 1.20 | 3.50 | |
| 1 | Ketokonazole | 7.0–20.0 | 1.8 ± 1.2 | 1.1 ± 1.3 | 0.991 | 2.50 | 6.10 |
| 2 | 7.0–20.0 | 1.9 ± 1.3 | 0.9 ± 1.2 | 0.992 | 2.70 | 6.80 | |
| Analytes | Peak # | Spiked Level (µg mL−1) | Found (a) (µg mL−1) | Accuracy (%), (RSD,%) | t |
|---|---|---|---|---|---|
| Econazole | 1 | 3.0 | 2.7 ± 0.3 | 91 (19) | 0.69 |
| 2 | 5.0 | 5.2 ± 0.6 | 104 (12) | 1.46 | |
| 1 | 3.0 | 2.8 ± 0.3 | 92 (17) | 1.98 | |
| 2 | 5.0 | 5.4 ± 0.2 | 108 (15) | 1.65 | |
| 1 | 3.0 | 2.7 ± 0.3 | 89 (12) | 0.66 | |
| Miconazole | 2 | 5.0 | 4.8 ± 0.4 | 95 (17) | 0.37 |
| 1 | 3.0 | 2.8 ± 0.8 | 93 (14) | 0.87 | |
| 2 | 5.0 | 5.0 ± 1.4 | 99 (18) | 0.22 | |
| 1 | 5.0 | 5.2 ± 0.8 | 104 (12) | 1.01 | |
| Terconazole | 2 | 10.0 | 9.5 ± 1.4 | 95 (17) | 2.27 |
| 1 | 5.0 | 5.4 ± 1.1 | 108 (15) | 2.39 | |
| 2 | 10.0 | 9.2 ± 1.5 | 92 (19) | 1.59 | |
| 1 | 7.0 | 8.1 ± 1.4 | 115 (12) | 0.01 | |
| Ketokonazole | 2 | 15.0 | 15.6 ± 2.8 | 104 (14) | 2.51 |
| 1 | 7.0 | 8.3 ± 1.6 | 119 (14) | 0.90 | |
| 2 | 15.0 | 16.2 ± 3.1 | 108 (16) | 1.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Bosco, C.; Bonoli, F.; Gentili, A.; Fanali, C.; D’Orazio, G. Chiral Nano-Liquid Chromatography and Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Antifungal Drugs in Milk. Molecules 2021, 26, 7094. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237094
Dal Bosco C, Bonoli F, Gentili A, Fanali C, D’Orazio G. Chiral Nano-Liquid Chromatography and Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Antifungal Drugs in Milk. Molecules. 2021; 26(23):7094. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237094
Chicago/Turabian StyleDal Bosco, Chiara, Flavia Bonoli, Alessandra Gentili, Chiara Fanali, and Giovanni D’Orazio. 2021. "Chiral Nano-Liquid Chromatography and Dispersive Liquid-Liquid Microextraction Applied to the Analysis of Antifungal Drugs in Milk" Molecules 26, no. 23: 7094. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237094