
Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol





Abstract
:1. Introduction
2. Experimental Procedures and Computational Methods
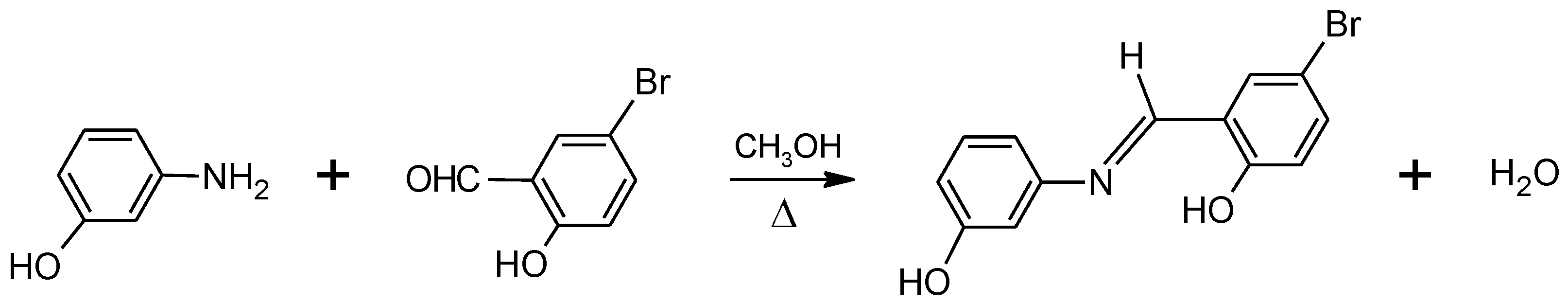
2.1. Synthesis
2.2. Matrix-Isolation Experiments
2.3. Theoretical Calculations
3. Results and Discussion
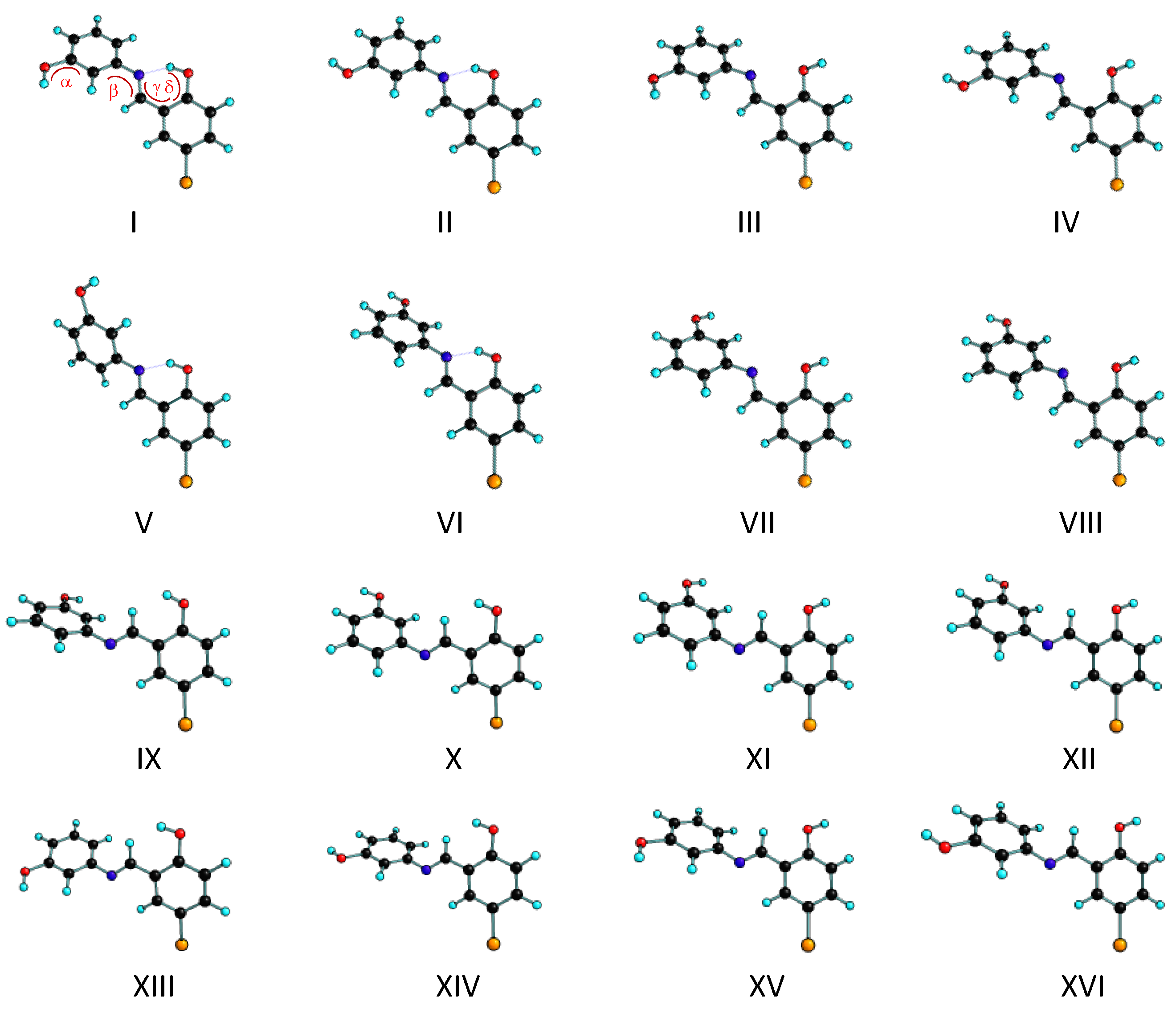
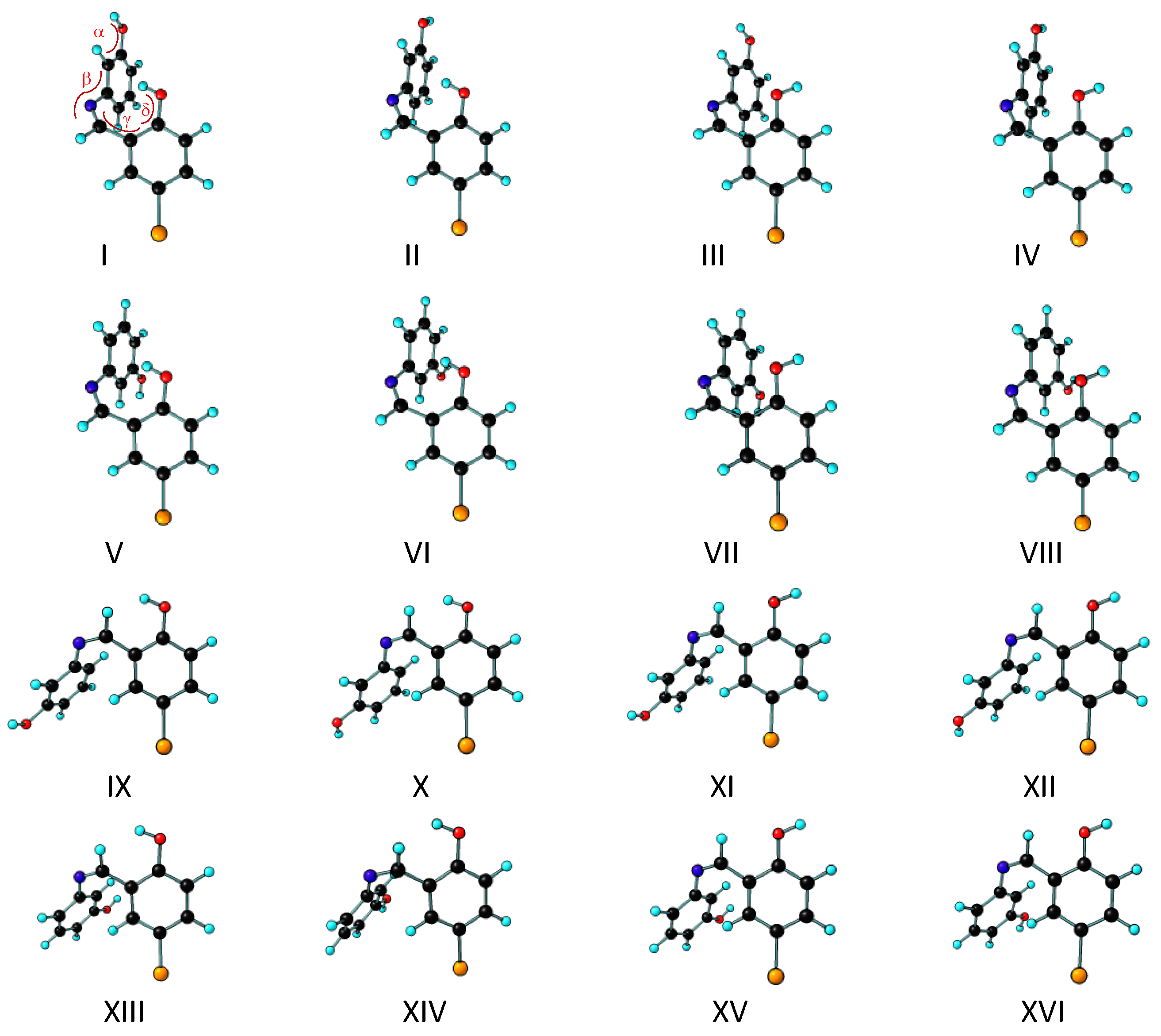
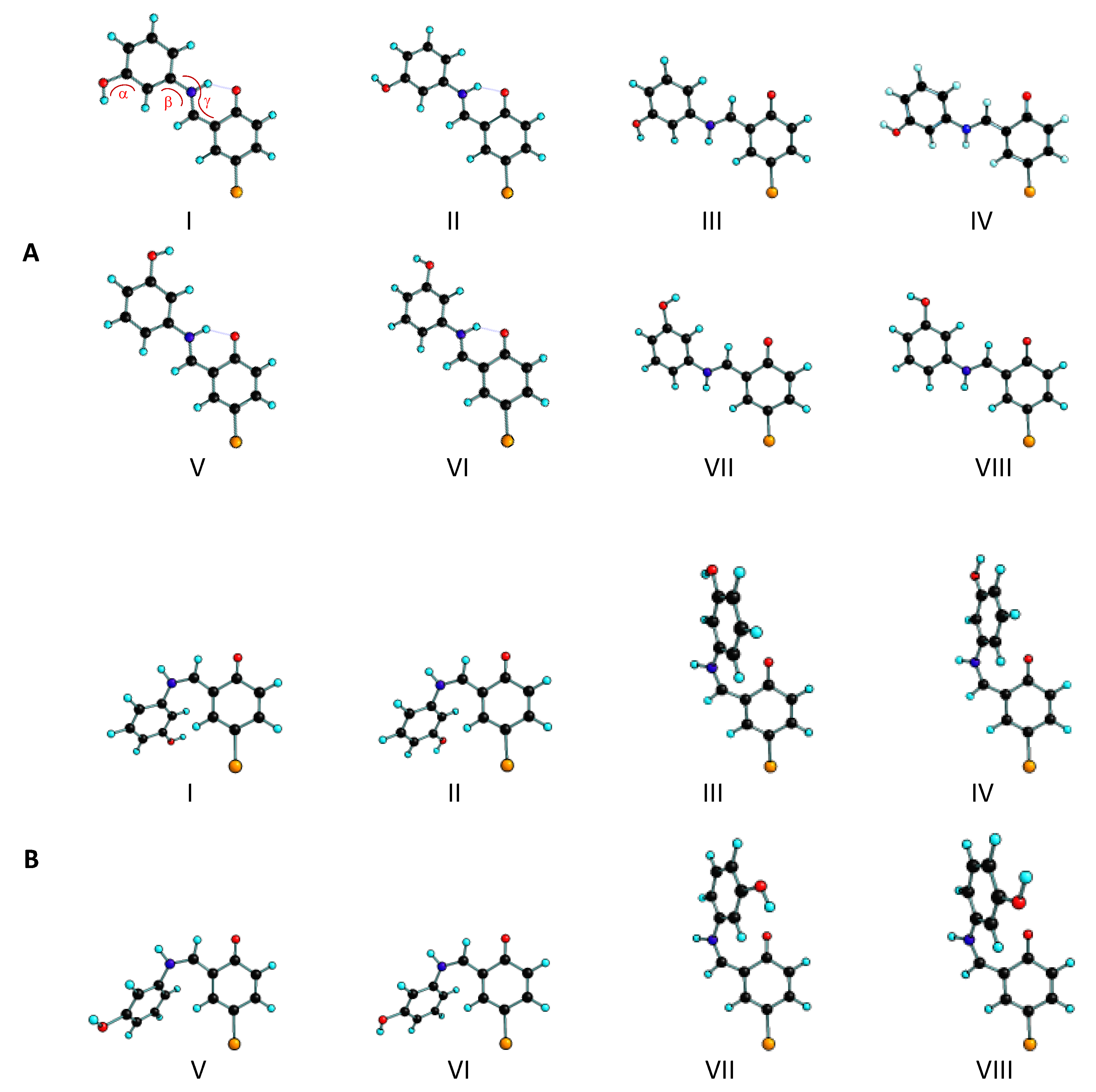
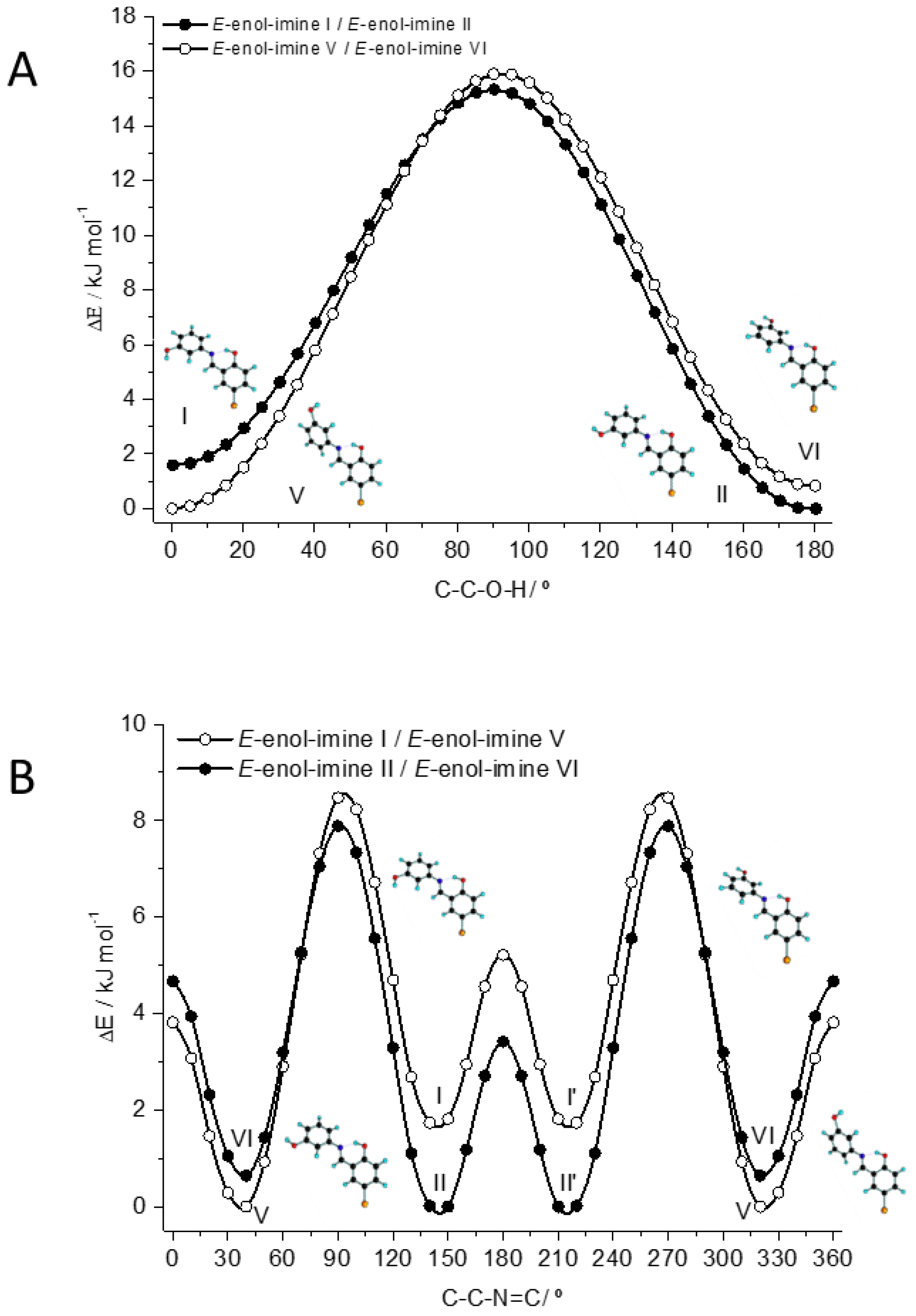
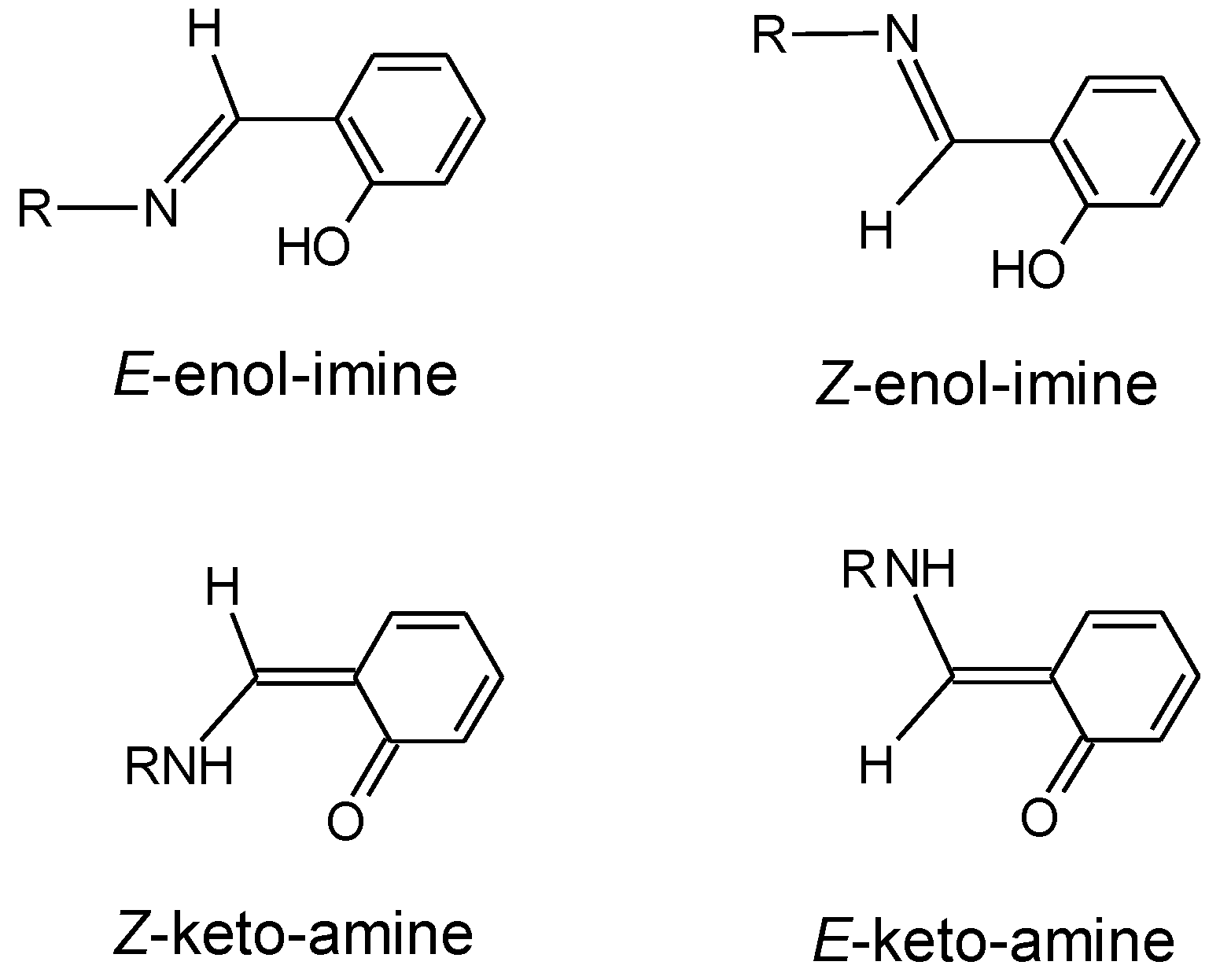
3.1. Structures and Conformational Landscapes of BHAP Isomeric Forms
3.2. Natural Bond Orbital Analysis
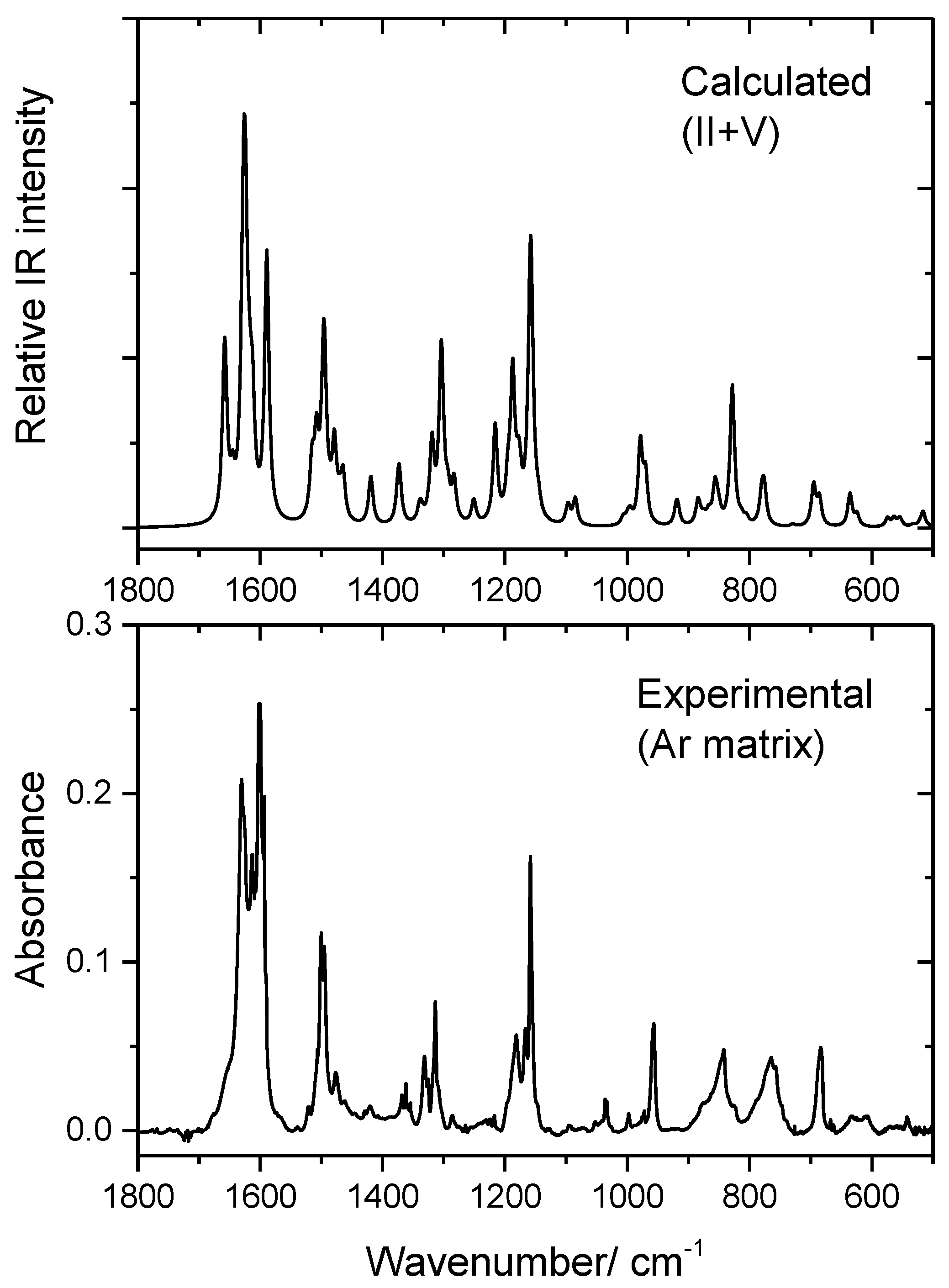
3.3. Infrared Spectrum of BHAP in an Ar Matrix
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, P.; Bhamidipati, M.; Coles, M.; Rao, D.V.G.L.N. Biological Nano-Ceramic Materials for Holographic Data Storage. Chem. Phys. Lett. 2004, 400, 506–510. [Google Scholar] [CrossRef]
- Yuan, W.; Sun, L.; Tang, H.; Wen, Y.; Jiang, G.; Huang, W.; Jiang, L.; Song, Y.; Tian, H.; Zhu, D.A. Novel Thermally Stable Spironaphthoxazine and Its Application in Rewritable High Density Optical Data Storage. Adv. Mater. 2005, 17, 156–160. [Google Scholar] [CrossRef]
- Yanez, C.O.; Andrade, C.D.; Yao, S.; Luchita, G.; Bondar, M.V.; Belfield, K.D. Photosensitive Polymeric Materials for Two-Photon 3D WORM Optical Data Storage Systems. ACS Appl. Mater. Interfaces 2009, 1, 2219–2229. [Google Scholar] [CrossRef]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and Thermochromism of Schiff Bases in the Solid State: Structural Aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef]
- Amimoto, K.; Kawato, T. Photochromism of Organic Compounds in the Crystal State. J. Photochem. Photobiol. C: Photochem. Rev. 2005, 6, 207–226. [Google Scholar] [CrossRef]
- Melloni, A.; Paccani, R.R.; Donati, D.; Zanirato, V.; Sinicropi, A.; Parisi, M.L.; Martin, E.; Ryazantsev, M.; Ding, W.J.; Frutos, L.M.; et al. Modeling, Preparation, and Characterization of a Dipole Moment Switch Driven by Z/E Photoisomerization. J. Am. Chem. Soc. 2010, 132, 9310–9319. [Google Scholar] [CrossRef] [PubMed]
- Staykov, A.; Watanabe, M.; Ishihara, T.; Yoshizawa, K. Photoswitching of Conductance through Salicylidene Methylamine. J. Phys. Chem. C 2014, 118, 27539–27548. [Google Scholar] [CrossRef]
- Dalapati, S.; Jana, S.; Guchhait, N. Anion Recognition by Simple Chromogenic and Chromo-Fluorogenic Salicylidene Schiff Base or Reduced-Schiff Base Receptors. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 129, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, J.; Yoon, J. Recent Advances in Development of Chiral Fluorescent and Colorimetric Sensors. Chem. Rev. 2014, 114, 4918–4959. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem. Rev. 2015, 115, 1597–1621. [Google Scholar] [CrossRef]
- Wang, Z.; Möhwald, H.; Gao, C. Preparation and Redox-Controlled Reversible Response of Ferrocene-Modified Poly(Allylamine Hydrochloride) Microcapsules. Langmuir 2011, 27, 1286–1291. [Google Scholar] [CrossRef]
- Wang, B.; Xu, C.; Xie, J.; Yang, Z.; Sun, S. pH Controlled Release of Chromone from Chromone-Fe3O4 Nanoparticles. J. Am. Chem. Soc. 2008, 130, 14436–14437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Qi, W.; Yan, X.; He, Q.; Cui, Y.; Wang, K.; Li, D.; Li, J. Proton Gradients Produced by Glucose Oxidase Microcapsules Containing Motor F0F1-ATPase for Continuous ATP Biosynthesis. J. Phys. Chem. B 2009, 113, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Avadanei, M.; Cozan, V.; Shova, S.; Paixão, J.A. Solid State Photochromism and Thermochromism of Two Related N-Salicylidene Anilines. Chem. Phys. 2014, 444, 43–51. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Pirbudak, G.; Berber, H.; Sıdır, İ. Study on the Electronic and Photophysical Properties of the Substitute-((2-phenoxybenzylidene)amino)phenol Derivatives: Synthesis, Solvatochromism, Electric Dipole Moments and DFT Calculations. J. Mol. Liq. 2017, 242, 1096–1110. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Berber, H.; Sıdır, İ. The Dipole Moments and Solvatochromism of ((4-(Benzyloxy)benzylidene)amino)phenol Compounds as Solvatochromic Materials. J. Solut. Chem. 2019, 48, 775–806. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Aslan, C.; Berber, H.; Sıdır, İ. The Electronic Structure, Solvatochromism, and Electric Dipole Moments of New Schiff Base Derivatives Using Absorbance and Fluorescence Spectra. Struct. Chem. 2019, 30, 835–851. [Google Scholar] [CrossRef]
- Minkin, V.I.; Tsukanov, A.V.; Dubonosov, A.D.; Bren, V.A. Tautomeric Schiff Bases: Iono-, Solvato-, Thermo- and Photochromism. J. Mol. Struct. 2011, 998, 179–191. [Google Scholar] [CrossRef]
- Pajak, J.; Maes, G.; De Borggraeve, W.M.; Boens, N.; Filarowski, A. Matrix-Isolation FT-IR and Theoretical Investigation of the Vibrational Properties of the Sterically Hindered Ortho-Hydroxy Acylaromatic Schiff Bases. J. Mol. Struct. 2007, 844–845, 83–93. [Google Scholar] [CrossRef]
- Filarowski, A.; Głowiaka, T.; Koll, A. Strengthening of the Intramolecular O⋯H⋯N Hydrogen Bonds in Schiff Bases as a Result of Steric Repulsion. J. Mol. Struct. 1999, 484, 75–89. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Głowiak, T. Structure and Hydrogen Bonding in Ortho-Hydroxy Ketimines. J. Mol. Struct. 2003, 644, 187–195. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Karpfen, A.; Wolschann, P. Intramolecular Hydrogen Bond in Molecular and Proton-Transfer Forms of Schiff Bases. Chem. Phys. 2004, 297, 323–332. [Google Scholar] [CrossRef]
- Król-Starzomska, I.; Filarowski, A.; Rospenk, M.; Koll, A.; Melikova, S. Proton Transfer Equilibria in Schiff Bases with Steric Repulsion. J. Phys. Chem. A 2004, 108, 2131–2138. [Google Scholar] [CrossRef]
- Kownacki, K.; Mordzinski, A.; Wilbrandt, R.; Grabowska, A. Laser-Induced Absorption and Fluorescence Studies of Photochromic Schiff Bases. Chem. Phys. Lett. 1994, 227, 270–276. [Google Scholar] [CrossRef]
- Ziółek, M.; Kubicki, J.; Maciejewski, A.; Naskręcki, R.; Grabowska, A. An Ultrafast Excited State Intramolecular Proton Transfer (ESPIT) and Photochromism of Salicylideneaniline (SA) and Its “Double” Analogue Salicylaldehyde Azine (SAA). A Controversial Case. Phys. Chem. Chem. Phys. 2004, 6, 4682–4689. [Google Scholar] [CrossRef]
- Sliwa, M.; Mouton, N.; Ruckebusch, C.; Poisson, L.; Idrissi, A.; Aloïse, S.; Potier, L.; Dubois, J.; Poizat, O.; Buntinx, G. Investigation of Ultrafast Photoinduced Processes for Salicylidene Aniline in Solution and Gas Phase: Toward a General Photo-Dynamical Scheme. Photochem. Photobiol. Sci. 2010, 9, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Asahi, T.; Masuhara, H.; Nakatani, K.; Sliwa, M. Photochromic Dynamics of Salicylidene Aniline in Solid State by Using Femtosecond Transient Absorption Spectroscopy. Mol. Cryst. Liq. Cryst. 2005, 431, 541–548. [Google Scholar] [CrossRef]
- Ziółek, M.; Kubicki, J.; Maciejewski, A.; Naskrȩcki, R.; Grabowska, A. Enol-Keto Tautomerism of Aromatic Photochromic Schiff Base N,N′-Bis(salicylidene)-p-phenylenediamine: Ground State Equilibrium and Excited State Deactivation Studied by Solvatochromic Measurements on Ultrafast Time Scale. J. Chem. Phys. 2006, 124, 124518. [Google Scholar] [CrossRef] [PubMed]
- Ziółek, M.; Burdziński, G.; Filipczak, K.; Karolczak, J.; Maciejewski, A. Spectroscopic and Photophysical Studies of the Hydroquinone Family of Photochromic Schiff Bases Analyzed over a 17-Orders-of-Magnitude Time Scale. Phys. Chem. Chem. Phys. 2008, 10, 1304–1318. [Google Scholar] [CrossRef]
- Grzegorzek, J.; Filarowski, A.; Mielke, Z. The Photoinduced Isomerization and Its Implication in the Photo-Dynamical Processes in Two Simple Schiff Bases Isolated in Solid Argon. Phys. Chem. Chem. Phys. 2011, 13, 16596–16605. [Google Scholar] [CrossRef]
- Dunkin, I.R. Matrix Isolation Techniques: A Practical Approach; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Fausto, R. (Ed.) Low Temperature Molecular Spectroscopy; NATO-ASI Series C483; Kluwer: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Reva, I.D.; Lopes Jesus, A.J.; Rosado, M.T.S.; Fausto, R.; Eusébio, M.E.; Redinha, J.S. Stepwise Conformational Cooling Towards a Single Isomeric State in the Four Internal Rotors System 1,2-Butanediol. Phys. Chem. Chem. Phys. 2006, 8, 5339–5349. [Google Scholar] [CrossRef] [Green Version]
- Rosado, M.T.S.; Lopes Jesus, A.J.; Reva, I.D.; Fausto, R.; Redinha, J.S. Conformational Cooling Dynamics in Matrix-Isolated 1,3-butanediol. J. Phys. Chem. A 2009, 113, 7499–7507. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, L.; Reva, I.; Nowak, M.J.; Fausto, R. Five Isomers of Monomeric Cytosine and Their Interconversions Induced by Tunable UV Laser Light. Phys. Chem. Chem. Phys. 2011, 13, 9676–9684. [Google Scholar] [CrossRef] [Green Version]
- Ismael, A.M.; Cristiano, M.L.S.; Fausto, R.; Gómez-Zavaglia, A. Tautomer Selective Photochemistry in 1-(Tetrazol-5-yl)ethanol. J. Phys. Chem. A 2010, 114, 13076–13085. [Google Scholar] [CrossRef]
- Nunes, C.M.; Pereira, N.A.M.; Reva, I.; Amado, P.S.M.; Cristiano, M.L.S.; Fausto, R. Bond-Breaking/Bond-Forming Reactions by Vibrational Excitation: Infrared-Induced Bidirectional Tautomerization of Matrix-Isolated Thiotropolone. J. Phys. Chem. Lett. 2020, 11, 8034–8039. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: https://gaussian.com/g09/ (accessed on 1 March 2021).
- Dennington, R.; Keith, T.; Milam, J. GaussView (Version 5.0); Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- ChemCraft (version 1.8)—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 1 March 2021).
- Avadanei, M.; Kuş, N.; Cozan, V.; Fausto, R. Structure and Photochemistry of N-Salicylidene-p-carboxyaniline Isolated in Solid Argon. J. Phys. Chem. A 2015, 119, 9121–9132. [Google Scholar] [CrossRef] [PubMed]
- Zgierski, M.Z.; Grabowska, A. Photochromism of Salicylideneaniline (SA). How the Photochromic Transient Is Created: A Theoretical Approach. J. Chem. Phys. 2000, 112, 6329–6337. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar] [CrossRef]
- Wiberg, K. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Martínez-Cifuentes, M.; Weiss-López, B.E.; Santos, L.S.; Araya-Maturana, R. Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones. Molecules 2014, 19, 9354–9368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, F.; Alkorta, I.; Elguero, J. Barriers about Double Carbon-Nitrogen Bond in Imine Derivatives (Aldimines, Oximes, Hydrazones, Azines). Croat. Chem. Acta 2009, 82, 173–183. Available online: https://hrcak.srce.hr/38652 (accessed on 1 March 2021).
- Reva, I.; Simão, A.; Fausto, R. Conformational Properties of Trimethyl Phosphate Monomer. Chem. Phys. Lett. 2005, 406, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Justino, L.L.G.; Reva, I.; Fausto, R. Thermally and Vibrationally Induced Conformational Isomerizations, Infrared Spectra, and Photochemistry of Gallic Acid in Low-Temperature Matrices. J. Chem. Phys. 2016, 145, 014304. [Google Scholar] [CrossRef] [Green Version]
- Reva, I.D.; Stepanian, S.G.; Adamowicz, L.; Fausto, R. Missing Conformers. Comparative Study of Conformational Cooling in Cyanoacetic Acid and Methyl Cyanoacetate Isolated in Low Temperature Inert Gas Matrixes. Chem Phys. Lett. 2003, 374, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Kuş, N.; Sharma, A.; Peña, I.; Bermúdez, M.C.; Cabezas, C.; Alonso, J.L.; Fausto, R. Conformers of β-Aminoisobutyric Acid Probed by Jet-Cooled Microwave and Matrix Isolation Infrared Spectroscopic Techniques. J. Chem. Phys. 2013, 138, 144305. [Google Scholar] [CrossRef]
- Pettersson, M.; Maçôas, E.M.S.; Khriachtchev, L.; Lundell, J.; Fausto, J.; Räsänen, M. Cis→Trans Conversion of Formic Acid by Dissipative Tunneling in Solid Rare Gases: Influence of Environment on the Tunneling Rate. J. Chem. Phys. 2002, 117, 9095–9098. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M. Rotational Isomerization of Small Carboxylic Acids Isolated in Argon Matrices: Tunnelling and Quantum Yields for the Photoinduced Processes. Phys. Chem. Chem. Phys. 2005, 7, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanbu, S.; Sekine, M.; Nakata, M. Hydrogen-Atom Tunneling in Isomerization around the C–O Bond of 2-Chloro-6-Fluorophenol in Low-Temperature Argon Matrixes. J. Phys. Chem. A 2011, 115, 9911–9918. [Google Scholar] [CrossRef]
- Kuş, N.; Sagdinc, S.; Fausto, R. Infrared Spectrum and UV-Induced Photochemistry of Matrix- Isolated 5-Hydroxyquinoline. J. Phys. Chem. A 2015, 119, 6296–6308. [Google Scholar] [CrossRef]
- Maçôas, E.M.S.; Fausto, R.; Pettersson, M.; Khriachtchev, L.; Räsänen, M. Infrared Induced Rotamerization of Oxalic Acid Monomer in an Argon Matrix. J. Phys. Chem. A 2000, 104, 6956–6961. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.; Fausto, R.; Lundell, J.; Pettersson, M.; Khriachtchev, L.; Räsänen, M. Conformational Analysis and Near-Infrared-Induced Rotamerization of Malonic Acid in an Argon Matrix. J. Phys. Chem. A 2000, 104, 11725–11732. [Google Scholar] [CrossRef] [Green Version]
- Duarte, L.; Giuliano, B.M.; Reva, I.; Fausto, R. Tautomers and UV-Induced Photoisomerization of a Strongly Intramolecularly H-Bonded Aromatic Azo-Dye: 1-(Cyclopropyl)Diazo-2-Naphthol. J. Phys. Chem. A 2013, 117, 10671–10680. [Google Scholar] [CrossRef]
- Rozenberg, M.; Fausto, R.; Reva, I. Variable Temperature FTIR Spectra of Polycrystalline Purine Nucleobases and Estimating Strengths of Individual Hydrogen Bonds. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2021, 251, 119323. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dihedral Angles | |||||
|---|---|---|---|---|---|
| Conformer | C–C=N–C | C–C–O–H α | C–C–N=C β | C–C–C=N ϒ | C–C–O–H δ |
| E-enol-imine | |||||
| I | −177.2 | 0.3 | 37.8 | 0.7 | −0.3 |
| II | −177.3 | 178.6 | 36.9 | 0.6 | −0.3 |
| III | −177.4 | 0.5 | 43.5 | 4.4 | 179.3 |
| IV | −177.4 | 179.1 | 43.2 | 4.3 | 179.1 |
| V | −177.1 | 0.2 | −144.5 | 0.6 | −0.1 |
| VI | −177.1 | −179.1 | −143.6 | 0.6 | −0.3 |
| VII | −177.0 | 0.2 | −140.9 | 2.7 | 178.7 |
| VIII | −177.2 | −178.3 | −139.2 | 2.5 | 179.4 |
| IX | −175.6 | 1.6 | 42.0 | −171.4 | 9.4 |
| X | −176.2 | 178.9 | 40.6 | −173.4 | 5.6 |
| XI | −177.3 | 0.2 | 39.6 | −178.5 | 178.7 |
| XII | −177.3 | 179.2 | 39.6 | −178.4 | 179.8 |
| XIII | −175.9 | −0.8 | −141.9 | −172.6 | 6.4 |
| XIV | −176.0 | −178.5 | −140.4 | −173.0 | 6.8 |
| XV | −177.1 | 0.1 | −144.1 | −178.9 | 179.6 |
| XVI | −177.2 | −178.8 | −142.3 | −179.3 | 179.4 |
| Z-enol-imine | |||||
| I | 7.2 | 0.6 | −128.6 | 38.8 | −23.5 |
| II | 6.7 | −179.5 | −126.9 | 39.1 | −21.1 |
| III | 5.5 | −1.4 | −133.1 | 47.2 | −175.0 |
| IV | 5.7 | −176.3 | −131.9 | 47.1 | −176.5 |
| V | 6.1 | −0.5 | 58.2 | 39.5 | −21.9 |
| VI | 6.3 | −179.4 | 58.6 | 38.9 | −21.7 |
| VII | 5.1 | 0.9 | 52.7 | 47.3 | −175.4 |
| VIII | 5.3 | −178.2 | 53.2 | 46.5 | −176.0 |
| IX | 7.8 | 0.8 | −127.1 | −148.7 | 8.2 |
| X | 7.3 | −179.4 | −124.7 | −148.5 | 7.8 |
| XI | 8.1 | 0.5 | −122.9 | −154.6 | −176.9 |
| XII | 7.6 | −179.2 | −120.7 | −154.4 | −176.6 |
| XIII | 7.2 | −1.5 | 59.3 | −148.4 | 6.6 |
| XIV | 7.3 | −178.7 | 60.9 | −148.7 | 9.3 |
| XV | 7.3 | −0.2 | 65.2 | −154.5 | −176.0 |
| XVI | 7.4 | −179.2 | 64.7 | −153.8 | −177.0 |
| Conformer | ΔEel | ΔE(0) | ΔG°298.15 | μ | ΔEel | ΔE(0) | ΔG298.15 |
|---|---|---|---|---|---|---|---|
| E-enol-imine | |||||||
| I | 1.69 | 1.51 | 0.70 | 1.36 | |||
| II | 0.10 | 0.15 | −0.51 | 3.35 | |||
| III | 56.77 | 54.10 | 51.30 | 2.78 | |||
| IV | 55.71 | 53.20 | 50.51 | 1.63 | |||
| V | 0.00 | 0.00 | 0.00 | 1.01 | |||
| VI | 0.84 | 0.67 | 0.67 | 3.53 | |||
| VII | 54.95 | 52.51 | 49.66 | 2.95 | |||
| VIII | 56.38 | 53.65 | 50.51 | 0.91 | |||
| IX | 47.18 | 44.45 | 41.31 | 2.43 | |||
| X | 45.47 | 42.88 | 38.97 | 3.33 | |||
| XI | 39.76 | 37.18 | 34.54 | 4.57 | |||
| XII | 38.89 | 36.58 | 34.25 | 3.20 | |||
| XIII | 44.90 | 42.38 | 39.57 | 1.83 | |||
| XIV | 46.28 | 43.51 | 40.72 | 3.88 | |||
| XV | 38.13 | 35.68 | 32.73 | 3.89 | |||
| XVI | 39.62 | 36.86 | 33.64 | 4.33 | |||
| Z-enol-imine | |||||||
| I | 62.99 | 61.92 | 61.52 | 0.80 | 0.25 | 0.29 | 0.46 |
| II | 63.25 | 61.94 | 61.44 | 2.72 | 0.51 | 0.31 | 0.37 |
| III | 73.30 | 71.33 | 70.23 | 2.93 | 10.56 | 9.70 | 9.16 |
| IV | 75.05 | 72.47 | 70.54 | 3.88 | 12.31 | 10.84 | 9.48 |
| V | 62.74 | 61.63 | 61.07 | 0.51 | 0.00 | 0.00 | 0.00 |
| VI | 63.12 | 61.73 | 60.93 | 2.80 | 0.38 | 0.10 | −0.14 |
| VII | 74.60 | 72.25 | 70.38 | 2.96 | 11.86 | 10.62 | 9.31 |
| VIII | 75.01 | 72.47 | 70.53 | 3.97 | 12.27 | 10.84 | 9.46 |
| IX | 70.42 | 68.65 | 66.04 | 1.66 | 7.68 | 7.02 | 4.98 |
| X | 70.67 | 68.82 | 66.26 | 1.82 | 7.93 | 7.19 | 5.20 |
| XI | 66.67 | 64.98 | 62.18 | 4.31 | 3.93 | 3.35 | 1.11 |
| XII | 67.09 | 65.26 | 62.41 | 3.92 | 4.35 | 3.63 | 1.34 |
| XIII | 71.27 | 69.42 | 66.59 | 2.17 | 8.53 | 7.79 | 5.52 |
| XIV | 70.42 | 68.58 | 65.80 | 0.72 | 7.68 | 6.95 | 4.73 |
| XV | 67.35 | 65.73 | 62.82 | 4.88 | 4.61 | 4.10 | 1.76 |
| XVI | 66.87 | 65.14 | 62.33 | 2.89 | 4.13 | 3.51 | 1.26 |
| Dihedral Angles | ||||
|---|---|---|---|---|
| Conformer | C–C=C–N | C–C–O–H α | C–C–N–C β | C–N–C=C ϒ |
| keto-amine forms structurally related to the E-enol-imine isomer | ||||
| I | 0.0 | 0.0 | 0.0 | 179.9 |
| II | 0.0 | −179.9 | −0.7 | 179.9 |
| III | 178.4 | 0.6 | 166.5 | 177.5 |
| IV | 179.0 | −179.3 | 169.9 | 178.4 |
| V | 0.0 | 0.4 | 175.1 | 179.6 |
| VI | 0.0 | −179.6 | 174.1 | 179.0 |
| VII | 179.1 | 1.6 | −8.7 | 178.6 |
| VIII | 178.7 | −179.0 | −9.0 | 178.6 |
| keto-amine forms structurally related to the Z-enol-imine isomer | ||||
| I | 176.0 | 4.7 | −44.8 | −16.3 |
| II | 177.1 | 179.5 | −47.3 | −15.7 |
| III | −14.9 | −3.7 | 153.1 | −24.5 |
| IV | −15.3 | −176.6 | 156.0 | −23.9 |
| V | 177.4 | −1.2 | 131.6 | −15.1 |
| VI | 176.6 | −177.9 | 136.9 | −16.1 |
| VII | −15.6 | 8.8 | −25.6 | −23.3 |
| VIII | −15.3 | 178.3 | −28.2 | −24.2 |
| Conformer | ΔEel | ΔE(0) | ΔG°298.15 | μ | ΔEel | ΔE(0) | ΔG298.15 | |
|---|---|---|---|---|---|---|---|---|
| keto-amine forms structurally related to the E-enol-imine isomer | ||||||||
| I | 19.69 | 18.94 | 16.35 | 3.49 | 2.75 | 2.66 | 2.39 | |
| II | 16.95 | 16.28 | 13.96 | 5.30 | 0.00 | 0.00 | 0.00 | |
| III | 61.49 | 61.03 | 56.80 | 4.74 | 2.95 | 2.86 | 3.82 | |
| IV | 58.54 | 58.17 | 52.98 | 5.67 | 0.00 | 0.00 | 0.00 | |
| V | 17.87 | 17.01 | 11.28 | 2.97 | 0.92 | 0.72 | –2.68 | |
| VI | 18.42 | 17.67 | 14.20 | 5.67 | 1.48 | 1.39 | 0.25 | |
| VII | 58.66 | 58.14 | 52.81 | 3.82 | 0.12 | –0.03 | –0.17 | |
| VIII | 59.08 | 58.45 | 52.93 | 6.41 | 0.54 | 0.28 | –0.05 | |
| keto-amine forms structurally related to the Z-enol-imine isomer | ||||||||
| I | 80.86 | 81.47 | 78.49 | 4.46 | 63.91 | 65.19 | 64.53 | |
| II | 82.00 | 82.43 | 79.19 | 6.89 | 65.05 | 66.15 | 65.23 | |
| III | 94.97 | 94.96 | 93.91 | 4.53 | 36.43 | 36.79 | 40.93 | |
| IV | 92.80 | 92.95 | 92.27 | 4.05 | 34.26 | 34.78 | 39.29 | |
| V | 82.94 | 83.08 | 79.48 | 5.39 | 65.99 | 66.80 | 65.52 | |
| VI | 81.05 | 81.44 | 78.61 | 6.29 | 64.09 | 65.16 | 64.66 | |
| VII | 91.88 | 92.47 | 92.00 | 3.91 | 33.34 | 34.30 | 39.02 | |
| VIII | 93.01 | 93.29 | 92.87 | 4.75 | 34.47 | 35.12 | 39.89 | |
| Atom | NBO Charge | Atom | NBO Charge | |
|---|---|---|---|---|
| C1 | 0.325 | O15 | −0.677 |  |
| C2 | −0.253 | Br16 | 0.061 | |
| C3 | −0.171 | O17 | −0.670 | |
| C4 | −0.256 | H18 | 0.220 | |
| C5 | 0.154 | H19 | 0.207 | |
| C6 | −0.278 | H20 | 0.209 | |
| N7 | −0.522 | H21 | 0.209 | |
| C8 | 0.162 | H22 | 0.170 | |
| C9 | −0.176 | H23 | 0.219 | |
| C10 | −0.175 | H24 | 0.223 | |
| C11 | −0.138 | H25 | 0.224 | |
| C12 | −0.182 | H26 | 0.507 | |
| C13 | −0.238 | H27 | 0.468 | |
| C14 | 0.376 |
| Coefficients (%) b | ||||
|---|---|---|---|---|
| Orbital | Occupancy (e) a | A | B | Description |
| σ(C1–C2) | 1.97297 | 50.75 | 49.25 | C1 sp1.68 + C2 sp1.92 |
| π(C1–C2) | 1.68279 | 49.24 | 50.76 | C1 sp3.35 + C2 p |
| σ(C1–C6) | 1.97708 | 50.10 | 49.90 | C1 sp1.60 + C6 sp1.85 |
| σ(C1–O17) | 1.99029 | 11.14 | 88.86 | C1 p + O17 p |
| σ(C2–C3) | 1.90762 | 49.96 | 50.04 | C2 sp1.89 + C3 sp1.79 |
| σ(C2–H18) | 1.92993 | 60.78 | 39.22 | C2 sp2.24 + H18 s |
| σ(C3–C4) | 1.97617 | 49.69 | 50.31 | C3 sp1.78 + C4 sp1.77 |
| π(C3–C4) | 1.68051 | 46.96 | 53.04 | C3 p + C4 p |
| σ(C3–H19) | 1.97978 | 60.44 | 39.56 | C3 sp2.54 + H19 s |
| σ(C4–C5) | 1.97155 | 49.10 | 50.90 | C4 sp1.87 + C5 sp1.65 |
| σ(C4–H20) | 1.97822 | 60.54 | 39.46 | C4 sp2.44 + H20 s |
| σ(C5–C6) | 1.96869 | 50.49 | 49.51 | C5 sp1.88 + C6 sp1.78 |
| π(C5–C6) | 1.66431 | 46.58 | 53.42 | C5 p + C6 p |
| σ(C5–N7) | 1.98468 | 39.31 | 60.69 | C5 sp2.65 + N7 sp1.30 |
| σ(C6–H21) | 1.97686 | 60.52 | 39.48 | C6 sp2.45 + H21 s |
| σ(N7–C8) | 1.98676 | 59.95 | 40.05 | N7 sp1.33 + C8 sp2.02 |
| π(N7–C8) | 1.92978 | 70.93 | 29.07 | N7 p + C8 p |
| σ(C8–C9) | 1.97187 | 48.56 | 51.44 | C8 sp1.82 + C9 sp2.11 |
| σ(C8–H22) | 1.97565 | 59.42 | 40.58 | C8 sp2.16 + H22 s |
| σ(C9–C10) | 1.96315 | 51.13 | 48.87 | C9 sp1.88 + C10 sp1.86 |
| π(C9–C10) | 1.64114 | 54.94 | 45.06 | C9 p + C10 p |
| σ(C9–C14) | 1.97181 | 51.09 | 48.91 | C9 sp2.02 + C14 sp1.71 |
| σ(C10–C11) | 1.97983 | 50.21 | 49.79 | C10 sp1.74 + C11 sp1.55 |
| σ(C10–H23) | 1.97719 | 61.05 | 38.95 | C10 sp2.49 + H23 s |
| σ(C11–C12) | 1.97972 | 50.32 | 49.68 | C11 sp1.59 + C12 sp1.82 |
| π(C11–C12) | 1.64989 | 56.19 | 43.81 | C11 p + C12 p |
| σ(C11–Br16) | 1.98466 | 49.53 | 50.47 | C11 sp3.51 + Br16 p |
| σ(C12–C13) | 1.96871 | 50.19 | 49.81 | C12 sp1.79 + C13 sp1.74 |
| σ(C12–H24) | 1.97873 | 61.21 | 38.79 | C12 sp2.47 + H24 s |
| σ(C13–C14) | 1.97688 | 49.33 | 50.67 | C13 sp1.91 + C14 sp1.71 |
| π(C13–C14) | 1.58461 | 54.83 | 45.17 | C13 p + C14 p |
| σ(C13–H25) | 1.97697 | 61.21 | 38.79 | C13 sp2.43 + H25 s |
| σ(C14–O15) | 1.99406 | 33.91 | 66.09 | C14 sp2.86 + O15 sp1.95 |
| σ(O15–H26) | 1.98451 | 77.97 | 22.03 | O15 sp2.91 + H26 s |
| σ(O17–H27) | 1.98736 | 73.72 | 26.28 | O17 sp3.76 + H27 s |
| Lp N7 | 1.60068 | p | ||
| Lp1 O15 | 1.97390 | sp1.46 | ||
| Lp2 O15 | 1.81108 | p | ||
| Lp1 Br16 | 1.99361 | s | ||
| Lp2 Br16 | 1.97619 | p | ||
| Lp3 Br16 | 1.94629 | p | ||
| Lp1 O17 | 1.97920 | sp1.24 | ||
| Lp2 O17 | 1.35923 | sp2.01 | ||
| Bond | BO | Bond | BO | Bond | BO | Bond | BO |
|---|---|---|---|---|---|---|---|
| C1–C2 | 1.35 | O17–H27 | 0.77 | C8–C9 | 1.14 | C14–C9 | 1.27 |
| C2–C3 | 1.45 | C2–H18 | 0.92 | C8–H22 | 0.92 | C14–O15 | 1.11 |
| C3–C4 | 1.43 | C3–H19 | 0.92 | C9–C10 | 1.33 | O15–H26 | 0.64 |
| C4–C5 | 1.36 | C4–H20 | 0.92 | C10–C11 | 1.45 | C10–H23 | 0.92 |
| C5–C6 | 1.36 | C6–H21 | 0.92 | C11–C12 | 1.35 | C11–Br16 | 1.03 |
| C6–C1 | 1.39 | C5–N7 | 1.07 | C12–C13 | 1.47 | C12–H24 | 0.92 |
| C1–O17 | 1.02 | N7–C8 | 1.70 | C13–C14 | 1.34 | C13–H25 | 0.92 |
| Exp. | Calculated | Calculated | ||||
|---|---|---|---|---|---|---|
| Ar Matrix b | V | Approximate Description c | II | Approximate Descriptionc | ||
| ν | ν | IIR | ν | IIR | ||
| 3642/3640 | 3622.9 | 66.48 | νOHp | 3625.1 | 84.3 | νOHp |
| 3573–3035 | 3033.1 | 536.05 | νOHbp | 3033.6 | 538.1 | νOHbp |
| 3021 | 3027.7 | 2.81 | νCHbp | 3028.4 | 4.2 | νCHbp |
| 3025.6 | 11.12 | νCHp | 3023.9 | 5.0 | νCHp | |
| 3015.9 | 4.44 | νCHp | 3019.4 | 2.0 | νCHp | |
| 3014.4 | 4.46 | νCHbp | 3014.8 | 4.0 | νCHbp | |
| 3005 | 3008.2 | 1.29 | νCHbp | 3009.9 | 1.4 | νCHbp |
| 2993/3005 | 2998.2 | 8.52 | νCHp | 3005.8 | 17.9 | νCHp |
| 2987/2975 | 2991.1 | 7.78 | νCHp | 2983.9 | 10.3 | νCHp |
| 2864/2846 | 2878.3 | 34.13 | νCHam | 2877.4 | 34.5 | νCHam |
| 1630/1626 | 1656.5 | 97.93 | νN=C; νCCam; δCHam | 1658.1 | 117.5 | νN=C; νCCam; δCHam |
| 1613 | 1644.6 | 23.95 | νCCbp; δOHbp | 1644.7 | 14.5 | νCCbp; δOHbp |
| 1602/1600 | 1623.9 | 272.72 | νCCp | 1627.3 | 245.1 | νCCp |
| 1617.6 | 80.99 | νCCp; δOHp | 1612.2 | 114.3 | νCCp; δOHp | |
| 1593 | 1588.5 | 173.04 | δOHbp; νCCbp; δCHbp; νN=C | 1589.0 | 141.4 | δOHbp; νCCbp; δCHbp; νN=C |
| 1521/1500 | 1514.8 | 63.04 | νCCp; δCHp; δOHp | 1507.4 | 87.0 | νCCp; δCHp; δOHp |
| 1495 | 1496.9 | 81.64 | δCHbp; δOHbp; νCCbp | 1494.6 | 156.1 | δCHbp; δOHbp; νCCbp |
| 1477 | 1478.2 | 87.27 | δCHp-bp; νCCp-bp; δOHp-bp | 1485.5 | 10.7 | δCHp-bp; νCCp-bp; δOHp-bp |
| 1461 | 1464.1 | 52.23 | νCCbp; δCHbp; δOHbp | 1469.6 | 15.5 | νCCbp; δCHbp; δOHbp |
| 1421 | 1418.2 | 25.92 | νCCbp; δCHbp; δOHbp; δCHam | 1419.1 | 31.2 | νCCbp; δCHbp; δOHbp; δCHam |
| 1368/1358 | 1371.9 | 40.64 | δCHam | 1373.8 | 34.5 | δCHam |
| 1339 | 1338.8 | 2.96 | δCHp; νCCp | 1338.8 | 16.5 | δCHp; νCCp |
| 1331/1326 | 1334.8 | 2.83 | νCCbp; δCHbp | 1334.9 | 5.0 | νCCbp; δCHbp |
| 1326.2 | 1.70 | δCCp; δCHp; δOHp | 1318.7 | 92.2 | δCCp; δCHp; δOHp | |
| 1314 | 1302.9 | 131.61 | νCObp; δCCbp; δCHbp | 1304.2 | 79.5 | νCObp; δCCbp; δCHbp |
| 1308/1285 | 1292.9 | 28.68 | νCOp; νC-N; δCCp; δCHp | 1282.7 | 47.5 | νCOp; νC-N; δCCp; δCHp |
| 1264 | 1249.7 | 9.74 | δCHbp | 1250.8 | 17.9 | δCHbp |
| 1224/1217 | 1215.4 | 56.05 | νCCam; δCHp-bp | 1215.4 | 57.5 | νCCam; δCHp-bp |
| 1196/1181 | 1186.9 | 169.23 | δOHp; δCHp | 1194.6 | 41.4 | δOHp; δCHp |
| 1166 | 1178.0 | 28.77 | δCHp; δOHp | 1175.7 | 36.2 | δCHp; δOHp |
| 1158 | 1158.5 | 160.77 | δCHp; νCp-N; νCOp | 1156.5 | 192.3 | δCHp; νCp-N; νCOp |
| 1147 | 1144.6 | 7.08 | δCHbp | 1144.6 | 8.9 | δCHbp |
| 1093 | 1095.7 | 13.00 | δCHp | 1097.4 | 10.8 | δCHp |
| 1073 | 1084.1 | 15.73 | νC-Br; δCCbp; δCHbp | 1085.2 | 15.9 | νC-Br; δCCbp; δCHbp |
| 998 | 1005.4 | 7.15 | δCCp; γCHam | 1007.9 | 2.2 | δCCp; γCHam |
| 994.4 | 10.83 | γCHam | 998.2 | 9.1 | γCHam | |
| 973 | 973.5 | 0.10 | γCHp | 970.2 | 1.7 | γCHp |
| 957 | 969.8 | 53.61 | δCCp; δCHp; νCOp | 978.2 | 94.9 | δCCp; δCHp; νCOp |
| 958.6 | 0.04 | γCHbp | 958.4 | 0.5 | γCHbp | |
| 897 | 919.5 | 17.42 | δCCbp; δCNC; δCCNam; νCBr | 917.1 | 16.2 | δCCbp; δCNC; δCCNam; νCBr |
| 875 | 883.7 | 9.64 | γCHp-bp | 884.3 | 17.9 | γCHp-bp |
| 880.0 | 5.20 | γCHp-bp | 874.1 | 8.2 | γCHp-bp | |
| 859 | 856.2 | 42.42 | γCCp; γCHp-bp; γOHbp | 866.3 | 12.8 | γCCp; γCHp |
| 848 | 848.8 | 4.55 | γOHbp | 851.8 | 16.9 | γCHbp; γOHbp |
| 844 | 827.8 | 83.95 | γCCbp; γCHbp; γOHbp | 828.0 | 80.9 | γCCbp; γCHbp; γOHbp |
| 824/805 | 813.2 | 7.16 | δCCbp; δCNC; δCCNam | 805.0 | 7.9 | δCCbp; δCNC; δCCNam |
| 765 | 780.4 | 20.54 | γCHp | 779.0 | 11.8 | γCHp; δCCbp |
| 758 | 774.9 | 6.42 | γCHp; δCCbp | 775.9 | 34.0 | γCHp |
| 717 | 729.0 | 1.13 | γCHbp; γCCbp | 728.8 | 1.5 | γCHbp; γCCbp |
| 685 | 695.7 | 25.80 | γCCp; δCCbp | 694.3 | 22.9 | γCCp; δCCbp |
| 686.0 | 16.75 | γCCp | 685.2 | 15.2 | γCCp | |
| 633 | 635.8 | 16.53 | γCCp; δCCbp; γCHp; νCBr | 635.4 | 22.2 | γCCp; δCCbp; γCHp; νCBr |
| 610 | 624.7 | 8.06 | γCCp; δCCbp; γCHp; νCBr | 623.8 | 5.7 | γCCp; δCCbp; γCHp; νCBr |
| 571/560 | 574.1 | 10.14 | δCCp-bp; δCNC; δCCNam | 563.8 | 9.9 | δCCp-bp; δCNC; δCCNam |
| 553 | 552.7 | 4.79 | γCCbp; γCHbp; γCHam | 555.2 | 6.1 | γCCbp; γCHbp; γCHam |
| 541 | 530.8 | 1.48 | δCCp | 533.7 | 1.3 | δCCp |
| 528/521 | 516.2 | 17.41 | δCNp | 520.8 | 2.6 | δCNp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sıdır, İ.; Gülseven Sıdır, Y.; Góbi, S.; Berber, H.; Fausto, R. Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol. Molecules 2021, 26, 2814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092814
Sıdır İ, Gülseven Sıdır Y, Góbi S, Berber H, Fausto R. Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol. Molecules. 2021; 26(9):2814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092814
Chicago/Turabian StyleSıdır, İsa, Yadigar Gülseven Sıdır, Sándor Góbi, Halil Berber, and Rui Fausto. 2021. "Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol" Molecules 26, no. 9: 2814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092814