
Transformation of Pectins into Non-Ionic or Anionic Surfactants Using a One-Pot and Cascade Mode Process


Abstract
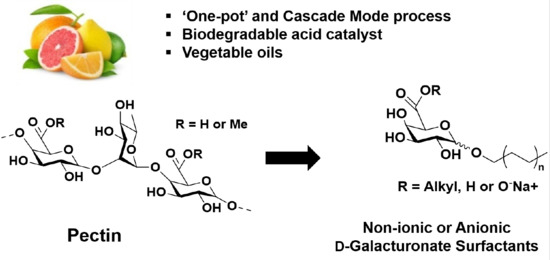
:
1. Introduction
2. Results
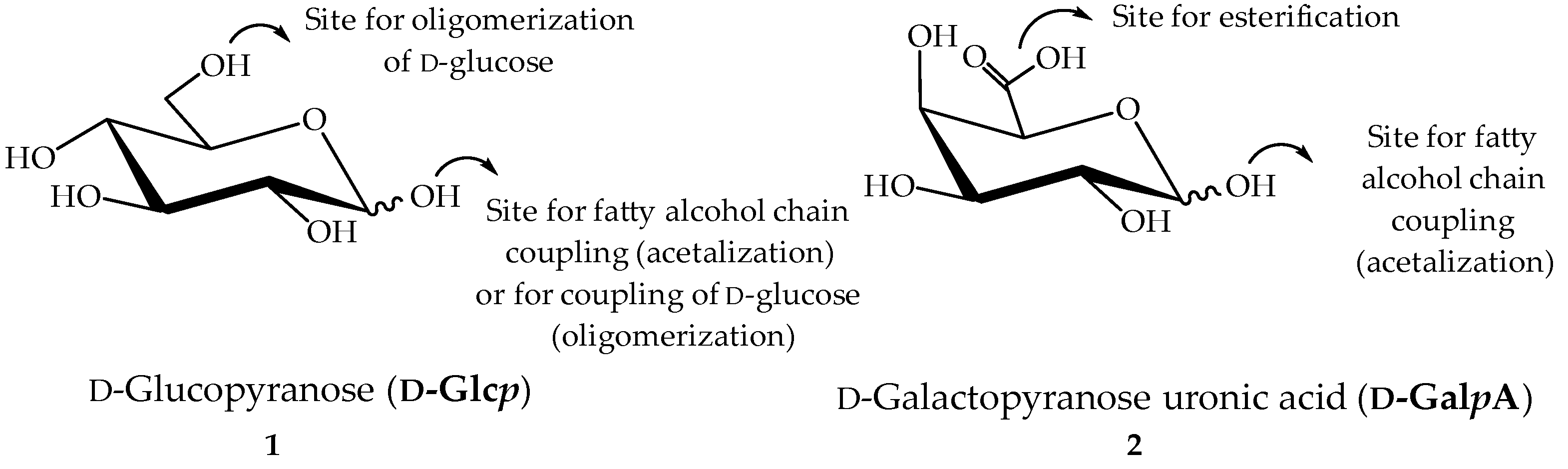
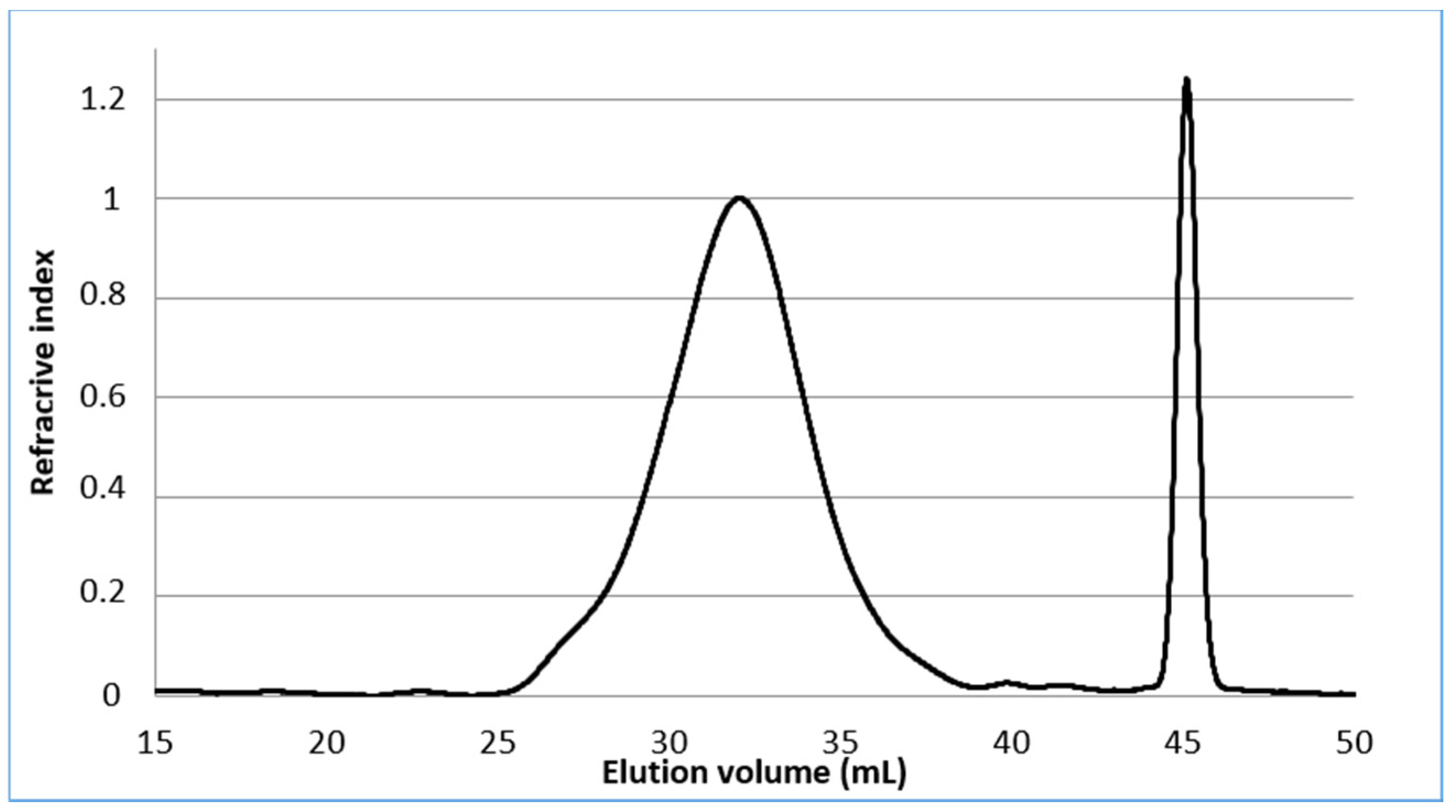
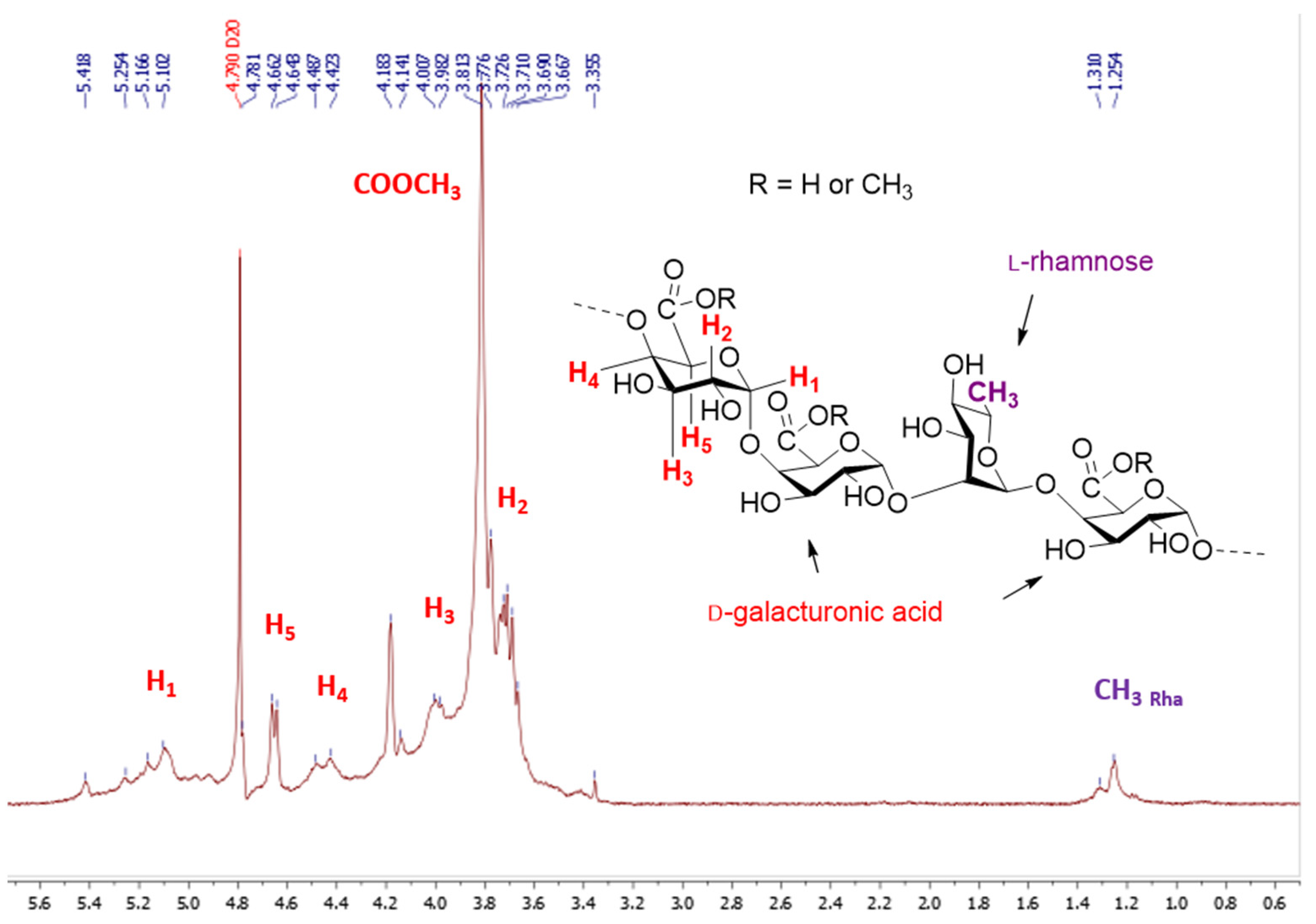
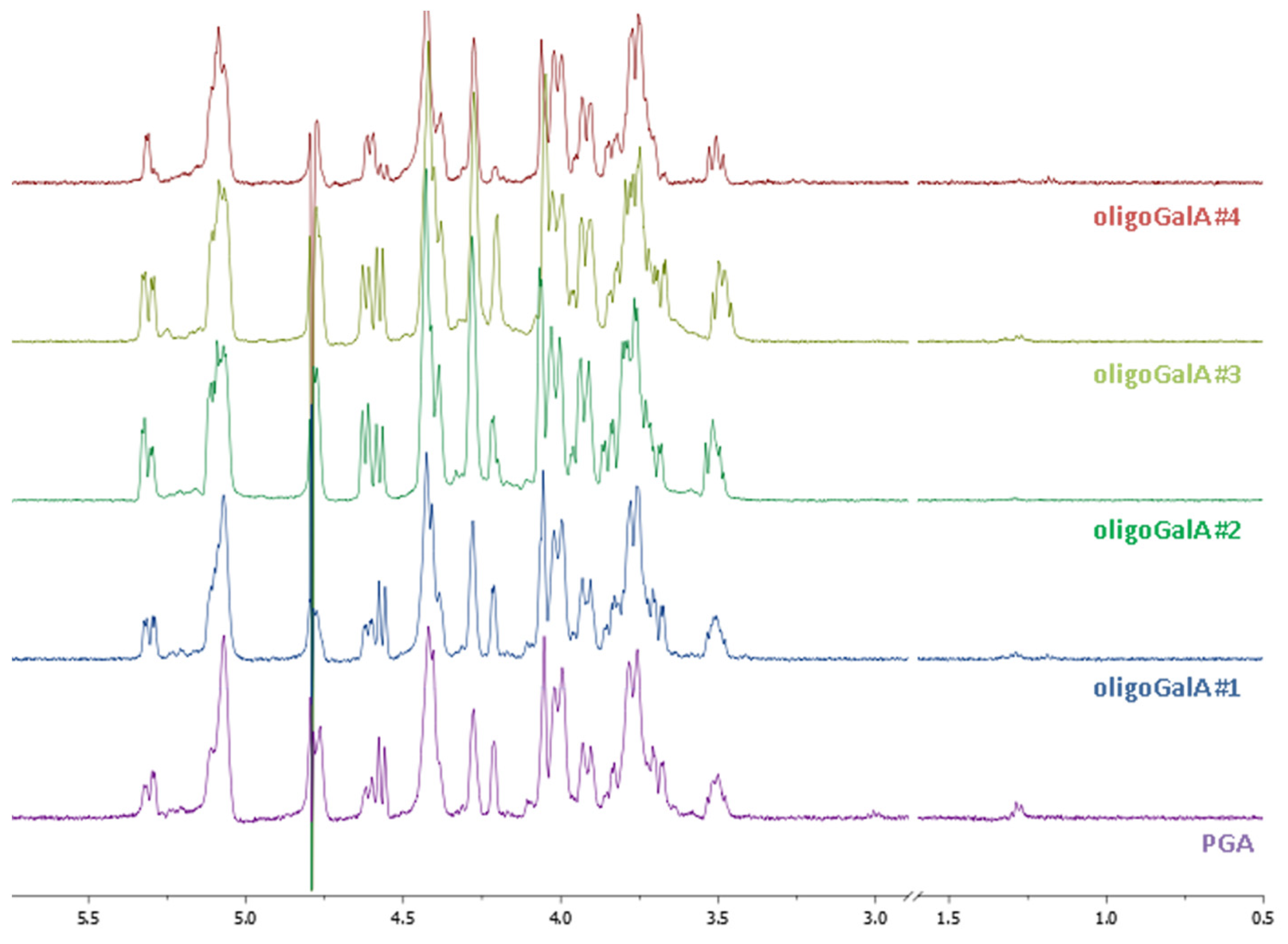
2.1. Characterisation of Pectin Starting Material
2.2. Synthesis of Galacturonate Derivatives
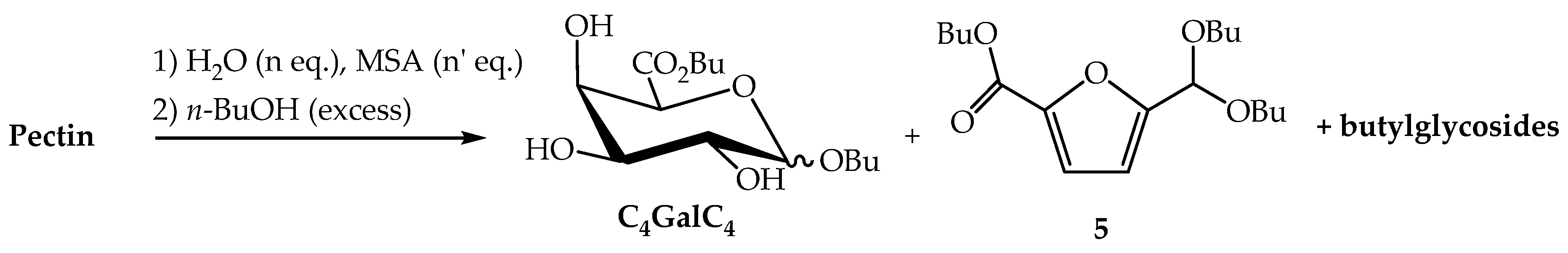
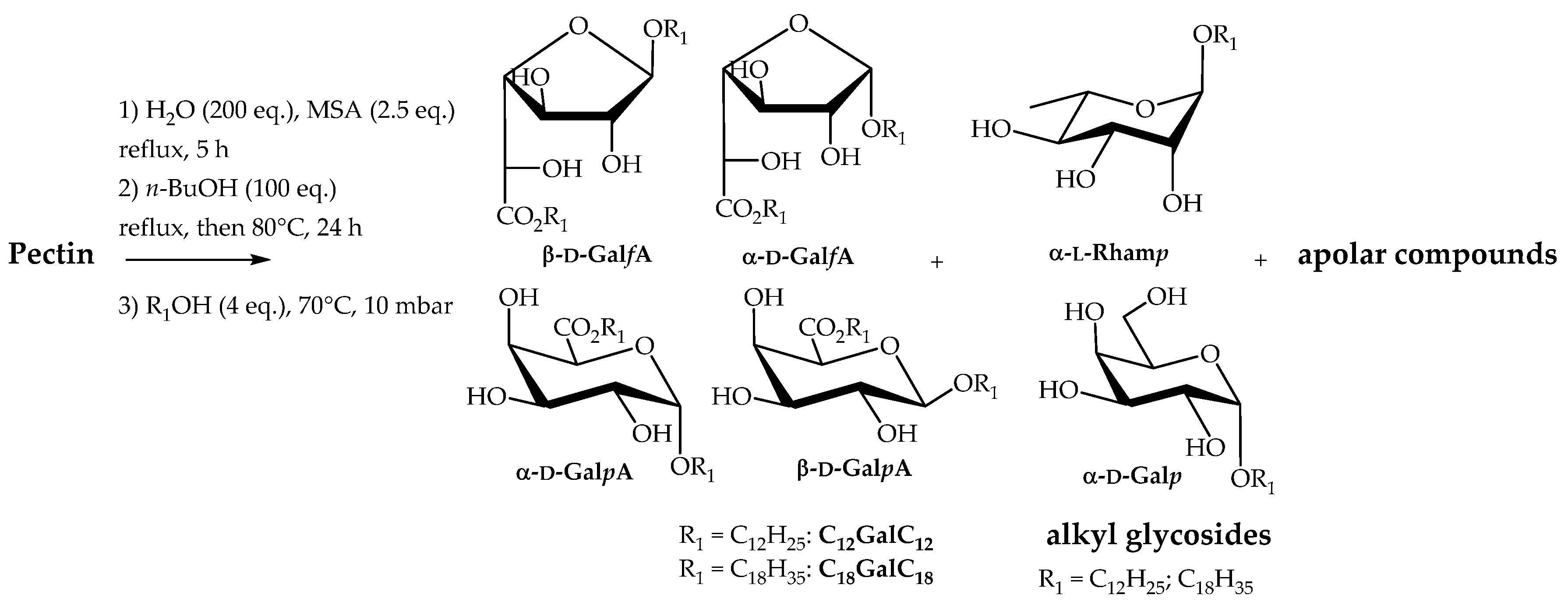
2.2.1. Direct Transformation of Pectin into n-Butyl (n-butyl ᴅ-galactosiduronates) C4GalC4
2.2.2. Direct Transformation of Pectin into Double-Tailed Surfactants
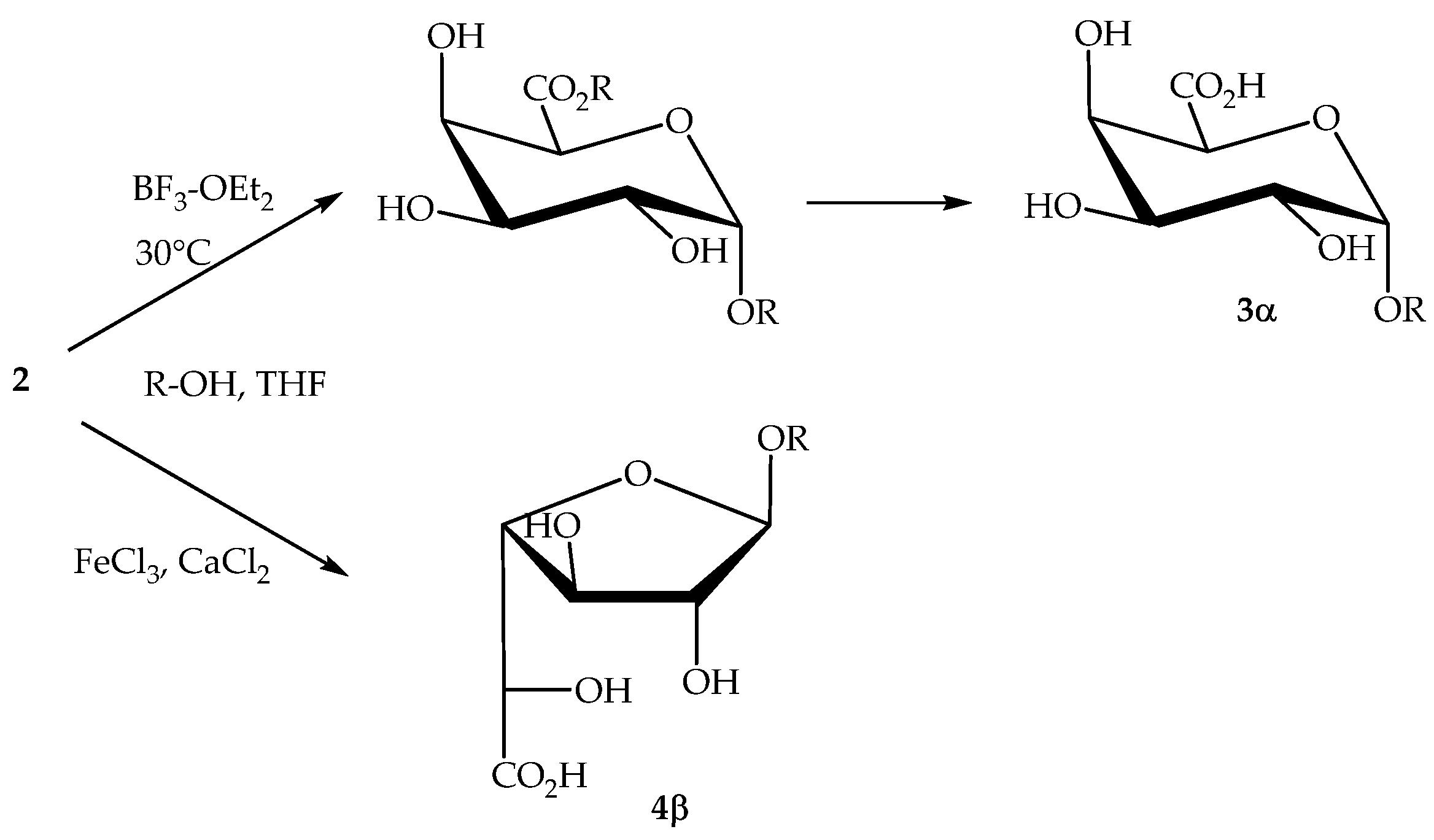
2.2.3. Direct Transformation of Pectin into Single-Tailed Surfactants
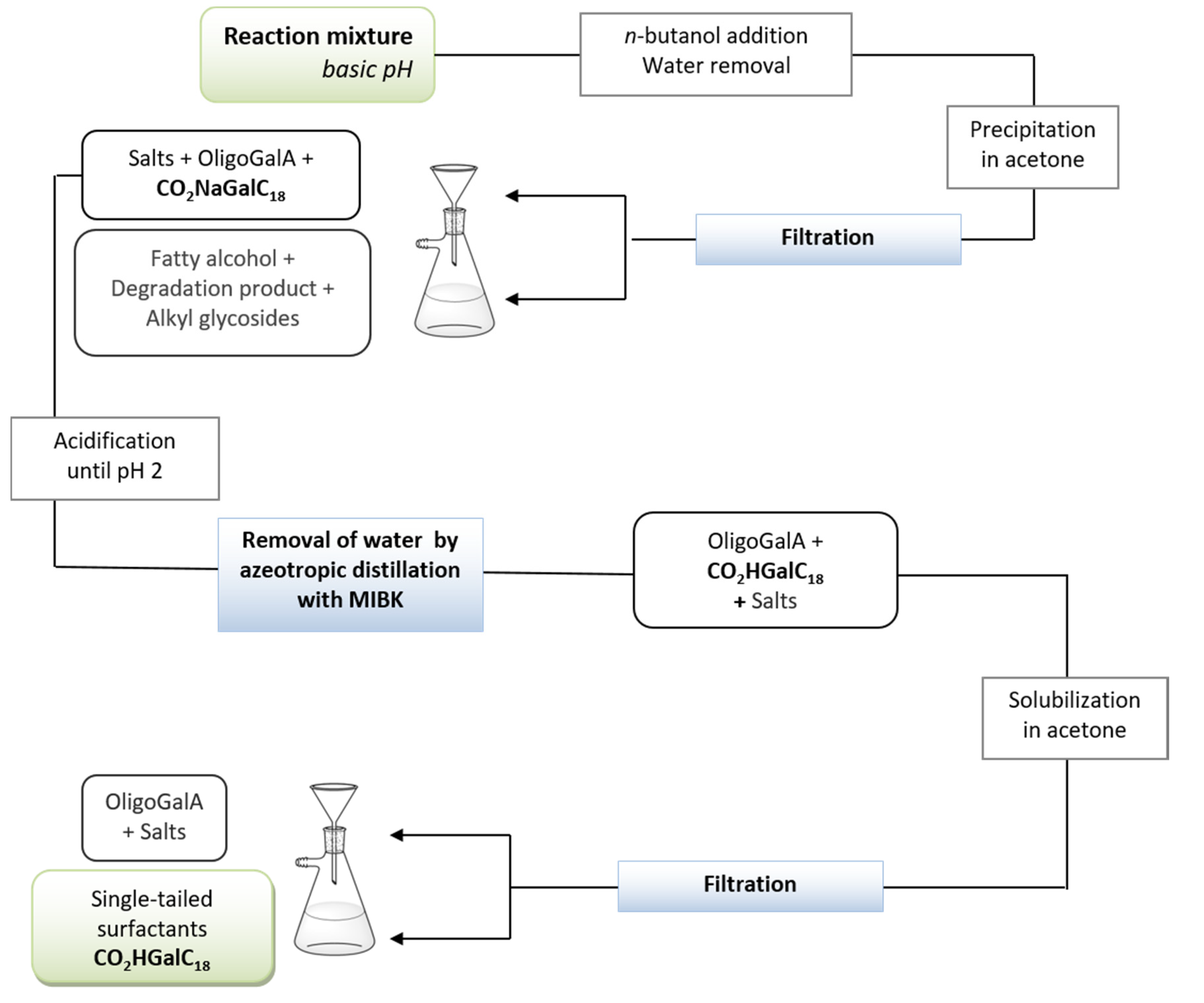
2.2.4. One-Pot Manufacturable Process for the Production of Single-Tailed Surfactants
2.3. Physico-Chemical Evaluations
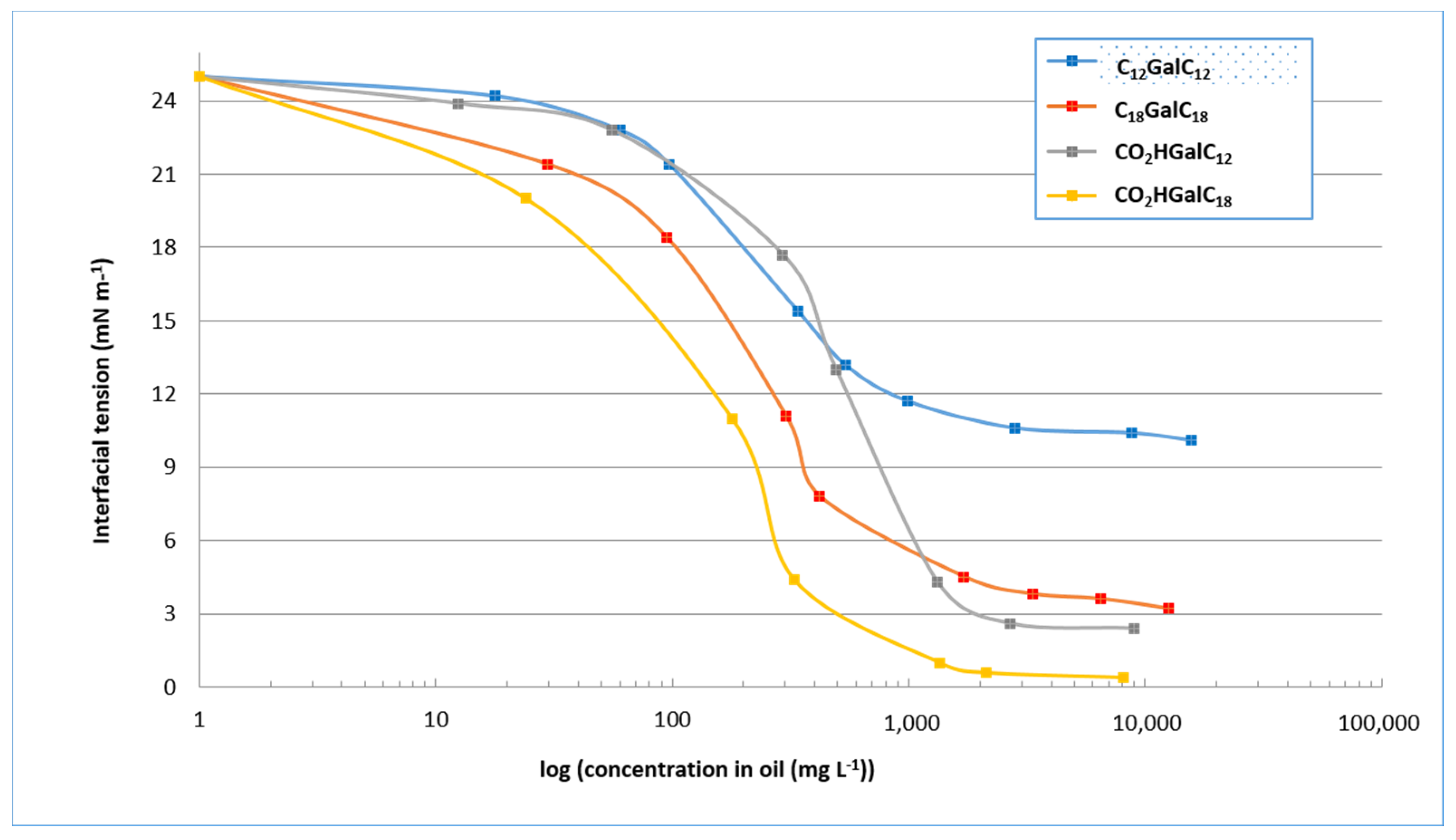
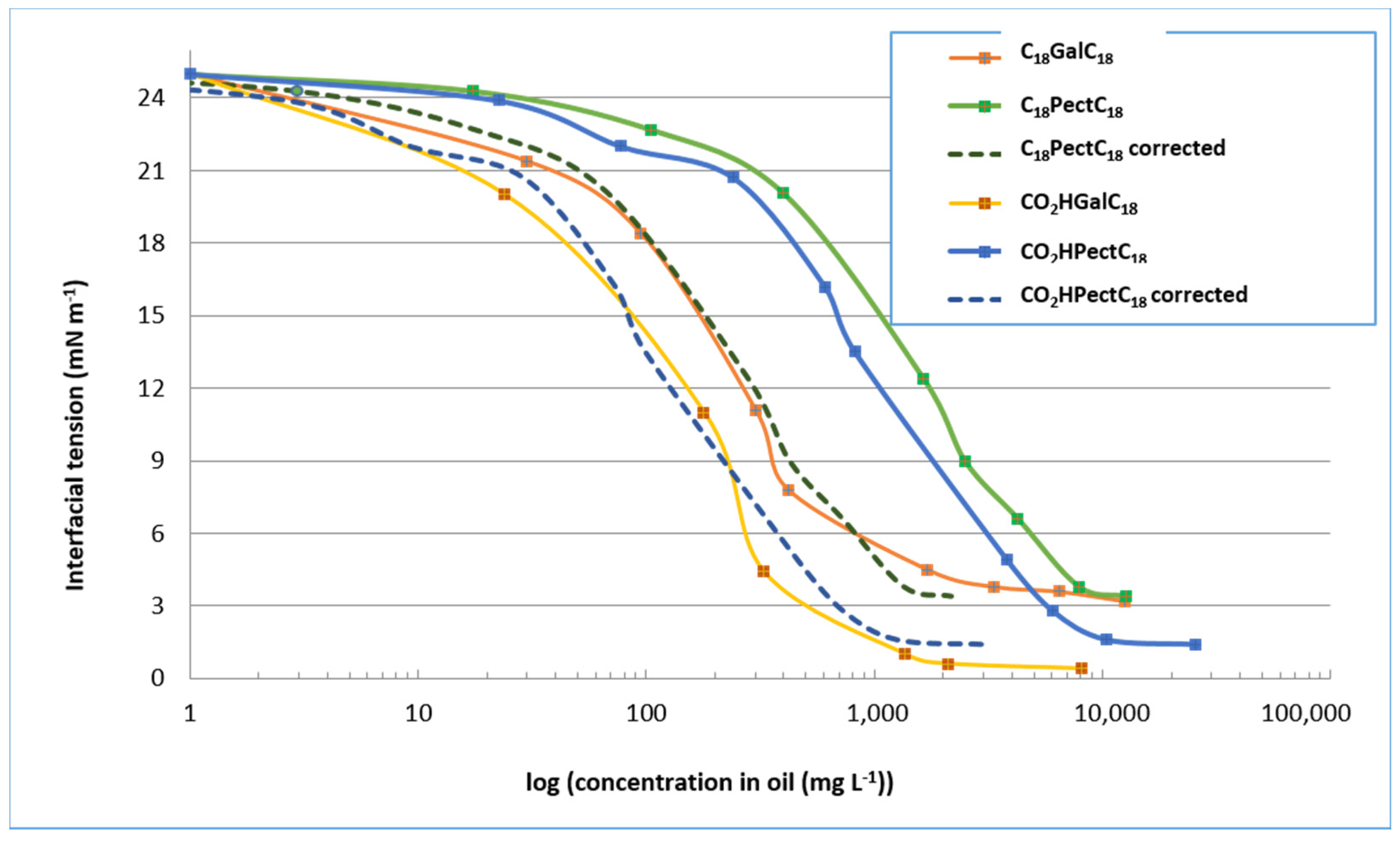
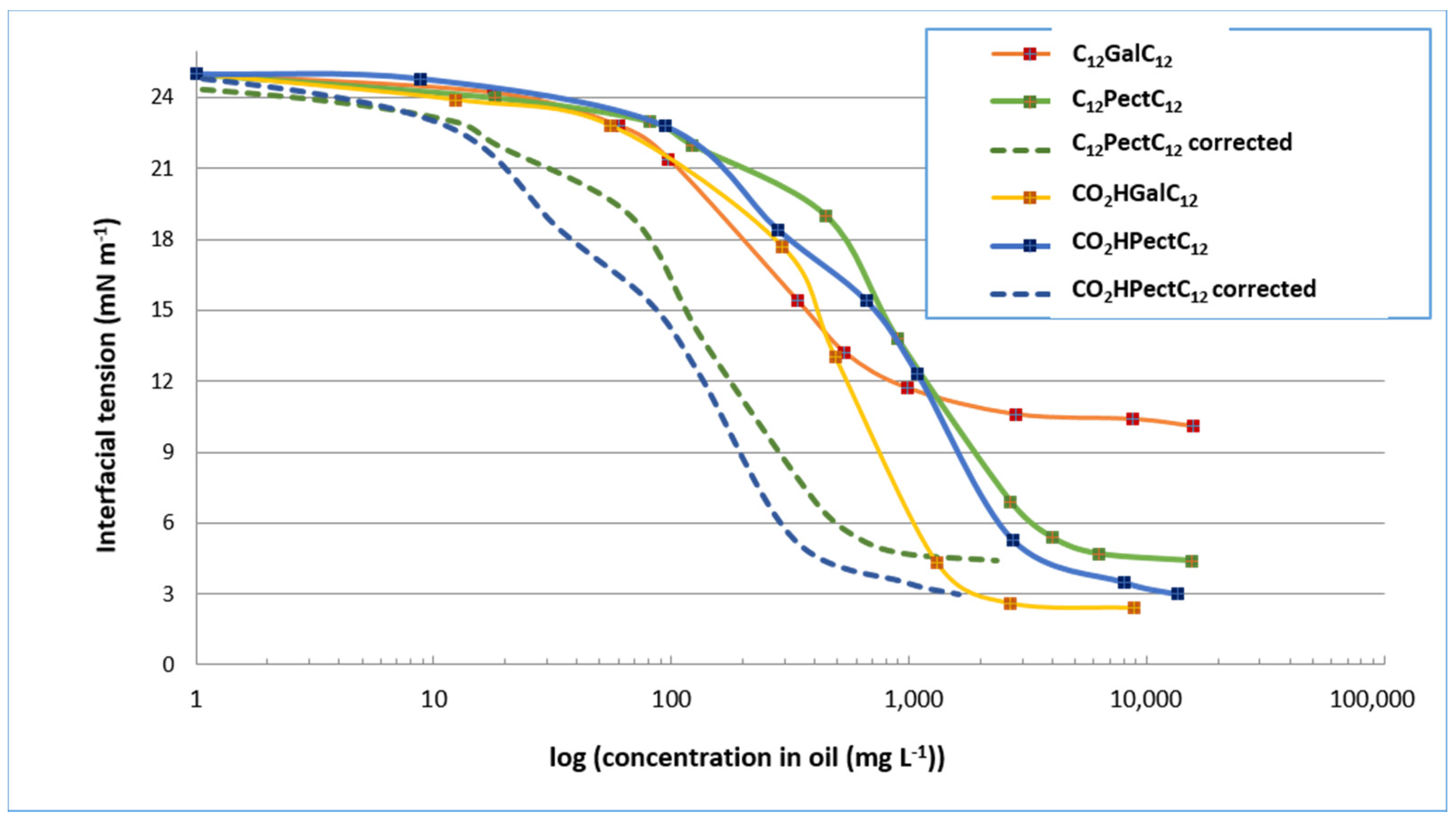
2.3.1. Measurements of Interfacial Tension
2.3.2. Measurements of Surface Tension
2.4. Ecotoxicity Studies
2.5. Biodegradability Studies
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Preparation of n-butyl (n-butyl-ᴅ-galactoside) uronate C4GalC4
4.1.2. Preparation of n-alkyl (n-alkyl-ᴅ-galactoside) uronates CnGalCn
4.1.3. Preparation of n-Alkyl-ᴅ-galactosiduronic acids CO2HGalCn
4.1.4. One-Pot Manufacturable Process of Producing CO2HGalC18
4.1.5. Preparation of n-Alkyl-ᴅ-galactosiduronic acid sodium salts CO2NaGalCn
4.2. Physico-Chemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Von Ribinsky, W.; Hill, K. Alkyl polyglycosides—Properties and applications of a new class of surfactants. Ang. Chem. Int. Edit. 1998, 37, 1328–1345. [Google Scholar] [CrossRef]
- Hill, K.; von Rybinsky, W.; Stoll, G. Alkyl Polyglucosides, Technology, Properties and Applications; John Wiley & Sons: Weinheim, Germany, 2008. [Google Scholar]
- Fischer, E. Ueber die glucoside der alkohole. Ber. Deutsch. Gesellschaft 1893, 26, 2400–2412. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, Y.; Sujiyama, J.; Chanzy, H.; Langan, P. Crystal structure and hydrogen bonding system in cellulose Iα from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef]
- Harmer, M.A.; Fan, A.; Liauw, A.A.; Kumar, R.K. A new route to high yield sugars from biomass: Phosphoric-sulfuric acid. Chem. Commun. 2009, 43, 6610–6612. [Google Scholar]
- Villendier, N.; Corma, A. One pot catalytic conversion of cellulose into biodegradable surfactant. Chem. Commun. 2010, 46, 4408–4410. [Google Scholar] [CrossRef]
- Pang, J.; Wang, A.; Zheng, M.; Zhang, T. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperature. Chem. Commun. 2010, 46, 6935–6937. [Google Scholar] [CrossRef]
- Puga, A.V.; Corma, A. Efficient production and separation of biodegradable surfactants from cellulose in 1-butyl-3-methylimidazolium chloride. ChemSusChem 2014, 7, 3362–3373. [Google Scholar] [CrossRef]
- Morales-delRosa, S.; Campos-Martin, J.M.; Fierro, J.L.G. Complete chemical hydrolysis of cellulose into fermentable sugars through ionic liquids and antisolvent pretreatments. ChemSusChem 2014, 7, 3467–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wang, J.; Cen, C.; Huang, Y.; Wang, A.; Zhang, T. Graphene oxide for cellulose hydrolysis: How it works as a highly active catalyst? Chem. Commun. 2014, 50, 3439–3442. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; Iborra, S. Martinez-Silvestre, S. Transformation of cellulose into nonionic surfactants using a one-pot catalytic process. ChemSusChem 2016, 9, 3492–3502. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.V.; Corma, A. Direct conversion of cellulose into alkyl glycoside surfactants. ChemistrySelect 2017, 2, 2495–2498. [Google Scholar] [CrossRef]
- Karam, A.; De Oliveira Vigier, K.; Marinkovic, S.; Estrine, B.; Oldani, C.; Jérôme, F. Conversion of cellulose into amphiphilic alkyl glycosides catalyzed by Aquivion, a perfluorosulfonic acid polymer. ChemSusChem 2017, 10, 3604–3610. [Google Scholar] [CrossRef] [PubMed]
- Bouxin, F.; Marinkovic, S.; Le Bras, J.; Estrine, B. Direct conversion of xylan into alkyl pentosides. Carbohydr. Res. 2010, 345, 2469–2473. [Google Scholar] [CrossRef]
- Wang, Y.; Renault, L.; Guégan, J.P.; Benvegnu, T. Direct conversion of agarose into alkyl mono- and disaccharide surfactants based on 3,6-anhydro L- and D-galactose. ChemistrySelect 2021, 6, 389–395. [Google Scholar] [CrossRef]
- Folley, P.; Kermanshahi, A.; Beachand, E.S.; Zimmerman, J.B. Derivation and synthesis of renewable surfactants. Chem. Soc. Rev. 2012, 41, 1499–1518. [Google Scholar] [CrossRef]
- Benvegnu, T.; Plusquellec, D.; Lemiègre, L. Surfactants from Renewable Sources: Synthesis and Applications. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, N.M.N., Gandini, A., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2008; pp. 153–178. [Google Scholar]
- Bertho, J.N.; Ferrières, V.; Plusquellec, D. A new synthesis of D-glycosiduronates from unprotected D-uronic acids. J. Chem. Soc. Chem. Commun. 1995, 13, 1391–1393. [Google Scholar] [CrossRef]
- Ferrières, V.; Bertho, J.N.; Plusquellec, D. A convenient synthesis of alkyl D-glycofuranosiduronic acids and alkyl D-glycofuranosides from unprotected carbohydrates. Carbohydr. Res. 1998, 311, 25–35. [Google Scholar] [CrossRef]
- Goodby, J.W.; Goertz, V.; Cowling, S.; Mackenzie, G.; Martin, P.; Plusquellec, D.; Benvegnu, T.; Boullanger, P.; Lafont, D.; Queneau, Y.; et al. Thermotropic liquid crystalline glycolipids. Chem. Soc. Rev. 2007, 36, 1971–2032. [Google Scholar] [CrossRef] [PubMed]
- Chubnová, I.; Sylla, B.; Nuger-Chauvin, C.; Daniellou, R.; Legentil, L.; Kralová, B.; Ferrières, V. Natural glycans and glycoconjugates as immunomodulating agents. Nat. Prod. Rep. 2011, 28, 937–952. [Google Scholar] [CrossRef]
- Pérez, S.; Rodriguez-Carvajal, M.A.; Doco, T. A complex plant cell wall polysaccharide: Rhamnogalacturonan II. A structure in quest of a function. Biochimie 2003, 85, 109–121. [Google Scholar] [CrossRef]
- Caffall, K.H.; Mohnen, D. The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr. Res. 2009, 344, 1879–1900. [Google Scholar] [CrossRef]
- Pheulpin, P.; Rossard, S.; Barbier, M.; Kovensky, J.; Courtois, B.; Courtois-Sambourg, J. Method for Producing Families of Galacturonic Acid Oligomers and Use Thereof. U.S. Patent WO2009004153A1, 8 January 2009. [Google Scholar]
- Ognyanov, M.; Remoroza, C.; Schols, H.A.; Georgiev, Y.; Kratchanova, M.; Kratchanov, C. Isolation and structure elucidation of pectic polysaccharide from rose hip fruits (Rosa canina L.). Carbohydr. Polym. 2016, 151, 803–811. [Google Scholar] [CrossRef]
- Wang, W.; Ma, X.; Jiang, P.; Hu, L.; Zhi, Z.; Chen, J.; Ding, T.; Ye, X.; Liu, D. Characterization of pectin from grapefruit peel: A comparison of ultrasound-assisted and conventional heating extractions. Food Hydrocoll. 2016, 61, 730–739. [Google Scholar] [CrossRef]
- Thibault, J.F. Automatisation du dosage des substances pectiques par la méthode au méta-hydroxydiphényl. Lebensm-Wiss. Technol. 1979, 12, 247–251. [Google Scholar]
- Ralet, M.C.; Crepeau, M.J.; Buchholt, H.C.; Thibault, J.F. Polyelectrolyte behaviour and calcium binding properties of sugar beet pectins differing in their degrees of methylation and acetylation. Biochem. Eng. J. 2003, 16, 191–201. [Google Scholar] [CrossRef]
- Kamerling, J.P.; Gerwig, G.J.; Vliegenthart, J.F.G.; Clamp, J.R. Characterization by gas-liquid chromatography-mass spectrometry and proton-magnetic-resonance spectroscopy of pertrimethylsilyl methyl glycosides obtained in the methanolysis of glycoproteins and glycopeptides. Biochem. J. 1975, 151, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Montreuil, J.; Bouquelet, S.; Debray, H.; Fournet, B.; Spik, G.; Strecker, G. Glycoproteins. In Carbohydrates Analysis: A Practical Approach; Chaplin, M.F., Kennedy, J.K., Eds.; IRL Press: Oxford, UK, 1986; pp. 143–204. [Google Scholar]
- Burana-osot, J.; Soonthornchareonnon, N.; Hosoyama, S.; Linhardt, R.J.; Toida, T. Partial depolymerization of pectin by a photochemical reaction. Carbohydr. Res. 2010, 345, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Gernon, M.D.; Wu, M.; Buszta, T.; Janney, P. Environmental benefits of methanesulfonic acid. Comparative properties and advantages. Green Chem. 1999, 1, 127–140. [Google Scholar] [CrossRef]
- Han, X.; Li, C.; Liu, X.; Xia, Q.; Wang, Y. Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over MnOx–CeO2 composite catalysts. Green Chem. 2017, 19, 996–1004. [Google Scholar] [CrossRef]
- Donoeva, B.; Masoud, N.; de Jongh, P.E. Carbon Support Surface Effects in the Gold-Catalyzed Oxidation of 5-Hydroxymethylfurfural. ACS Catal. 2017, 7, 4581–4591. [Google Scholar] [CrossRef]
- Aouda, M.; Al-Maamari, T.; Al-Salmi, F. Intramolecular and intermolecular ion-dipole interactions in sodiumn lauryl ether sulfates (SLES) self-aggregation and mixed micellization with Tritan X-100. Colloid Surface A 2009, 335, 55–61. [Google Scholar] [CrossRef]
- Deshpande, S.; Shiau, B.J.; Wade, D.; Sabatini, D.A.; Harwell, J.H. Surfactant selection for enhancing ex situ soil washing. Water Res. 1999, 33, 351–360. [Google Scholar] [CrossRef]
- El-Sukkary, M.M.A.; Syed, N.A.; Aiad, I.; El-Azab, W.I.M. Synthesis and characterization of some alkyl polyglycosides surfactants. J. Surfactants Deterg. 2008, 11, 129–137. [Google Scholar] [CrossRef]
- OECD. OECD Guideline for Testing of Chemicals. Freshwater Alga and Cyanobacteria, Growth Inhibition Test; OECD/OCDE 201; OECD: Paris, France, 2011. [Google Scholar]
- OECD. OECD Guideline for Testing of Chemicals. Daphnia sp., Acute Immobilisation Test; OECD/OCDE 202; OECD: Paris, France, 2004. [Google Scholar]
- OECD. OECD Guideline for Testing of Chemicals. Fish, Acute Toxicity Test; OECD/OCDE 203; OECD: Paris, France, 1992. [Google Scholar]
- Spitsbergen, J.M.; Kent, M.L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research-advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87. [Google Scholar]
- OECD. OECD Guideline for Testing of Chemicals. Readily Biodegradability; OECD/OCDE 301; OECD: Paris, France, 1992. [Google Scholar]
- Roussel, M.; Benvegnu, T.; Lognoné, V.; Le Deit, H.; Laurent, I.; Plusquellec, D. Synthesis and physico-chemical properties of novel biocompatible alkyl D-mannopyranosiduronate surfactants derived from alginate. Eur. J. Org. Chem. 2005, 14, 3085–3094. [Google Scholar]
- Sari-Chmayssem, N.; Pessel, F.; Guégan, J.P.; Taha, S.; Benvegnu, T. Direct and one-pot conversion of polyguluronates and alginates into alkyl-L-guluronamide-based surfactant compositions. Green Chem. 2016, 18, 6573–6585. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
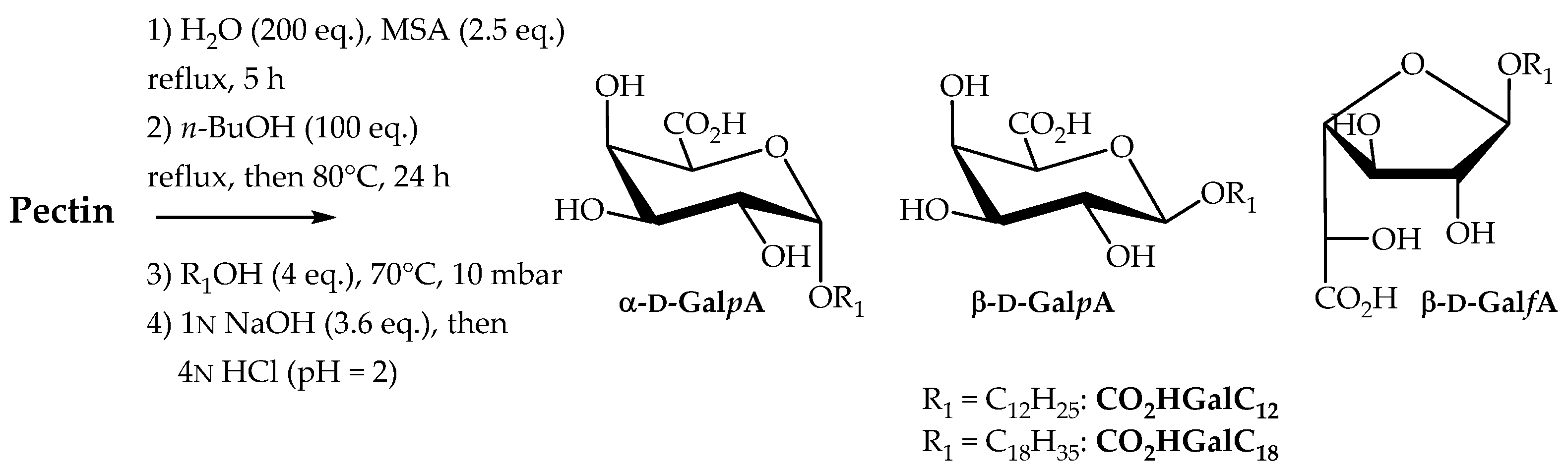{kind=link}
| Entry | First Step | Second Step | Yields * (%) | |||
|---|---|---|---|---|---|---|
| Hydrolysis | n eq. water | n′ eq. MSA | Reaction Conditions | C4GalC4 | 5 | |
| 1 | Reflux 5 h | 1000 | 2.5 | BuOH 100 eq., reflux 24 h BuOH 100 eq., reflux 48 h BuOH 100 eq., reflux 24 h BuOH 100 eq., 80 °C 24 h BuOH 100 eq., 80 °C 24 h BuOH 50 eq., 80 °C 24 h BuOH 100 eq., 80 °C 24 h | 39 | 27 |
| 2 | Reflux 5 h | 1000 | 2.5 | 41 | 36 | |
| 3 | Reflux 24 h | 1000 | 2.5 | 53 | 24 | |
| 4 | Reflux 5 h | 1000 | 2.5 | 76 | 6 | |
| 5 | Reflux 5 h | 200 | 2.5 | 21 | 1 | |
| 6 | Reflux 5 h | 200 | 2.5 | 18 | 2 | |
| 7 | Reflux 5 h | 200 | 1.1 | 11 | 3 | |
| Compound | Mass Composition (%) | Molar Composition of CnGalCn (%) * | ||||||
|---|---|---|---|---|---|---|---|---|
| Apolar Products | Fatty Alcohol | CnGalCn | Alkyl Glycosides | αf | βf | αp | βp | |
| C12PectC12 | 5 | 74 | 15 | 6 | 7 | 38 | 46 | 9 |
| C18PectC18 | 6 | 71 | 17 | 6 | 10 | 40 | 44 | 6 |
| Compound | Mass Composition (%) | Molar Composition of CO2HGalCn (%) * | ||||||
|---|---|---|---|---|---|---|---|---|
| Apolar products | Fatty Alcohol | CO2HGalCn | Alkyl Glycosides | αf | βf | αp | βp | |
| CO2HPectC12 | 5 | 77 | 12 | 6 | - | 13 | 76 | 11 |
| CO2HPectC18 | 5 | 77 | 13 | 5 | - | 19 | 68 | 13 |
| Compound | 𝛄CMC (mN m−1) | CMC (mg L−1) | CMC (mmol L−1) |
|---|---|---|---|
| Na+COO−GalC8 | 23 | 25.4 | 77.5 |
| Na+COO−GalC10 | 23 | 8.0 | 22.5 |
| Na+COO−GalC12 | 27 | 7.0 | 18.2 |
| Na+COO−GalC18 | 32 | 1.6 | 3.6 |
| Sodium Lauryl Ether Sulfate (SLES) | 35 | 0.3 | 0.8 |
| Sodium Dodecyl Sulfate (SDS) | 35 | 2.4 | 8.4 |
| Alkyl Polyglucosides C12 | 32 | - | 0.3 |
| Category Acute 1 (very toxic) | |
| CL50 96 h (for the fish) | ≤1 mg L−1 and/or |
| CE50 48 h (for the shellfish) | ≤1 mg L−1 and/or |
| CEr50 72 h (for the seaweed) | ≤1 mg L−1 |
| Category Acute 2 (toxic) | |
| CL50 96 h (for the fish) | >1 but ≤10 mg L−1 and/or |
| CE50 48 h (for the shellfish) | >1 but ≤10 mg L−1 and/or |
| CEr50 72 h (for the seaweed) | >1 but ≤10 mg L−1 |
| Category Acute 3 (poorly toxic) | |
| CL50 96 h (for the fish) | >10 but ≤100 mg L−1 and/or |
| CE50 48 h (for the shellfish) | >10 but ≤100 mg L−1 and/or |
| CEr50 72 h (for the seaweed) | >10 but ≤100 mg L−1 |
| Above 100 mg L−1, the substance is considered as non-toxic | |
| Test | Effect | Toxicological Descriptor * | CO2HGalC18 |
|---|---|---|---|
| Microalgae | Rate of growth | CEr50-72 h | Non-toxic at 100 mg L−1 |
| CEr20-72 h | Non-toxic at 100 mg L−1 | ||
| CEr10-72 h | Non-toxic at 100 mg L−1 | ||
| NOEC-72 h | ≥100 mg L−1 | ||
| Daphnia | Immobilisation | CE50-24 h | Non-toxic at 100 mg L−1 |
| CE50-48 h | Non-toxic at 100 mg L−1 | ||
| Fish | Mortality | CL50-96 h | Non-toxic at 100 mg L−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milliasseau, D.; Jeftić, J.; Pessel, F.; Plusquellec, D.; Benvegnu, T. Transformation of Pectins into Non-Ionic or Anionic Surfactants Using a One-Pot and Cascade Mode Process. Molecules 2021, 26, 1956. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071956
Milliasseau D, Jeftić J, Pessel F, Plusquellec D, Benvegnu T. Transformation of Pectins into Non-Ionic or Anionic Surfactants Using a One-Pot and Cascade Mode Process. Molecules. 2021; 26(7):1956. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071956
Chicago/Turabian StyleMilliasseau, Damien, Jelena Jeftić, Freddy Pessel, Daniel Plusquellec, and Thierry Benvegnu. 2021. "Transformation of Pectins into Non-Ionic or Anionic Surfactants Using a One-Pot and Cascade Mode Process" Molecules 26, no. 7: 1956. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26071956