
Preparation of High-Purity Ammonium Tetrakis(pentafluorophenyl)borate for the Activation of Olefin Polymerization Catalysts

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
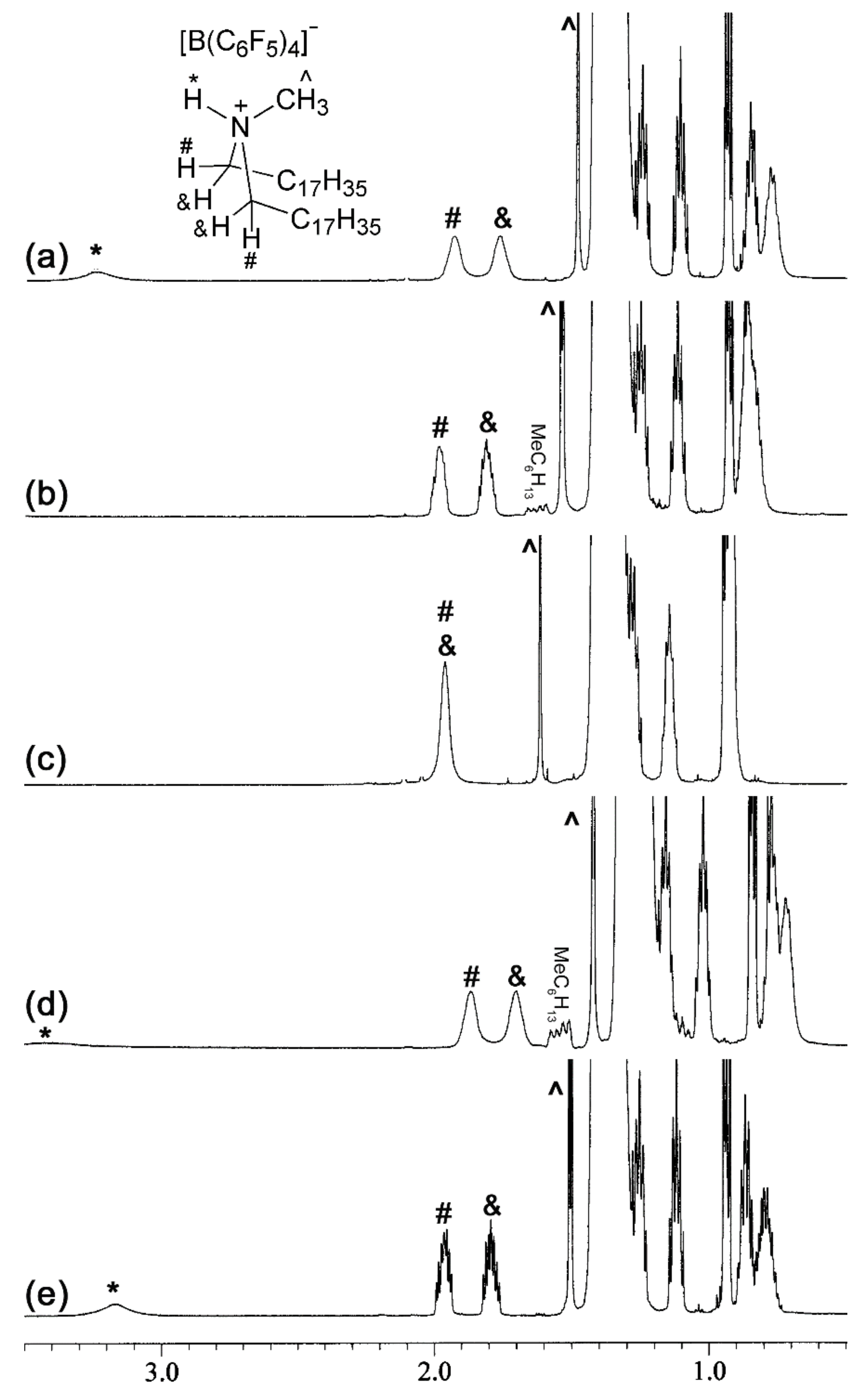
2.1. Preparation of [Me(C18H37)2N-H]+[B(C6F5)4]−
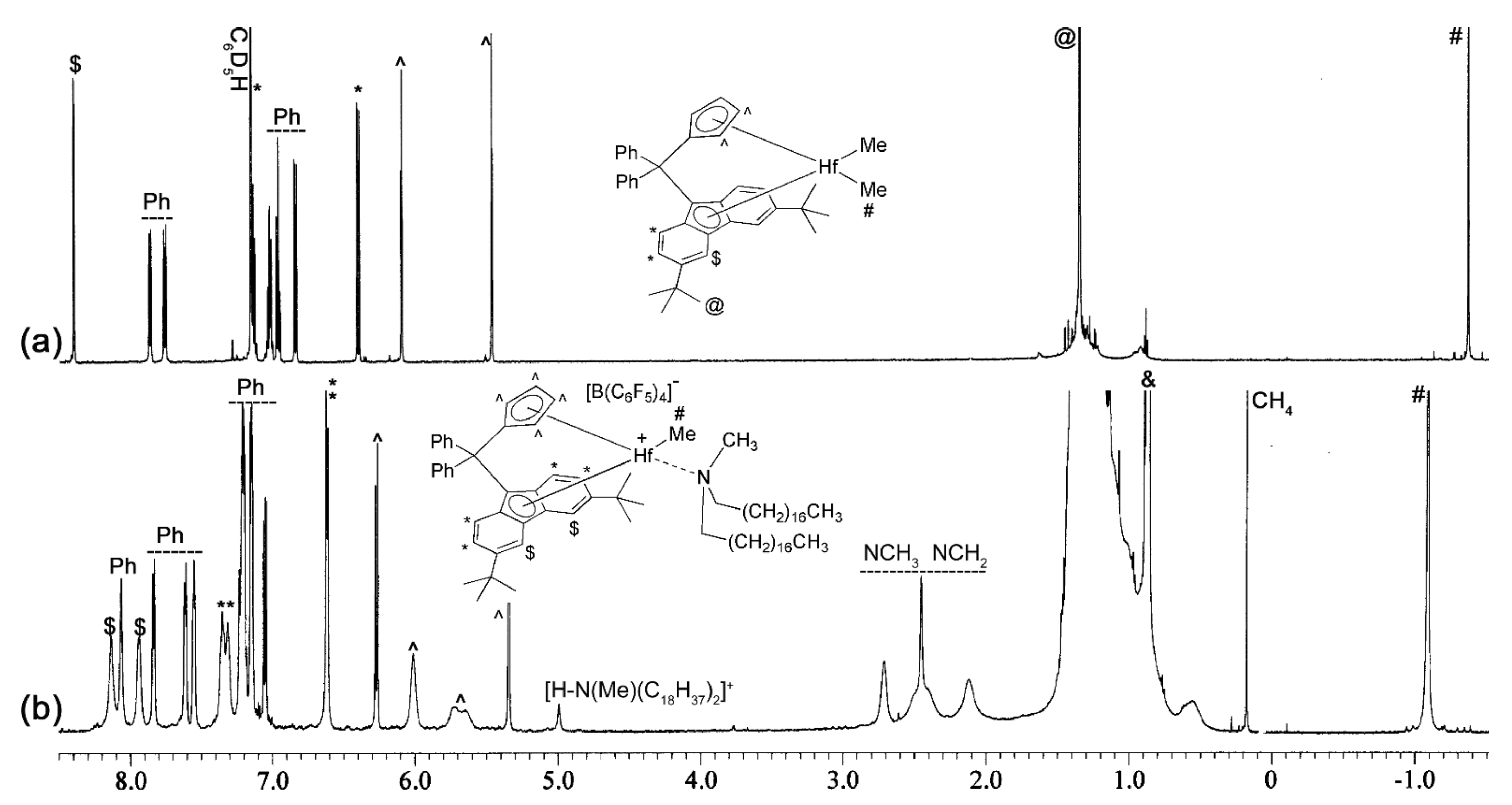
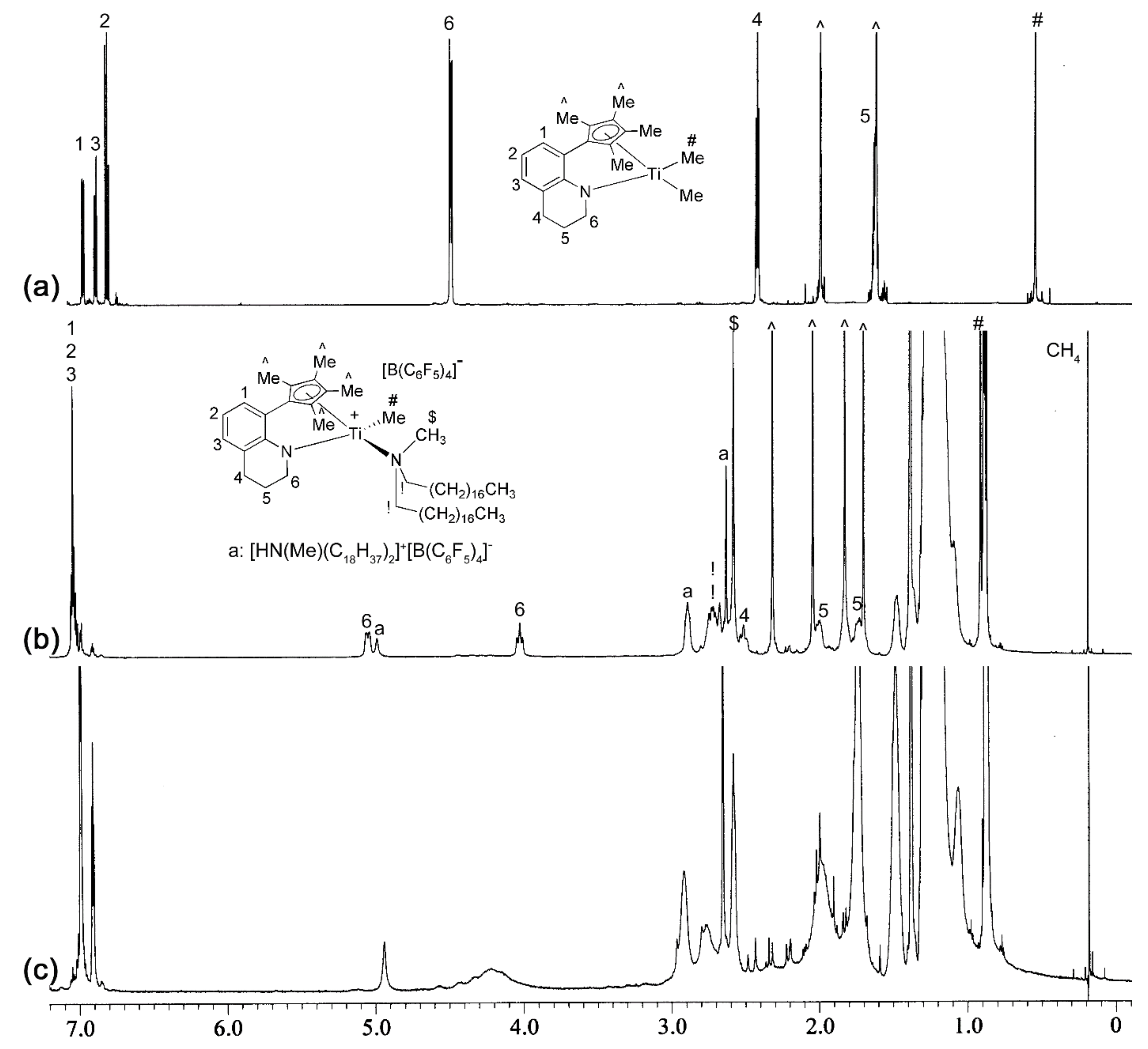
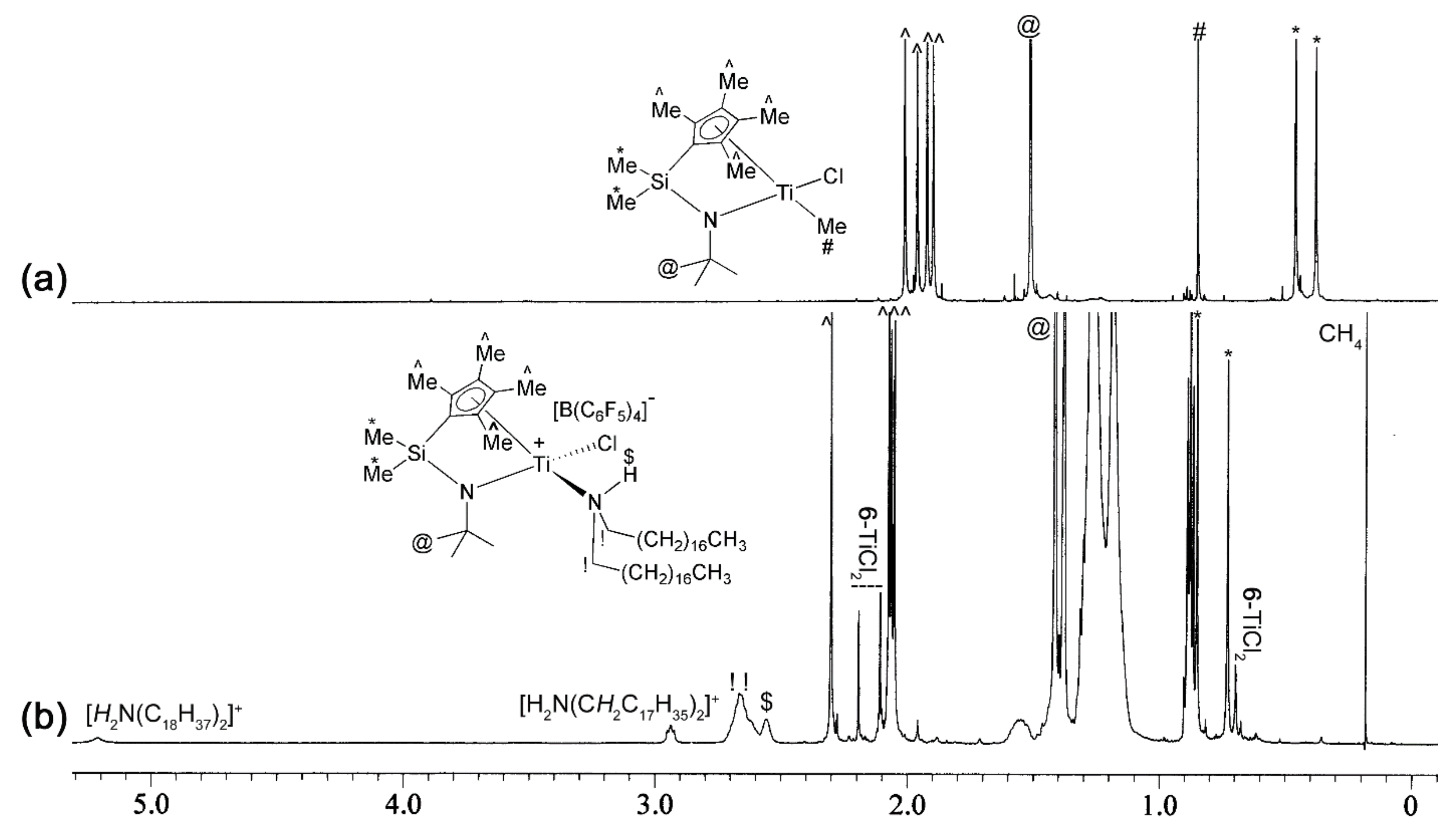
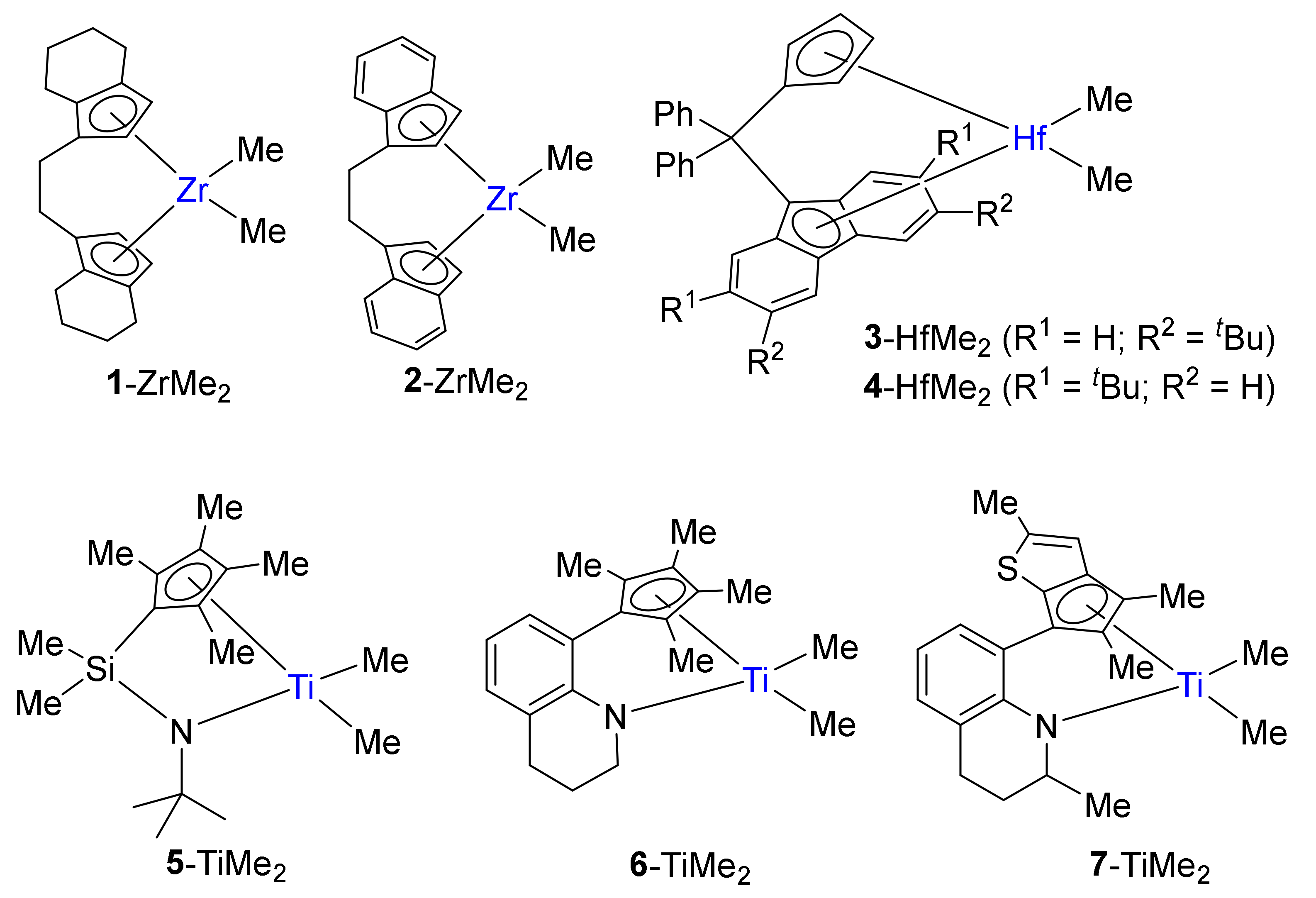
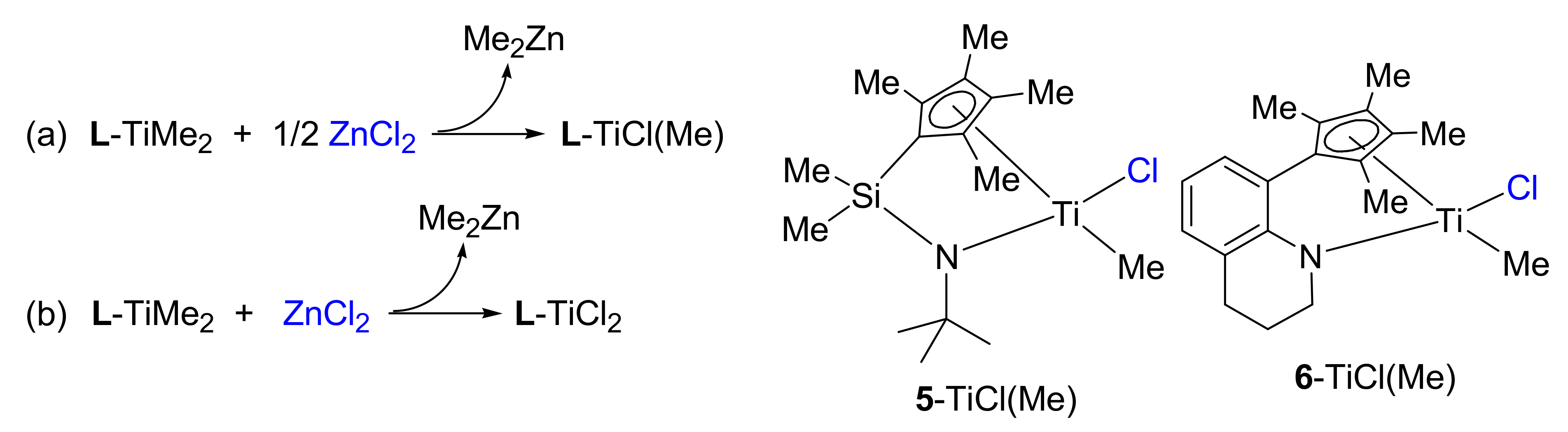
2.2. Preparation of L-M(Me)2 and L-MCl(Me)-Type Complexes for Activation Studies
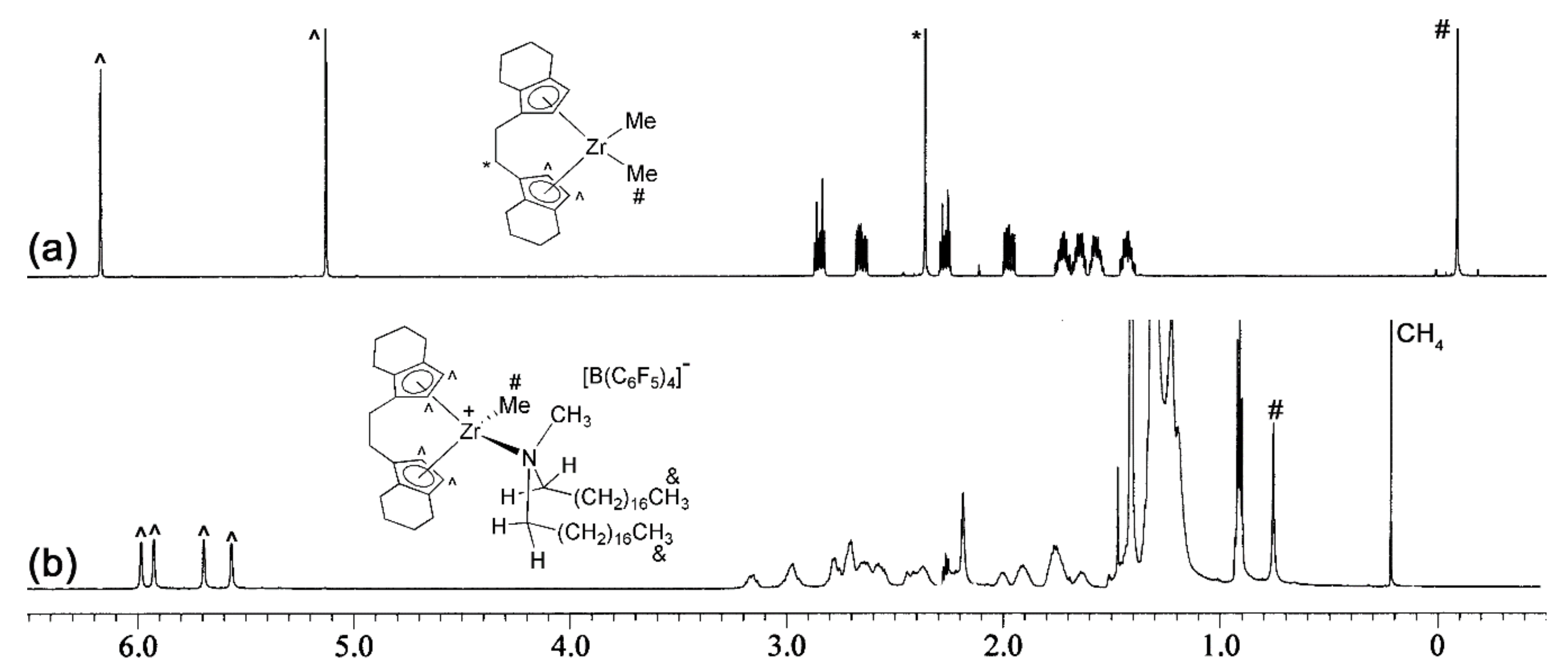
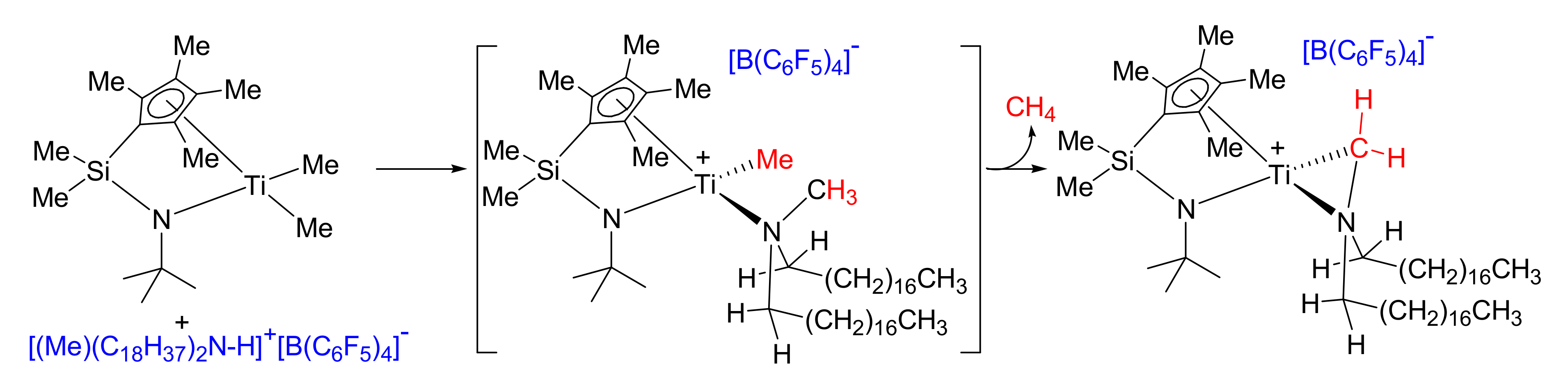
2.3. Activation Reactions
2.4. Polymerization Studies
3. Materials and Methods
3.1. Preparation of ([Me(C18H37)2N-H]+[B(C6F5)4]−, [(C12H25)3N-H]+[B(C6F5)4]−, and [(C18H37)2NH2]+[B(C6F5)4]−
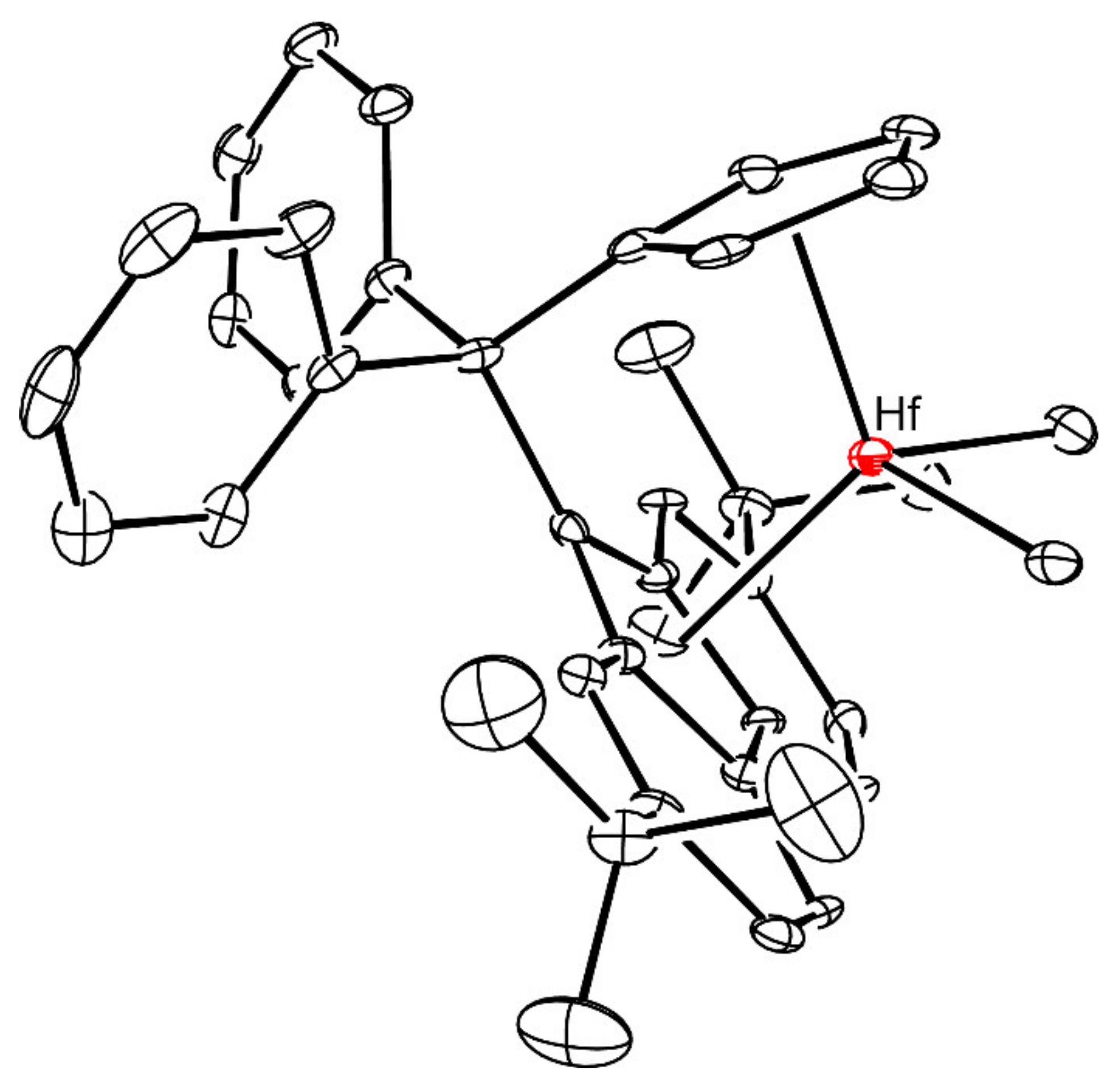
3.2. Preparation of 4-HfMe2 and 3-HfMe2
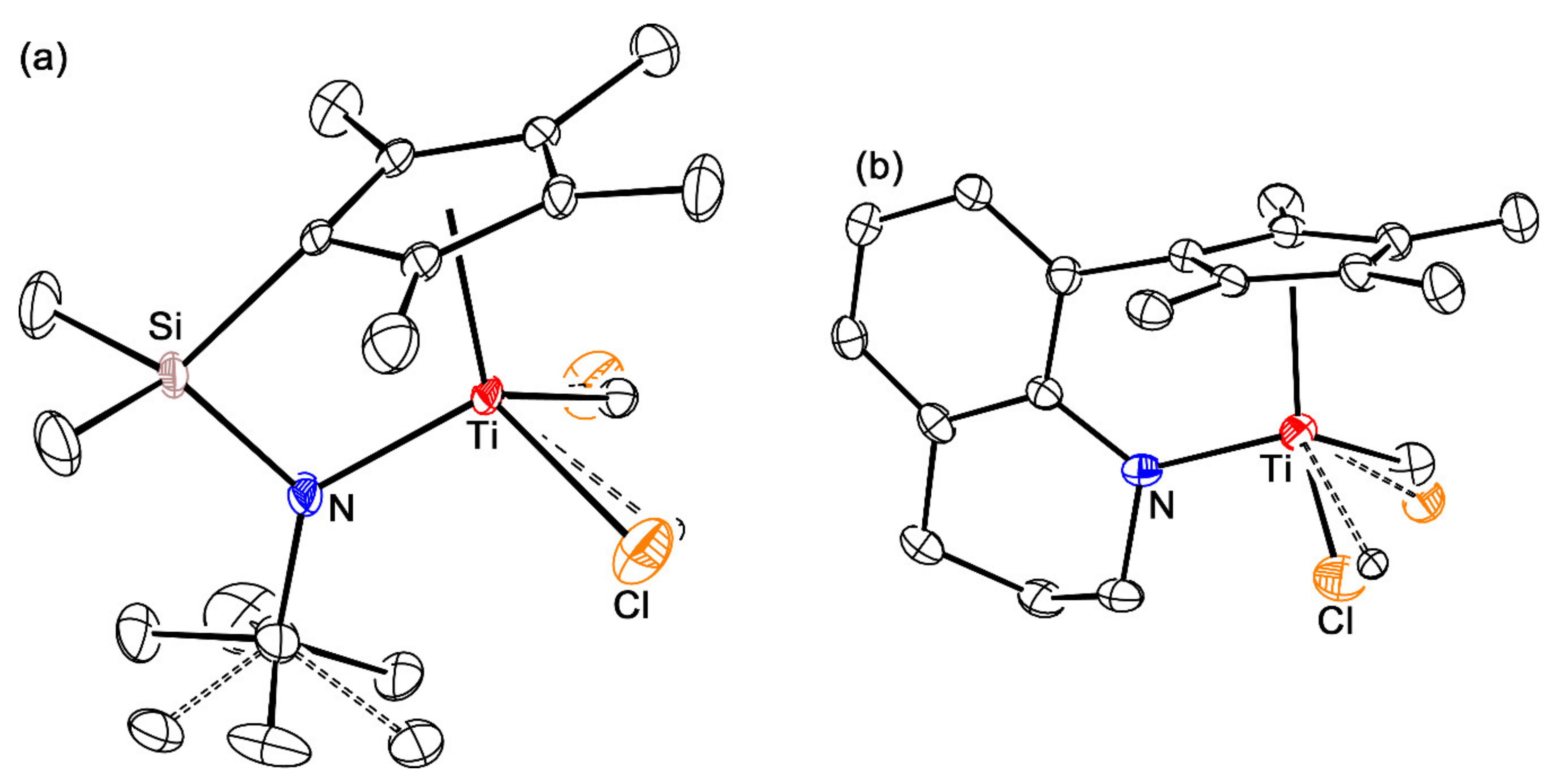
3.3. Preparation of 5-TiCl(Me) and 6-TiCl(Me)
3.4. A Representative Activation Reaction
3.5. A Representative Polymerization Procedure (Entry 4)
3.6. X-ray Crystallography
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kaminsky, W. Discovery of methylaluminoxane as cocatalyst for olefin polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Vinogradov, A.A. Heterocycle-fused cyclopentadienyl metal complexes: Heterocene synthesis, structure and catalytic applications. Coord. Chem. Rev. 2021, 426, 213515. [Google Scholar] [CrossRef]
- Yuan, S.-F.; Yan, Y.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent advancements in N-ligated group 4 molecular catalysts for the (co)polymerization of ethylene. Coord. Chem. Rev. 2020, 411, 213254. [Google Scholar] [CrossRef]
- Antonov, A.A.; Bryliakov, K.P. Post-metallocene catalysts for the synthesis of ultrahigh molecular weight polyethylene: Recent advances. Eur. Polym. J. 2021, 142, 110162. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Suo, H.; Silva, M.D.F.C.G.D.; Pombeiro, A.J.L.; Sun, W.H. Recent developments in vanadium-catalyzed olefin coordination polymerization. Coord. Chem. Rev. 2020, 416, 213332. [Google Scholar] [CrossRef]
- Liang, T.; Goudari, S.B.; Chen, C. A simple and versatile nickel platform for the generation of branched high molecular weight polyolefins. Nat. Commun. 2020, 11, 372. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Chen, C. Emerging palladium and nickel catalysts for copolymerization of olefins with polar monomers. Angew. Chem. Int. Ed. 2019, 58, 7192–7200. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Boereboom, J.M.; Bulo, R.E.; Weckhuysen, B.M. Insights into the activation of silica-supported metallocene olefin polymerization catalysts by methylaluminoxane. Catal. Today 2019, 334, 223–230. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Macchioni, A.; Busico, V.; Ehm, C. BHT-Modified MAO: Cage Size Estimation, Chemical Counting of Strongly Acidic Al Sites, and Activation of a Ti-Phosphinimide Precatalyst. ACS Catal. 2019, 9, 2996–3010. [Google Scholar] [CrossRef]
- Resconi, L.; Bossi, S.; Abis, L. Study on the role of methylalumoxane in homogeneous olefin polymerization. Macromolecules 1990, 23, 4489–4491. [Google Scholar] [CrossRef]
- Bae, S.M.; Jeong, S.M.; Baek, J.W.; Lee, H.J.; Kim, H.; Yoon, Y.; Chung, S.; Lee, B.Y. Dinuclear metallocene complexes for high-performance supported catalysts. Eur. Polym. J. 2021, 144, 110243. [Google Scholar] [CrossRef]
- Dai, S.; Chen, C. A Self-supporting strategy for gas-phase and slurry-phase ethylene polymerization using late-transition-metal catalysts. Angew. Chem. Int. Ed. 2020, 59, 14884–14890. [Google Scholar] [CrossRef]
- Chien, J.C.W.; Tsai, W.M.; Rausch, M.D. Isospecific polymerization of propylene catalyzed by rac-ethylenebis(indenyl)methylzirconium cation. J. Am. Chem. Soc. 1991, 113, 8570–8571. [Google Scholar] [CrossRef]
- Liu, D.; Wang, M.; Chai, Y.; Wan, X.; Cui, D. Self-activated coordination polymerization of alkoxystyrenes by a yttrium precursor: Stereocontrol and mechanism. ACS Catal. 2019, 9, 2618–2625. [Google Scholar] [CrossRef]
- Nomura, K.; Pengoubol, S.; Apisuk, W. Synthesis of ultrahigh molecular weight polymers containing reactive functionality with low pdis by polymerizations of long-chain α-olefins in the presence of their nonconjugated dienes by Cp*TiMe2(O-2,6-iPr2C6H3)–borate catalyst. Polymers 2020, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, T.H.; Baek, J.W.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Lee, B.Y. Extremely active ethylene tetramerization catalyst avoiding the use of methylaluminoxane: [iPrN{P(C6H4-p-SiR3)2}2CrCl2]+[B(C6F5)4]−. ChemCatChem 2019, 11, 4351–4359. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, H.M.; Park, H.S.; Kim, S.D.; Kwon, S.J.; Tahara, A.; Nagashima, H.; Lee, B.Y. MAO-free and extremely active catalytic system for ethylene tetramerization. Appl. Organometall. Chem. 2019, 33, e4829. [Google Scholar] [CrossRef]
- Stennett, T.E.; Haddow, M.F.; Wass, D.F. Avoiding MAO: Alternative activation methods in selective ethylene oligomerization. Organometallics 2012, 31, 6960–6965. [Google Scholar] [CrossRef]
- Sian, L.; Macchioni, A.; Zuccaccia, C. Understanding the role of metallocenium ion-pair aggregates on the rate of olefin insertion into the metal–carbon bond. ACS Catal. 2020, 10, 1591–1606. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G. Computational assessment of counterion effect of borate anions on ethylene polymerization by zirconocene and hafnocene catalysts. Organometallics 2020, 39, 2068–2079. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Wang, Y.; Pickens, D.B.; Motta, A.; Wang, Q.J.; Chung, Y.-W.; Lohr, T.L.; Marks, T.J. Highly branched polyethylene oligomers via group IV-catalysed polymerization in very nonpolar media. Nat. Catal. 2019, 2, 236–242. [Google Scholar] [CrossRef]
- Nakashima, T.; Nakayama, Y.; Shiono, T.; Tanaka, R. Neutral, noncoordinating, and hydrocarbon-soluble protic cocatalyst for olefin polymerization. ACS Catal. 2021, 11, 865–870. [Google Scholar] [CrossRef]
- Jia, L.; Yang, X.; Ishihara, A.; Marks, T.J. Protected (Fluoroaryl)borates as effective counteranions for cationic metallocene polymerization catalysts. Organometallics 1995, 14, 3135–3137. [Google Scholar] [CrossRef]
- Faler, C.A.; Whalley, M.T.; Gadorn, J.R. A Process To Make Non-Coordinating Anion Type Activators in Aliphatic and Alicyclic Hydrocarbon Solvents. WO 2,019,210,029A1, 31 October 2019. [Google Scholar]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Vittoria, A.; Macchioni, A.; Busico, V.; Ehm, C. Methylaluminoxane’s molecular cousin: A well-defined and “complete” al-activator for molecular olefin polymerization catalysts. ACS Catal. 2021, 11, 4464–4475. [Google Scholar] [CrossRef]
- Romanato, P.; Duttwyler, S.; Linden, A.; Baldridge, K.K.; Siegel, J.S. Intramolecular halogen stabilization of silylium ions directs gearing dynamics. J. Am. Chem. Soc. 2010, 132, 7828–7829. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.S.; Kim, J.G.; Kim, C.S.; Lee, B.Y. Preparation of “constrained geometry” titanium complexes of [1,2]azasilinane framework for ethylene/1-octene copolymerization. Molecules 2017, 22, 258. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Muhammad, N.; Hussain, S.; Jamil, M.I.; Uddin, A.; Aziz, T.; Tufail, M.K.; Guo, Y.; Wei, T.; Rasool, G.; et al. Kinetic and thermal study of ethylene and propylene homo polymerization catalyzed by ansa-zirconocene activated with alkylaluminum/borate: Effects of alkylaluminum on polymerization kinetics and polymer structure. Polymers 2021, 13, 268. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Pengoubol, S.; Apisuk, W. Synthesis of ultrahigh molecular weight polymers with low PDIs by polymerizations of 1-decene, 1-dodecene, and 1-tetradecene by Cp*TiMe2(O-2,6-iPr2C6H3)–borate catalyst. Molecules 2019, 24, 1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cueny, E.S.; Johnson, H.C.; Anding, B.J.; Landis, C.R. Mechanistic Studies of hafnium-pyridyl amido-catalyzed 1-octene polymerization and chain transfer using quench-labeling methods. J. Am. Chem. Soc. 2017, 139, 11903–11912. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, C.; Macchioni, A.; Busico, V.; Cipullo, R.; Talarico, G.; Alfano, F.; Boone, H.W.; Frazier, K.A.; Hustad, P.D.; Stevens, J.C.; et al. Intra- and intermolecular NMR studies on the activation of arylcyclometallated hafnium pyridyl-amido olefin polymerization precatalysts. J. Am. Chem. Soc. 2008, 130, 10354–10368. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Kim, C.S.; Kim, S.D.; Kwon, S.J.; Lee, H.M.; Kim, T.H.; Jeon, J.Y.; Lee, B.Y. Biaxial chain growth of polyolefin and polystyrene from 1,6-hexanediylzinc species for triblock copolymers. Macromolecules 2017, 50, 6606–6616. [Google Scholar] [CrossRef]
- Ehm, C.; Mingione, A.; Vittoria, A.; Zaccaria, F.; Cipullo, R.; Busico, V. High-throughput experimentation in olefin polymerization catalysis: Facing the challenges of miniaturization. Ind. Eng. Chem. Res. 2020, 59, 13940–13947. [Google Scholar] [CrossRef]
- Robert, K.R.; VanderLende, D.D. Highly Soluble Olefin Polymerization Catalyst Activator. U.S. Patent 5,919,983A, 6 July 1999. [Google Scholar]
- Lee, J.C.; Park, K.L.; Bae, S.M.; Lee, H.J.; Baek, J.W.; Lee, J.; Sa, S.; Shin, E.J.; Lee, K.S.; Lee, B.Y. Styrene moiety-carrying diorganozinc compound preparation for polystyrene-poly(ethylene-co-1-hexene)-polystyrene triblock copolymer production. Macromolecules 2020, 53, 7274–7284. [Google Scholar] [CrossRef]
- Lee, H.J.; Baek, J.W.; Kim, T.J.; Park, H.S.; Moon, S.H.; Park, K.L.; Bae, S.M.; Park, J.; Lee, B.Y. Synthesis of long-chain branched polyolefins by coordinative chain transfer polymerization. Macromolecules 2019, 52, 9311–9320. [Google Scholar] [CrossRef]
- Baek, J.W.; Kwon, S.J.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Shin, E.J.; Lee, K.S.; Lee, B.Y. Preparation of half- and post-metallocene hafnium complexes with tetrahydroquinoline and tetrahydrophenanthroline frameworks for olefin polymerization. Polymers 2019, 11, 1093. [Google Scholar] [CrossRef] [Green Version]
- Christman, W.E.; Morrow, T.J.; Arulsamy, N.; Hulley, E.B. Absolute estimates of PdII(η2-Arene) C–H acidity. Organometallics 2018, 37, 2706–2715. [Google Scholar] [CrossRef]
- Lee, J.Y.; Mathur, R.S. Process For Producing Tetrakis(Fluoroaryl)Borate Salts. WO 2,005,105,816A1, 10 November 2005. [Google Scholar]
- Park, J.T.; Woo, B.W.; Yoon, S.C.; Shim, S.C. An efficient synthetic method of ansa-zirconocene dimethyl complexes via Me2ZrCl2. J. Organometall. Chem. 1997, 535, 29–32. [Google Scholar] [CrossRef]
- Wu, C.J.; Lee, S.H.; Yun, H.; Lee, B.Y. Ortho Lithiation of tetrahydroquinoline derivatives and its use for the facile construction of polymerization catalysts. Organometallics 2007, 26, 6685–6687. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, J.H.; Song, B.G.; Yoon, S.W.; Go, M.J.; Lee, J.; Lee, B.Y. Preparation of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl titanium complexes for ethylene/α-olefin copolymerization. Catalysts 2013, 3, 104–124. [Google Scholar] [CrossRef] [Green Version]
- Grandini, C.; Camurati, I.; Guidotti, S.; Mascellani, N.; Resconi, L.; Nifant’ev, I.E.; Kashulin, I.A.; Ivchenko, P.V.; Mercandelli, P.; Sironi, A. Heterocycle-fused indenyl silyl amido dimethyl titanium complexes as catalysts for high molecular weight syndiotactic amorphous polypropylene. Organometallics 2004, 23, 344–360. [Google Scholar] [CrossRef]
- Resconi, L.; Camurati, I.; Grandini, C.; Rinaldi, M.; Mascellani, N.; Traverso, O. Indenyl-amido titanium and zirconium dimethyl complexes: Improved synthesis and use in propylene polymerization. J. Organometall. Chem. 2002, 664, 5–26. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Marks, T.J. “Constrained geometry” dialkyl catalysts. Efficient syntheses, C−H bond activation chemistry, monomer−dimer equilibration, and α-olefin polymerization catalysis. Organometallics 1997, 16, 3649–3657. [Google Scholar] [CrossRef]
- Rocchigiani, L.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Self-aggregation tendency of zirconocenium ion pairs which model polymer-chain-carrying species in aromatic and aliphatic solvents with low polarity. Chem. Eur. J. 2008, 14, 6589–6592. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Bellachioma, G.; Ciancaleoni, G.; Macchioni, A.; Zuccaccia, D.; Zuccaccia, C. Synthesis, characterization, interionic structure, and self-aggregation tendency of zirconaaziridinium salts bearing long alkyl chains. Organometallics 2011, 30, 100–114. [Google Scholar] [CrossRef]
- Rocchigiani, L.; Macchioni, A.; Zuccaccia, C. NMR Studies on the dynamic behavior of zirconaaziridinium ion pairs in solution. Organometallics 2012, 31, 4076–4079. [Google Scholar] [CrossRef]
- Kumawat, J.; Gupta, V.K. Single to multiple site behavior of metallocenes through C–H activation for olefin polymerization: A mechanistic insight from DFT. ACS Catal. 2020, 10, 1704–1715. [Google Scholar] [CrossRef]
- Kwon, S.J.; Baek, J.W.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Shin, E.J.; Lee, K.S.; Lee, B.Y. Preparation of pincer hafnium complexes for olefin polymerization. Molecules 2019, 24, 1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccaccia, C.; Tensi, L.; Kuhlman, R.L.; Gies, A.P.; Macchioni, A. C–H Activation and olefin insertion as sources of multiple sites in olefin polymerization catalyzed by CpAlkylHf(IV) complexes. ACS Catal. 2017, 7, 563–567. [Google Scholar] [CrossRef]
- Laur, E.; Louyriac, E.; Dorcet, V.; Welle, A.; Vantomme, A.; Miserque, O.; Brusson, J.-M.; Maron, L.; Carpentier, J.-F.; Kirillov, E. Substitution effects in highly syndioselective styrene polymerization catalysts based on single-component allyl ansa-lanthanidocenes: An experimental and theoretical study. Macromolecules 2017, 50, 6539–6551. [Google Scholar] [CrossRef]
- Cueny, E.S.; Johnson, H.C.; Landis, C.R. Selective quench-labeling of the hafnium-pyridyl amido-catalyzed polymerization of 1-octene in the presence of trialkyl-aluminum chain-transfer reagents. ACS Catal. 2018, 8, 11605–11614. [Google Scholar] [CrossRef]
- Park, J.H.; Do, S.H.; Cyriac, A.; Yun, H.; Lee, B.Y. Preparation of half-metallocenes of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl ligands for ethylene/α-olefin copolymerization. Dalton Trans. 2010, 39, 9994–10002. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
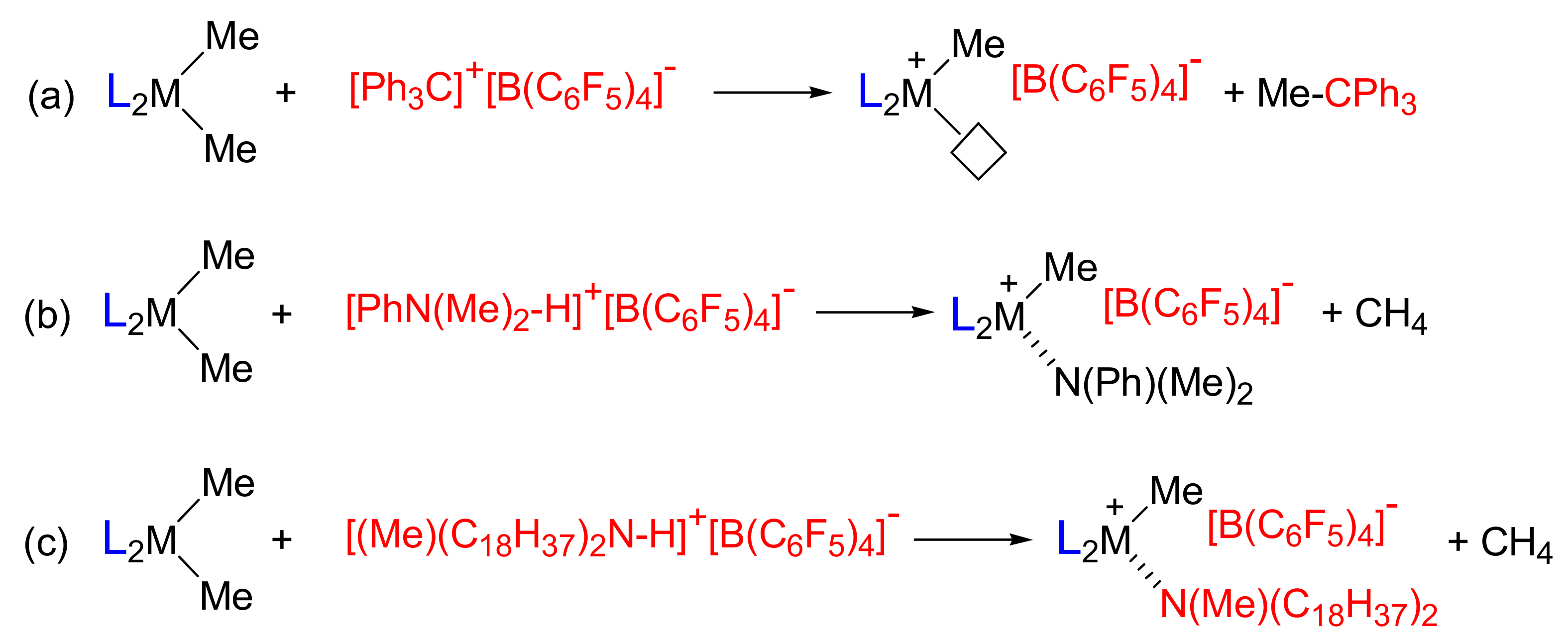{kind=link}
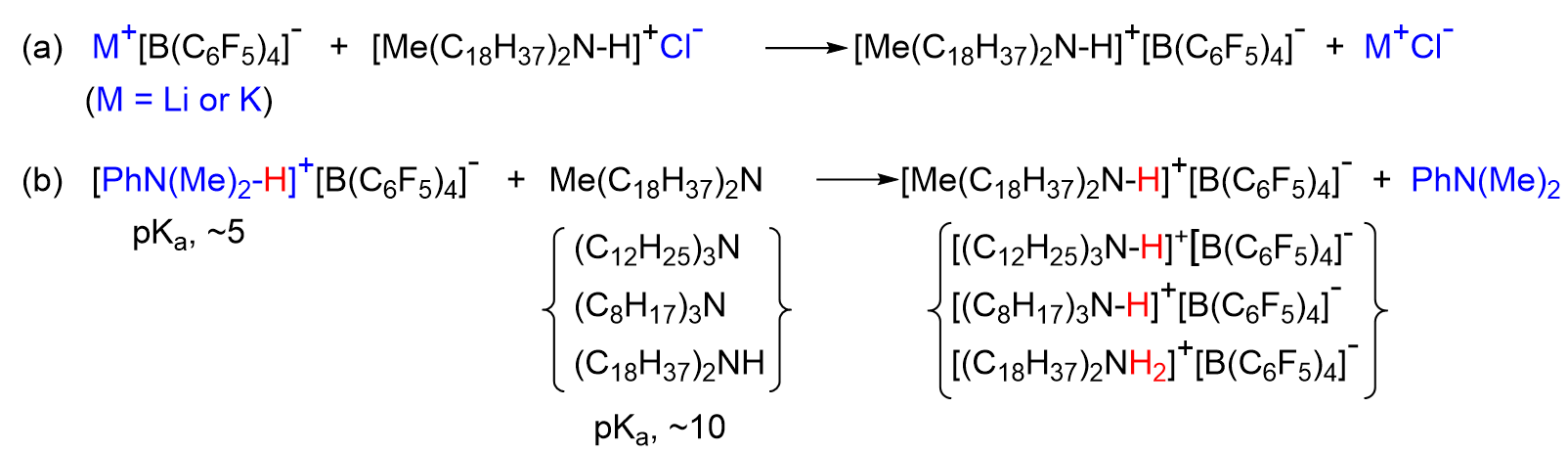{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Temperature b (°C) | Yield (g) | FOctc (mol%) | Mwd (kDa) | Mw/Mn |
|---|---|---|---|---|---|---|
| 1 | [1-Zr(Me)(NMe(C18H37)2)]+[B(C6F5)4]− | 65–116–94 | 9.3 | 18 | 32 | 13 |
| 2 | [3-Hf(Me)(NMe(C18H37)2)]+[B(C6F5)4]− | 82–164–91 | 5.2 | 33 | 716 | 39 |
| 3 | [4-Hf(Me)(NMe(C18H37)2)]+[B(C6F5)4]− | 78–161–91 | 5.0 | 34 | 814 | 56 |
| 4 e | [4-Hf(Me)(NMe(C18H37)2)]+[B(C6F5)4]− | 65–159–95 | 5.4 | 35 | 696 | 78 |
| 5 | [5-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 65–141–82 | 4.9 | 35 | 150 | 13 |
| 6 e | [5-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 65–126–83 | 6.2 | 29 | 103 | 7.3 |
| 7 f | [5-TiCl(N(H)(C18H37)2)]+[B(C6F5)4]− | 89–115–99 | 4.5 | 25 | 211 | 3.6 |
| 8 | [6-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 81–102–95 | 4.4 | 27 | 269 | 2.3 |
| 9 e | [6-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 80–124–91 | 5.3 | 29 | 237 | 2.5 |
| 10 g | [6-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 71–129–83 | 5.8 | 31 | 211 | 3.5 |
| 11 | [6-Ti(N(C18H37)2)]+[B(C6F5)4]− | - | 0 | - | ||
| 12 f | [6-TiCl(N(H)(C18H37)2)]+[B(C6F5)4]− | 65–129–82 | 4.2 | 32 | 213 | 2.7 |
| 13 h | [7-Ti(Me)(NMe(C18H37)2)]+[B(C6F5)4]− | 65–104–79 | 5.6 | 35 | 209 | 2.9 |
| 14 | [7-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 80–104–94 | 4.1 | 24 | 343 | 2.5 |
| 15 e | [7-Ti(CH2N(C18H37)2)]+[B(C6F5)4]− | 65–119–85 | 5.3 | 29 | 216 | 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-J.; Baek, J.-W.; Seo, Y.-H.; Lee, H.-C.; Jeong, S.-M.; Lee, J.; Lee, C.-G.; Lee, B.-Y. Preparation of High-Purity Ammonium Tetrakis(pentafluorophenyl)borate for the Activation of Olefin Polymerization Catalysts. Molecules 2021, 26, 2827. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092827
Lee H-J, Baek J-W, Seo Y-H, Lee H-C, Jeong S-M, Lee J, Lee C-G, Lee B-Y. Preparation of High-Purity Ammonium Tetrakis(pentafluorophenyl)borate for the Activation of Olefin Polymerization Catalysts. Molecules. 2021; 26(9):2827. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092827
Chicago/Turabian StyleLee, Hyun-Ju, Jun-Won Baek, Yeong-Hyun Seo, Hong-Cheol Lee, Sun-Mi Jeong, Junseong Lee, Chong-Gu Lee, and Bun-Yeoul Lee. 2021. "Preparation of High-Purity Ammonium Tetrakis(pentafluorophenyl)borate for the Activation of Olefin Polymerization Catalysts" Molecules 26, no. 9: 2827. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092827