
New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies

 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Biological Testing
2.2.1. Cell Proliferation Studies
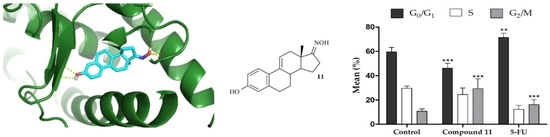
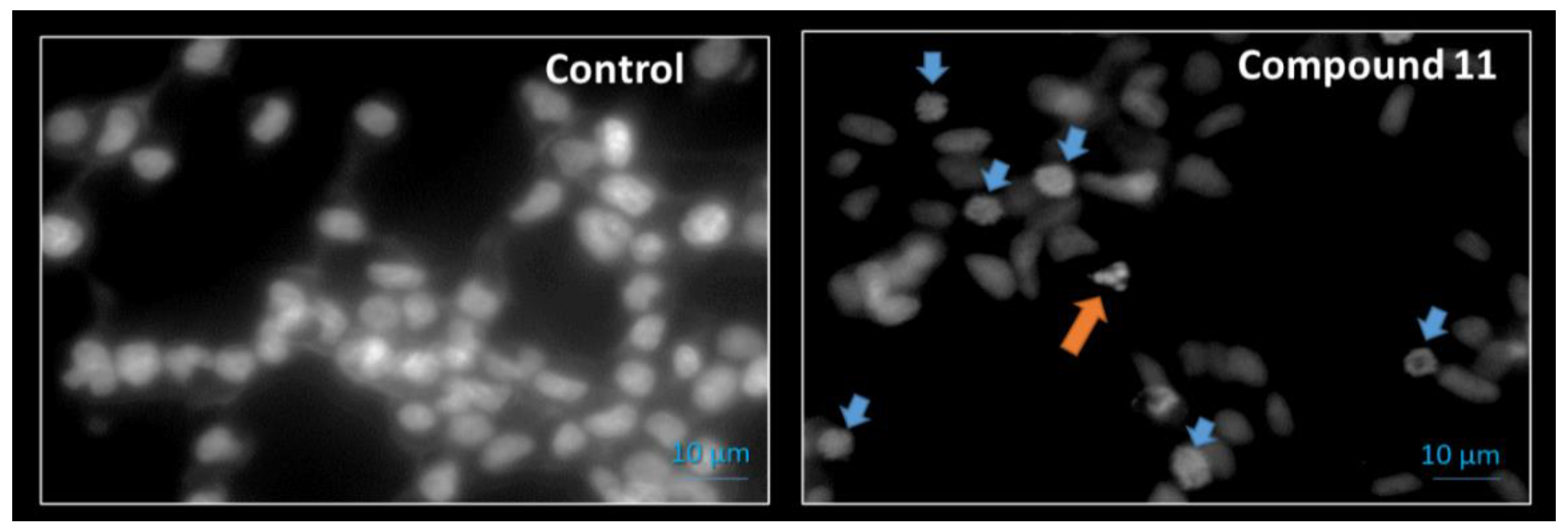
2.2.2. Cell Survival, Cell Cycle Distribution Evaluation and Hoechst 33,258 Staining
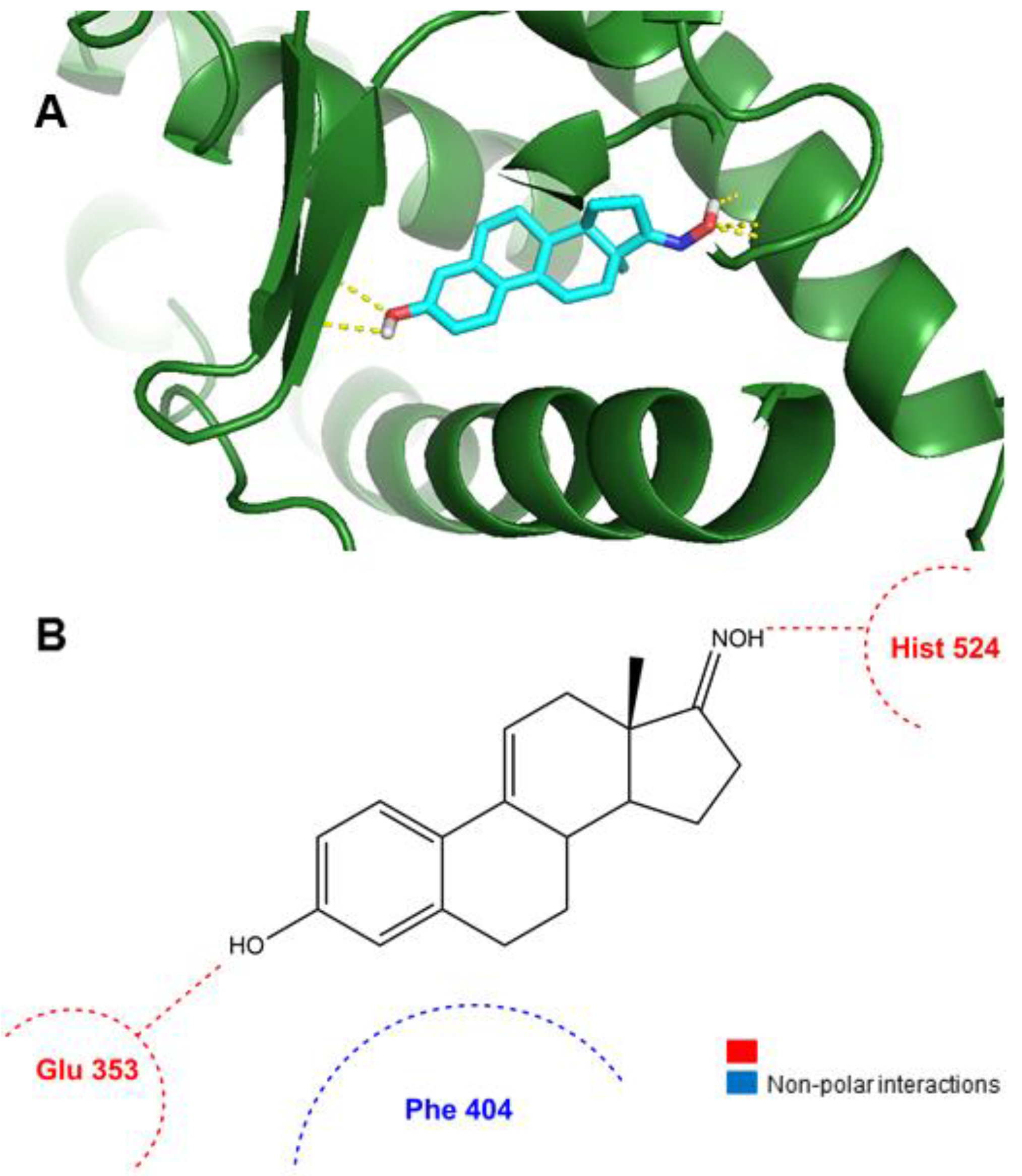
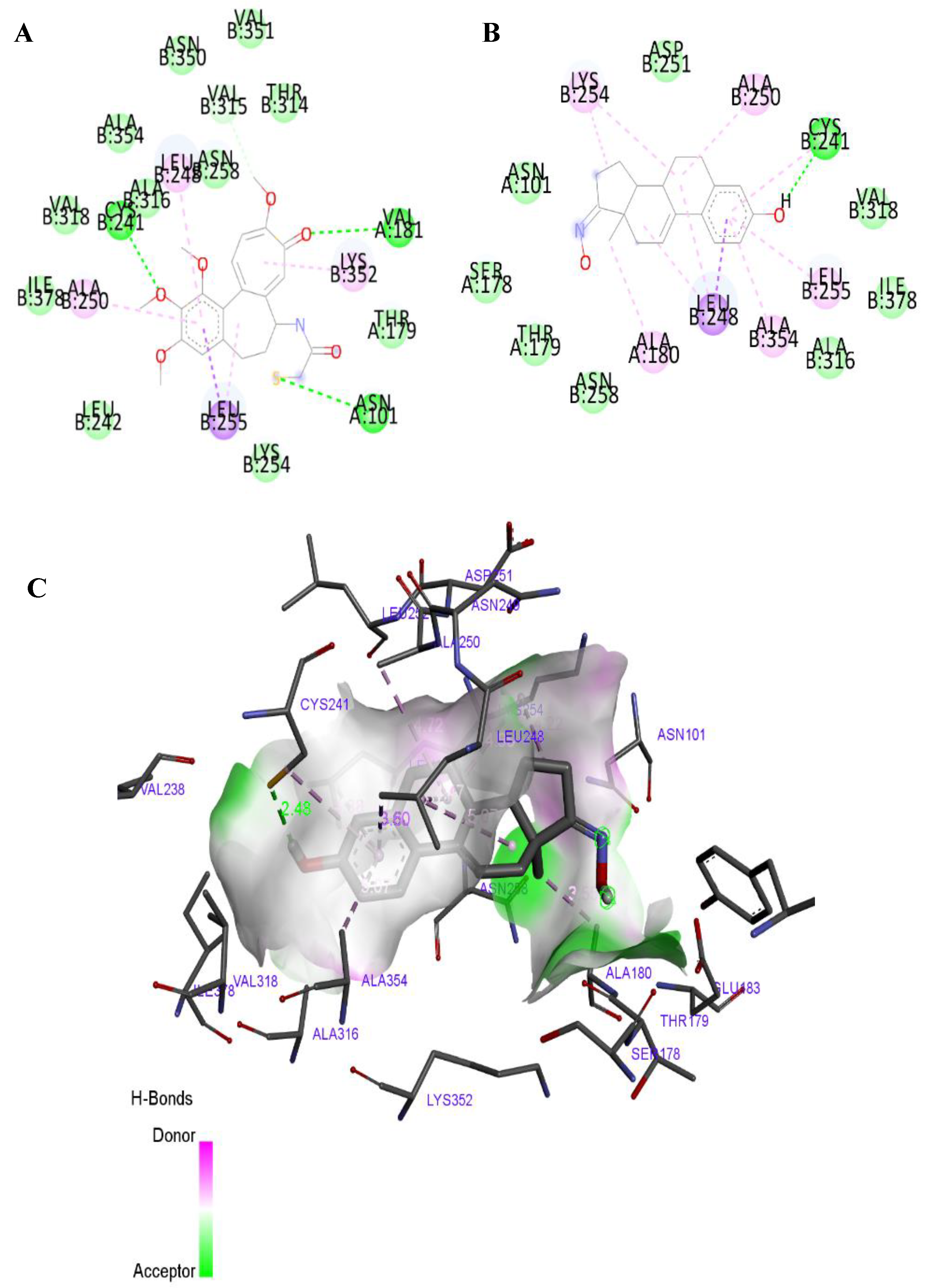
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Procedures for the Synthesis of Intermediates—Compounds 3, 4, 7, 8 and 9
Synthesis of 3-hydroxy-2-nitroestra-1,3,5(10)-trien-17-one (3) and 3-hydroxy-2,4-dinitroestra-1,3,5(10)-trien-17-one (4)
3-Hydroxy-2-nitroestra-1,3,5(10)-trien-17-one (3)
3-Hydroxy-2,4-dinitroestra-1,3,5(10)-trien-17-one (4)
3.1.2. General Procedure for the Synthesis of Oximes
Synthesis of 17-hydroxyiminoestra-1,3,5(10)-trien-3-ol (2)
Synthesis of 17-hydroxyimino-2-nitroestra-1,3,5(10)-trien-3-ol (5)
Synthesis of 17-hydroxyimino-2,4-dinitroestra-1,3,5(10)-trien-3-ol (6)
Synthesis of 17-hydroxyimino-2,4-diiodooestra-1,3,5(10)-trien-3-ol (10)
Synthesis of 17-hydroxyimino-3-hydroxyestra-1,3,5(10),9(11)-tetraen-3-ol (11)
Synthesis of 17-hydroxyimino-2,4-dibromo-3-hydroxyestra-1,3,5(10)-trien-3-ol (12)
3.2. Biology
3.2.1. Cell Culture
3.2.2. Preparation of Stock Solutions
3.2.3. Antiproliferative Activities against Six Cell Lines
3.2.4. E-Screening Assay in T47-D Cells
3.2.5. Analysis of LNCaP Cells Viability by Flow Cytometry
3.2.6. Cell Cycle Distribution of LNCaP Cells
3.2.7. Fluorescence Microscopy in LNCaP Cells after DNA Staining
3.2.8. Data Analysis
3.3. Molecular Docking Studies
3.3.1. Preparation of Proteins for Molecular Docking
3.3.2. Preparation of Ligands
3.3.3. Grid Parameters
3.3.4. Docking Simulations
3.3.5. Validation of the Molecular Docking Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, R.L.; Watts, C.K.W.; Musgrove, E.A. Cell cycle control by steroid hormones in breast cancer: Implications for endocrine resistance. Endocr. Relat. Cancer 1995, 2, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.; Nunez, L.; Peixinho, S.; Jiménez, C. Isolation and Synthesis of the First Natural 6-Hydroximino 4-en-3-one- Steroids from the Sponges. Tetrahedron Lett. 1997, 38, 1833–1836. [Google Scholar] [CrossRef]
- Deive, N.; Rodriguez, J.; Jiménez, C. Synthesis of Cytotoxic 6E-Hydroximino-4-ene Steroids: Structure/Activity Studies. J. Med. Chem. 2001, 44, 2612–2618. [Google Scholar] [CrossRef]
- Ãbele, E.; Lukevics, E. Recent Advances in the Chemistry of Oximes. Org. Prep. Proced. Int. 2000, 32, 235–264. [Google Scholar] [CrossRef]
- Canário, C.; Silvestre, S.; Falcão, A.; Alves, G. Steroidal Oximes: Useful Compounds with Antitumor Activities. Curr. Med. Chem. 2018, 25, 660–686. [Google Scholar] [CrossRef]
- Hejaz, H.A.M.; Purohit, A.; Mahon, M.F.; Reed, M.J.; Potter, B.V.L. Synthesis and Biological Activity of the Superestrogen (E)-17-Oximino-3-O-sulfamoyl-1,3,5(10)-estratriene: X-ray Crystal Structure of (E)-17-Oximino-3-hydroxy-1,3,5(10)-estratriene. J. Med. Chem. 1999, 42, 3188–3192. [Google Scholar] [CrossRef]
- Allan, G.M.; Lawrence, H.R.; Cornet, J.; Bubert, C.; Fischer, D.S.; Vicker, N.; Smith, A.; Tutill, H.J.; Purohit, A.; Day, J.M.; et al. Modification of Estrone at the 6, 16 and 17 Positions: Novel Potent Inhibitors of 17β-Hydroxysteroid Dehydrogenase Type 1. J. Med. Chem. 2006, 49, 1325–1345. [Google Scholar] [CrossRef]
- Hartmann, R.W.; Hector, M.; Haidar, S.; Ehmer, P.B.; Reichert, W.; Jose, J. Synthesis and Evaluation of Novel Steroidal Oxime Inhibitors of P450 17 (17alfa-Hydroxylase/C17-20-Lyase) and 5alfa-Reductase Types 1 and 2. J. Med. Chem. 2000, 43, 4266–4277. [Google Scholar] [CrossRef]
- Pokhrel, M.; Ma, E. Synthesis and screening of aromatase inhibitory activity of substituted C19 steroidal 17-oxime analogs. Molecules 2011, 16, 9868–9885. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.S.; Allan, G.M.; Bubert, C.; Vicker, N.; Smith, A.; Tutill, H.J.; Purohit, A.; Wood, L.; Packham, G.; Mahon, M.F.; et al. E-Ring Modified Steroids as Novel Potent Inhibitors of 17β-Hydroxysteroid Dehydrogenase Type 1. J. Med. Chem. 2005, 48, 5749–5770. [Google Scholar] [CrossRef]
- Leese, M.P.; Leblond, B.; Newman, S.P.; Purohit, A.; Reed, M.J.; Potter, B.V.L. Anti-cancer activities of novel D-ring modified 2-substituted estrogen-3-O-sulfamates. J. Steroid Biochem. Mol. Biol. 2005, 94, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef]
- Berényi, Á.; Minorics, R.; Iványi, Z.; Ocsovszki, I.; Ducza, E.; Thole, H.; Mernyák, E.; Frank, É.; Schneider, G.; Zupkó, I. Synthesis and investigation of the anticancer effects of estrone-16-oxime ethers in vitro. Steroids 2013, 78, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wendlandt, A.E.; Yelton, S.M.; Lou, D.; Watt, D.S.; Noonan, D.J. Synthesis and functional analysis of novel bivalent estrogens. Steroids 2010, 75, 825–833. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; He, H.-M.; Katzenellenbogen, J.A.; Lin, C.M.; Hamel, E. Synthesis, antitubulin and antimitotic activity, and cytotoxicity of analogs of 2-methoxyestradiol, an endogenous mammalian metabolite of estradiol that inhibits tubulin polymerization by binding to the colchicine binding site. J. Med. Chem. 1995, 38, 2041–2049. [Google Scholar] [CrossRef]
- Lao, K.; Wang, Y.; Chen, M.; Zhang, J.; You, Q. Design, synthesis and biological evaluation of novel 2-methoxyestradiol analogs as dual selective estrogen receptor modulators (SERMs) and antiangiogenic agents. Eur. J. Med. Chem. 2017, 139, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Klein, C.D.; Hartmann, R.W.; Chemie, M.; Saarlandes, U.; Box, P.O.; Saarbrücken, D. Synthesis and Evaluation of Steroidal Hydroxamic Acids as Inhibitors of P450 17 (17alfa-Hydroxylase/C17-20-Lyase). Arch. Pharm. Pharm. Med. Chem. 2001, 334, 138–140. [Google Scholar] [CrossRef]
- Kim, S.; Ma, E. Synthesis of Pregnane Derivatives, Their Cytotoxicity on LNCap and PC-3 Cells, and Screening on 5α-Reductase Inhibitory Activity. Molecules 2009, 14, 4655–4668. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, Y.; Ma, E. Synthesis and 5α-reductase inhibitory activity of C₂₁ steroids having 1,4-diene or 4,6-diene 20-ones and 4-azasteroid 20-oximes. Molecules 2012, 17, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, M.I.; Alam, M.S.; Yousuf, S.; Wu, Y.-C.; Lin, A.-S.; Shaheen, F. Pregnenolone derivatives as potential anticancer agents. Steroids 2011, 76, 1554–1559. [Google Scholar] [CrossRef]
- Holland, H.L.; Kumaresan, S.; Tan, L.; Njar, V.C.O. Synthesis of 6-Hydroximino-3-oxo Steroids, a New Class of Aromatase Inhibitor. J. Chem. Soc Perkin Trans 1 1992, 585–587. [Google Scholar] [CrossRef]
- Cushman, M.; He, H.M.; Katzenellenbogen, J.A.; Varma, R.K.; Hamel, E.; Lin, C.M.; Ram, S.; Sachdeva, Y.P. Synthesis of analogs of 2-methoxyestradiol with enhanced inhibitory effects on tubulin polymerization and cancer cell growth. J. Med. Chem. 1997, 40, 2323–2334. [Google Scholar] [CrossRef]
- Rzheznikov, V.M.; Golubovskaya, L.E.; Minailova, O.N.; Osetrova, I.P.; Smirnova, Z.S. Steroidal Nitrates: Synthesis and Antitumor Activity of of 9α, 11β-Dihydroxyestra-1,3,5(10)-triene 11-nitrates. Pharm. Chem. J. 2003, 37, 10–12. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, L.; Hernández-Linares, M.G.; Escobar, M.L.; López-Muñoz, H.; Zenteno, E.; Fernández-Herrera, M.A.; Guerrero-Luna, G.; Carrasco-Carballo, A.; Sandoval-Ramírez, J. Antiproleferative, Cytotoxic and Apoptotic Activity of Steroidal Oximes in Cervicouterine Cell Lines. Molecules 2016, 21, E1533. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.J.; Li, G.; Gao, J.M. Synthesis and cytotoxicity of novel steroidal C-20 oxime ester derivatives from 16-DPA. Arab. J. Chem. 2019, 12, 2084–2090. [Google Scholar] [CrossRef] [Green Version]
- Canário, C.; Matias, M.; de Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. Δ9,11-Estrone derivatives as potential antiproliferative agents: Synthesis, in vitro biological evaluation and docking studies. Comptes Rendus Chim. 2020, 201–217. [Google Scholar] [CrossRef]
- Stéphan, E.; Zen, R.; Authier, L.; Jaouen, G. Improved synthesis of a protected 11-oxoestrone. Steroids 1995, 60, 809–811. [Google Scholar] [CrossRef]
- Stubenrauch, G.; Knuppen, R. Convenient large scale preparation of catechol estrogens. Steroids 1976, 28, 733–741. [Google Scholar] [CrossRef]
- Saikia, L.; Baruah, J.; Thakur, A. A rapid, convenient, solventless green approach for the synthesis of oximes using grindstone chemistry. Org. Med. Chem. Lett. 2011, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.R.; Sung, G.H.; Kim, J.J.; Yoon, Y.J. A development of rapid, practical and selective process for preparation of z-oximes. J. Korean Chem. Soc. 2013, 57, 295–299. [Google Scholar] [CrossRef]
- Jadhav, A.D.; Gade, E.H.; Angarkhe, B.L.; Durrani, A. An Efficient One Pot Synthesis of Oxime by Classical Method. Int. J. Chem. Phys. Sci. 2018, 7, 12–16. [Google Scholar]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- Ayan, D.; Maltais, R.; Roy, J.; Poirier, D. A new nonestrogenic steroidal inhibitor of 17β-hydroxysteroid dehydrogenase type I blocks the estrogen-dependent breast cancer tumor growth induced by estrone. Mol. Cancer Ther. 2012, 11, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Benítez, F.; Roy, J.; Maltais, R.; Poirier, D. Impact of androstane A- and D-ring inversion on 17β-hydroxysteroid dehydrogenase type 3 inhibitory activity, androgenic effect and metabolic stability. Bioorganic Med. Chem. 2017, 25, 2065–2073. [Google Scholar] [CrossRef]
- Palomino, E.; Heef, M.J.; Horwitz, J.P.; Polin, L.; Brooks, S.C. Skeletal conformations and receptor binding of some 9,11-modified estradiols. J. Steroid Biochem. Mol. Biol. 1994, 50, 75–84. [Google Scholar] [CrossRef]
- Andruska, N.; Mao, C.; Cherian, M.; Zhang, C.; Shapiro, D.J. Evaluation of a Luciferase-based Reporter Assay as a Screen for Inhibitors of Estrogen-ERα-induced Proliferation of Breast Cancer Cells. J Biomol Screen 2012, 17, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Robert, S. DiPaola To Arrest or Not To G2-M Cell-Cycle Arrest. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar]
- Latif, A.D.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, Z.P.; Zupkó, I.; Hunyadi, A. Synthesis and in vitro antitumor activity of naringenin oxime and oxime ether derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef] [Green Version]
- Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wolfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; et al. Synthesis of trans-16-triazolyl-13 α-methyl-17-estradiol diastereomers and the effects of structural modifications on their in vitro antiproliferative activities. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Hayal, T.B.; Kıratlı, B.; Şişli, H.B.; Demirci, S.; Şahin, F.; Doğan, A. Design and synthesis of phenylpiperazine derivatives as potent anticancer agents for prostate cancer. Chem. Biol. Drug Des. 2019, 94, 1584–1595. [Google Scholar] [CrossRef]
- Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A time to kill: Targeting apoptosis in cancer. Int. J. Mol. Sci. 2015, 16, 2942–2955. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, F.; Finn, P.; La Thangue, N.B. The cell cycle, chromatin and cancer: Mechanism-based therapeutics come of age. Drug Discov. Today 2003, 8, 793–802. [Google Scholar] [CrossRef]
- Toné, S.; Sugimoto, K.; Tanda, K.; Suda, T.; Uehira, K.; Kanouchi, H.; Samejima, K.; Minatogawa, Y.; Earnshaw, W.C. Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp. Cell Res. 2007, 313, 3635–3644. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.P.H.; Horwitz, S.B. Taxol®: The first microtubule stabilizing agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, F.; Billstrom, A.; Hartley-Asp, B. Estramustine Depolymerizes Microtubules by Binding to Tubulin. Cancer Res. 1993, 53, 4573–4581. [Google Scholar]
- Lima, W.E.A.; Pereira, A.F.; Castro, A.A.; Cunha, E.F.F.; Ramalho, T.C. Flexibility in the Molecular Design of Acetylcholinesterase Reactivators: Probing Representative Conformations by Chemometric Techniques and Docking/QM Calculations. Lett. Drug Des. Discov. 2016, 13, 360–371. [Google Scholar] [CrossRef]
- Giacoppo, J.O.S.; França, T.C.C.; Kuča, K.; da Cunha, E.F.F.; Abagyan, R.; Mancini, D.T.; Ramalho, T.C. Molecular modeling and in vitro reactivation study between the oxime BI-6 and acetylcholinesterase inhibited by different nerve agents. J. Biomol. Struct. Dyn. 2015, 33, 2048–2058. [Google Scholar] [CrossRef]
- de Paula, R.L.; de Almeida, J.S.F.D.; Cavalcante, S.F.A.; Gonçalves, A.S.; Simas, A.B.C.; Franca, T.C.C.; Valis, M.; Kuca, K.; Nepovimova, E.; Granjeiro, J.M. Molecular modeling and in vitro studies of a neutral oxime as a potential reactivator for acetylcholinesterase inhibited by paraoxon. Molecules 2018, 23, 2954. [Google Scholar] [CrossRef] [Green Version]
- Day, J.M.; Tutill, H.J.; Purohit, A.; Reed, M.J. Design and validation of specific inhibitors of 17β-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 2008, 15, 665–692. [Google Scholar] [CrossRef] [Green Version]
- Dorléans, A.; Gigant, B.; Ravelli, R.B.G.; Mailliet, P.; Mikol, V.; Knossow, M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. USA 2009, 106, 13775–13779. [Google Scholar] [CrossRef] [Green Version]
- Bueno, O.; Gallego, J.E.; Martins, S.; Andrea, E.P.; Gago, F.; Gómez-sanjuan, A.; Camarasa, M.; Barasoain, I.; Steinmetz, M.O.; Díaz, J.F.; et al. High-affinity ligands of the colchicine domain in tubulin based on a structure-guided design. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.S.; Raghuvanshi, D.S.; Hasanain, M.; Alam, S.; Sarkar, J.; Mitra, K.; Khan, F.; Negi, A.S. Recent Advances in chemistry and pharmacology of 2-methoxyestradiol: An anticancer investigational drug. Steroids 2016, 110, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Santaniello, E.; Ravasi, M.; Ferraboschi, P. A-Ring nitration of estrone. J. Org. Chem. 1983, 48, 739–740. [Google Scholar] [CrossRef]
- Bose, A.; Sanjoto, W.P.; Villarreal, S.; Aguilar, H.; Banik, B.K. Novel nitration of estrone by metal nitrates. Tetrahedron Lett. 2007, 48, 3945–3947. [Google Scholar] [CrossRef]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Guzman, F.G.; Higashiyama, T.; Pangborn, W.; Osawa, Y.; Ghosh, D. Structure of human estrone sulfatase suggests functional roles of membrane association. J. Biol. Chem. 2003, 278, 22989–22997. [Google Scholar] [CrossRef] [Green Version]
- Aka, J.A.; Mazumdar, M.; Chen, C.-Q.; Poirier, D.; Lin, S.-X. 17β-Hydroxysteroid Dehydrogenase Type 1 Stimulates Breast Cancer By Dihydrotestosterone Inactivation in Addition To Estradiol Production. Mol. Endocrinol. 2010, 24, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 | NHDF |
|---|---|---|---|---|---|---|
| 1 | 41.93 | ND | ND | 29.53 | 42.69 | 61.82 |
| 2 | 26.65 | 29.26 | ND | 16.94 | ND | 43.88 |
| 5 | 16.75 | ND | ND | 12.32 | 18.51 | ND |
| 10 | ND | ND | ND | 28.00 | ND | ND |
| 11 | 25.63 | 43.45 | 3.59 | 18.35 | 24.33 | 30.84 |
| 12 | ND | ND | ND | 21.94 | ND | ND |
| 5-FU | 1.71 | 0.54 | 7.79 | 1.78 | 1.31 | 3.61 |
| Compound | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 |
|---|---|---|---|---|---|
| 1 | 1.47 | ND | ND | 2.09 | 1.45 |
| 2 | 1.65 | 1.50 | ND | 2.59 | ND |
| 11 | 1.20 | 0.71 | 8.59 | 1.68 | 1.27 |
| 5-FU | 2.11 | 6.69 | 0.46 | 2.03 | 2.76 |
| Compound | Lowest Energy (kcal.mol−1) | |||
|---|---|---|---|---|
| ERα | ST | 17β−HSD1 | β−Tubulin | |
| 1 | −10.3 b | −6.2 b | −8.1 b | −9.0 |
| 2 | −9.7 | −6.7 | −8.1 | −8.9 |
| 3 | −8.8 | −7.0 | −8.2 | −8.7 |
| 4 | −6.6 | −7.0 | −8.1 | −8.5 |
| 5 | −7.9 | −6.9 | −8.3 | −8.9 |
| 6 | −5.3 | −6.8 | −8.1 | −8.4 |
| 7 | −4.3 b | −6.0 b | −7.7 b | −8.4 |
| 8 | −10.9 b | −5.9 b | −8.2 b | −9.0 |
| 9 | −6.8 b | −6.3 b | −8.1 b | −9.0 |
| 10 | −5.8 | −6.6 | −7.7 | −8.5 |
| 11 | −10.5 | −6.5 | −8.3 | −9.1 |
| 12 | −5.0 | −6.3 | −7.6 | −8.5 |
| E2 | −9.9 a | − | − | − |
| N−acetyl−D−glucosamine | − | −7.2 a | − | − |
| 5α−Dihydrotestosterone | − | − | −8.3 a | − |
| Colchicine | − | − | − | −8.4 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canário, C.; Matias, M.; Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies. Molecules 2021, 26, 2687. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092687
Canário C, Matias M, Brito V, Santos AO, Falcão A, Silvestre S, Alves G. New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies. Molecules. 2021; 26(9):2687. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092687
Chicago/Turabian StyleCanário, Catarina, Mariana Matias, Vanessa Brito, Adriana O. Santos, Amílcar Falcão, Samuel Silvestre, and Gilberto Alves. 2021. "New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies" Molecules 26, no. 9: 2687. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092687