
Enhancement of Haloperidol Binding Affinity to Dopamine Receptor via Forming a Charge-Transfer Complex with Picric Acid and 7,7,8,8-Tetracyanoquinodimethane for Improvement of the Antipsychotic Efficacy

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
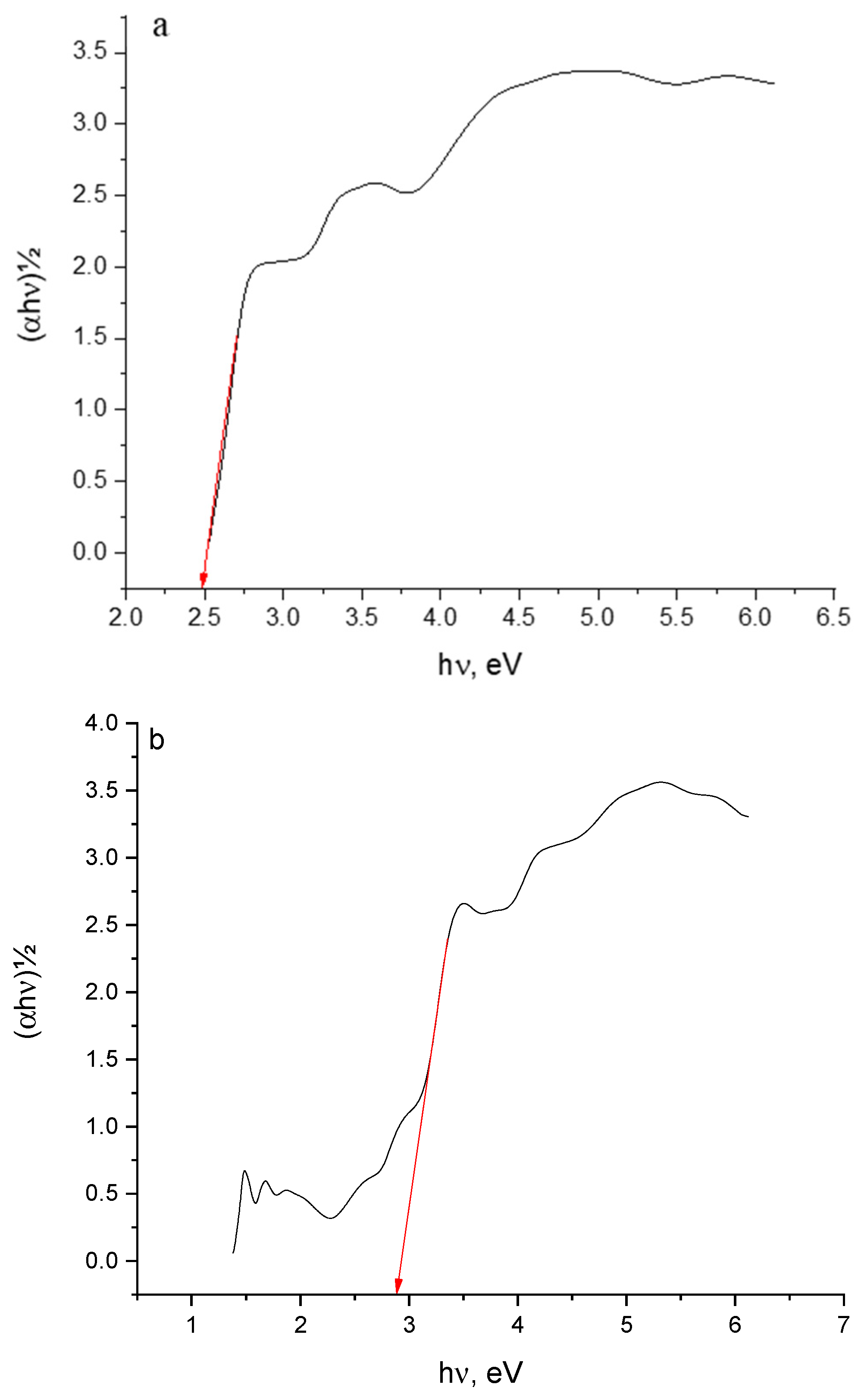
2. Results and Discussion
2.1. Preface
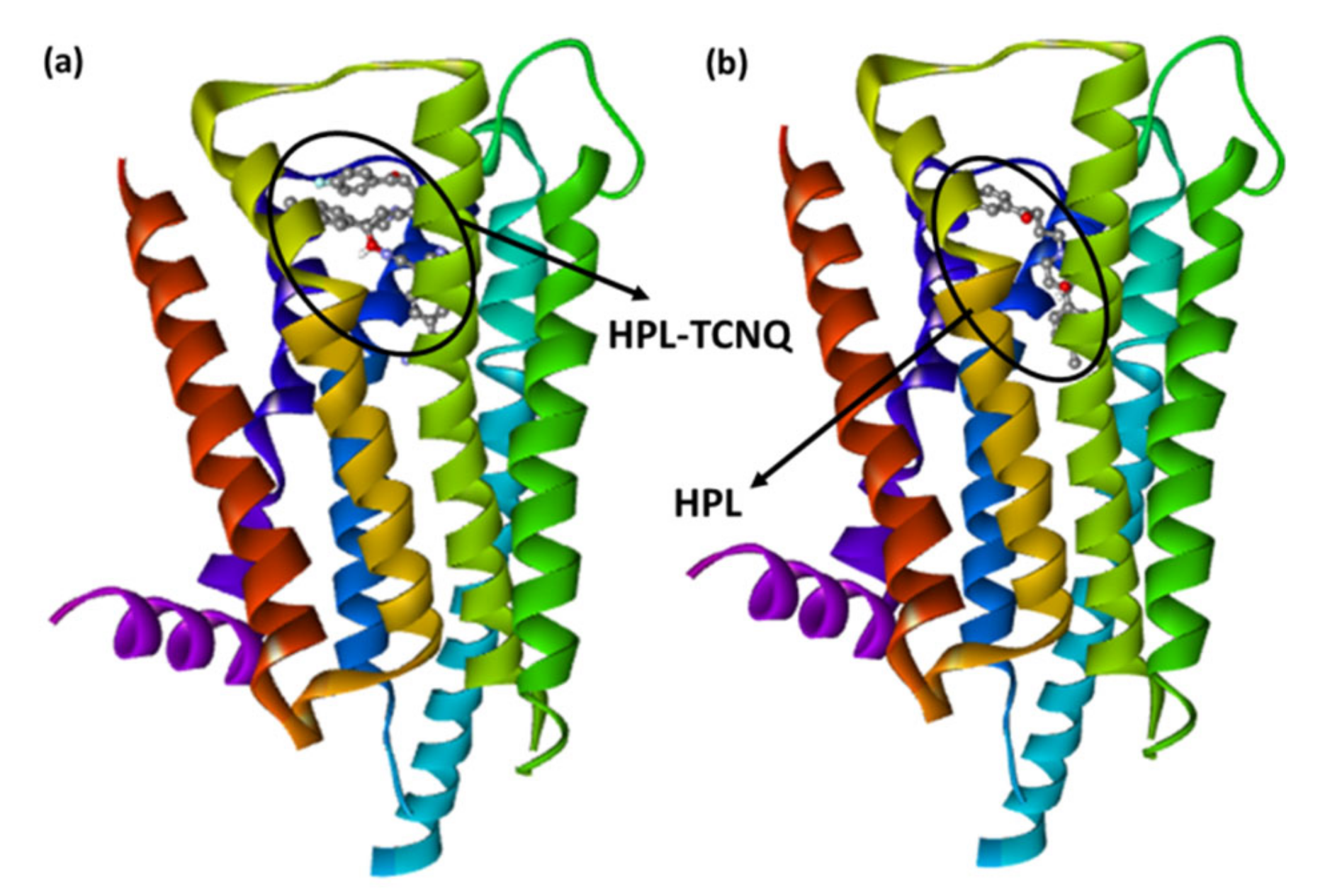
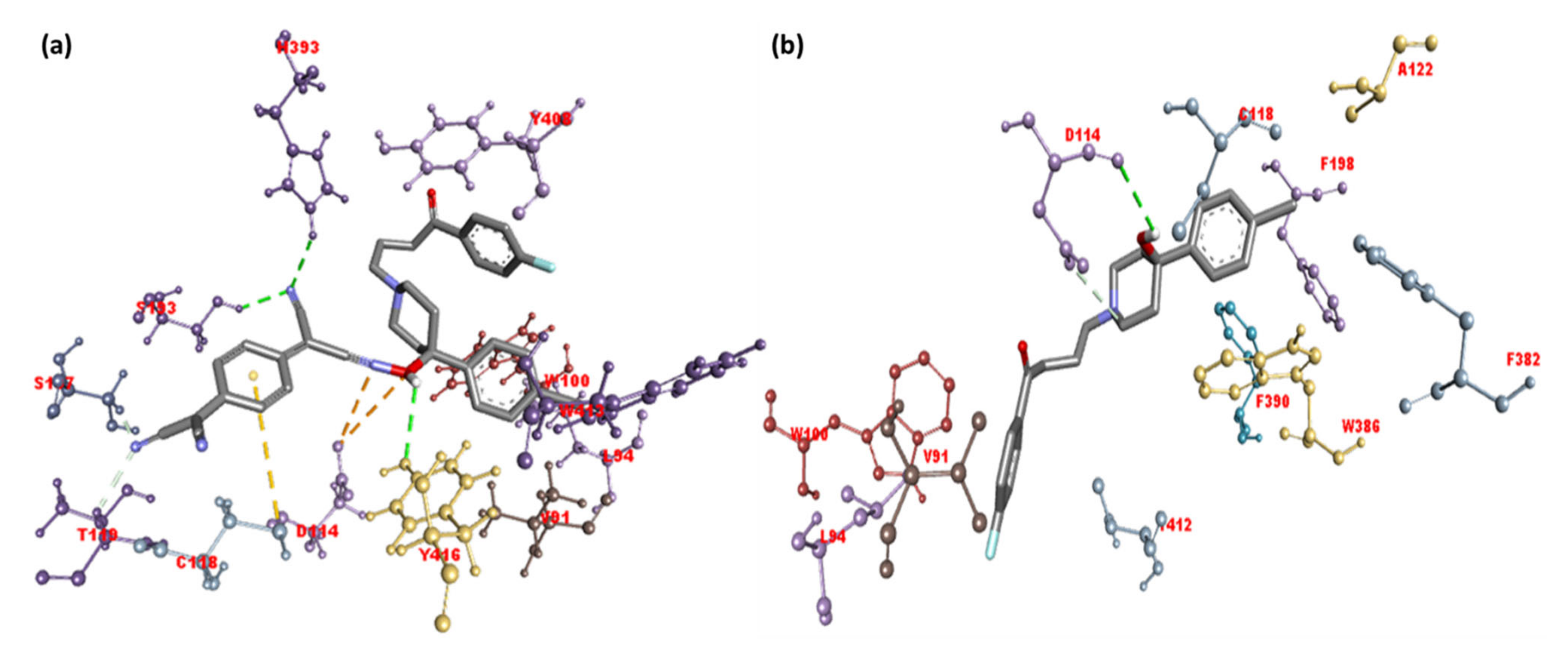
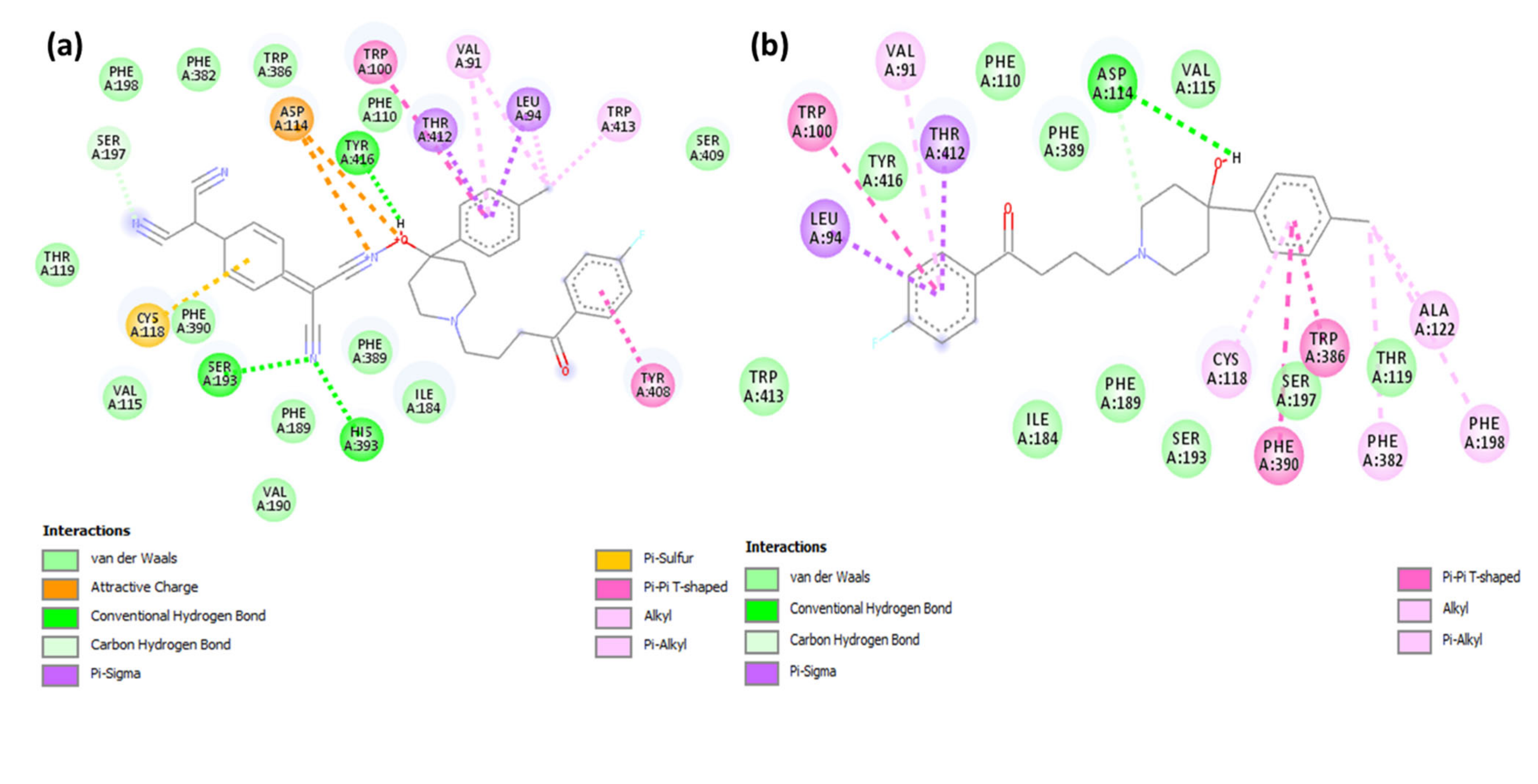
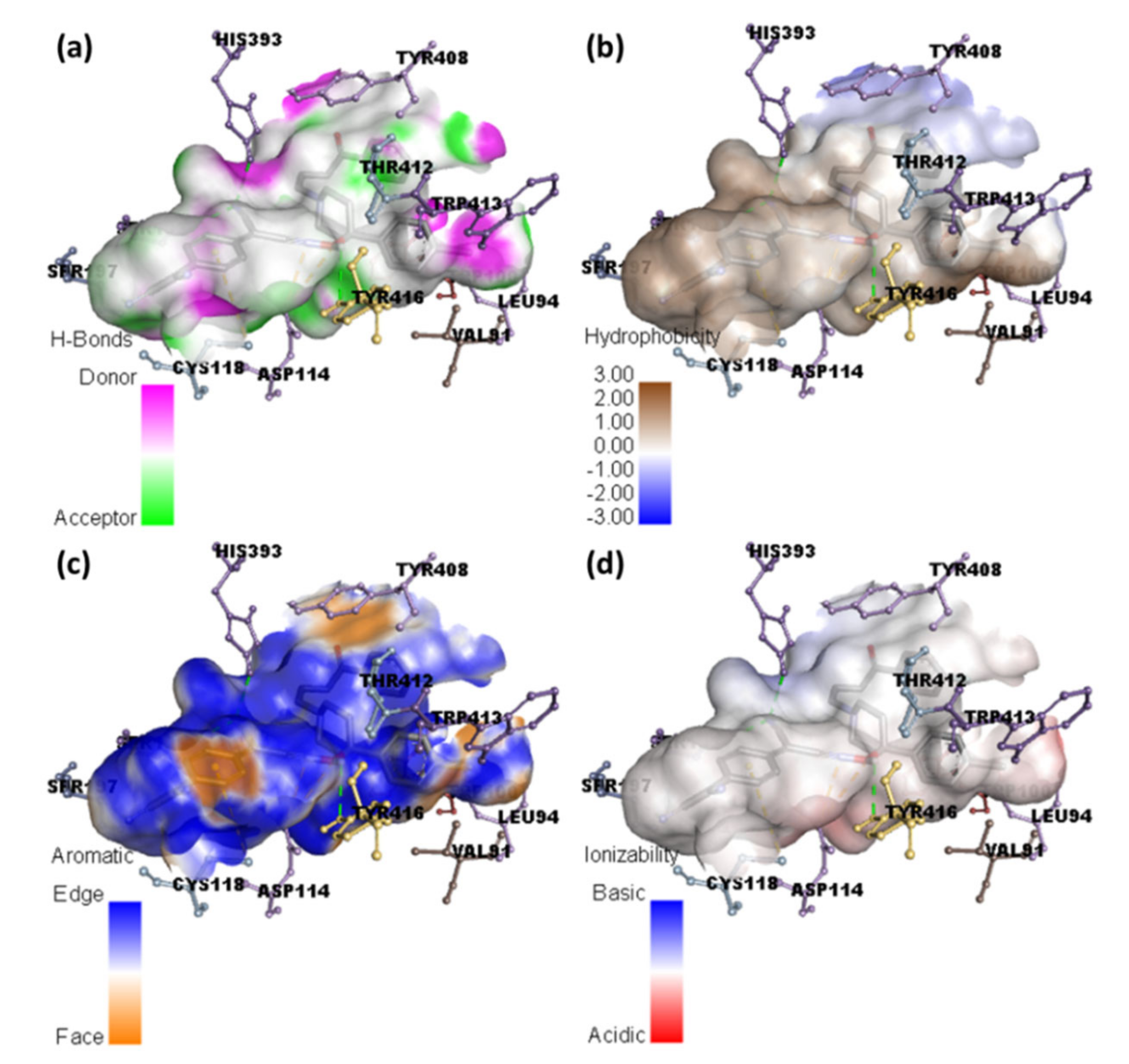
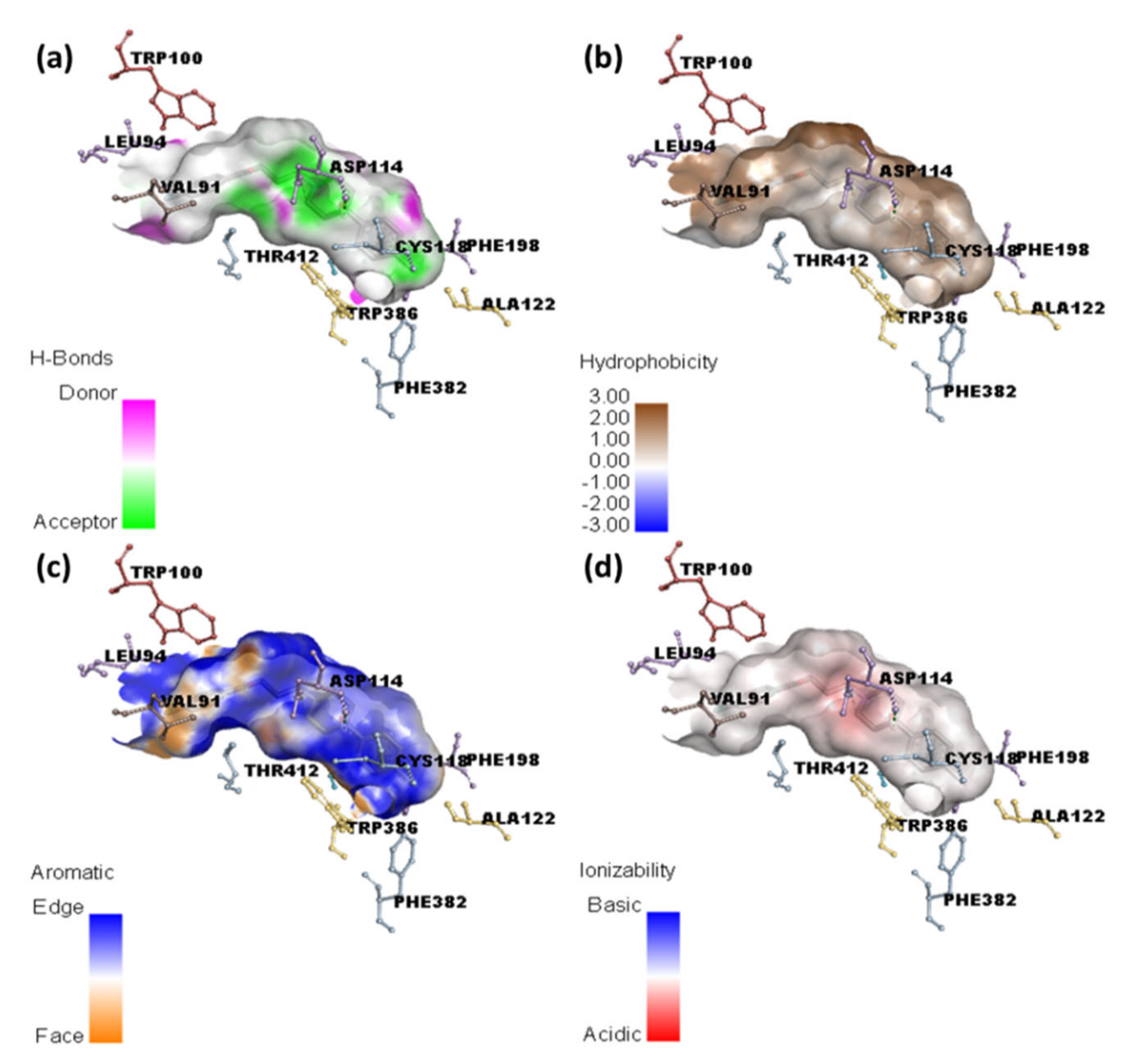
2.2. Molecular Docking Studies
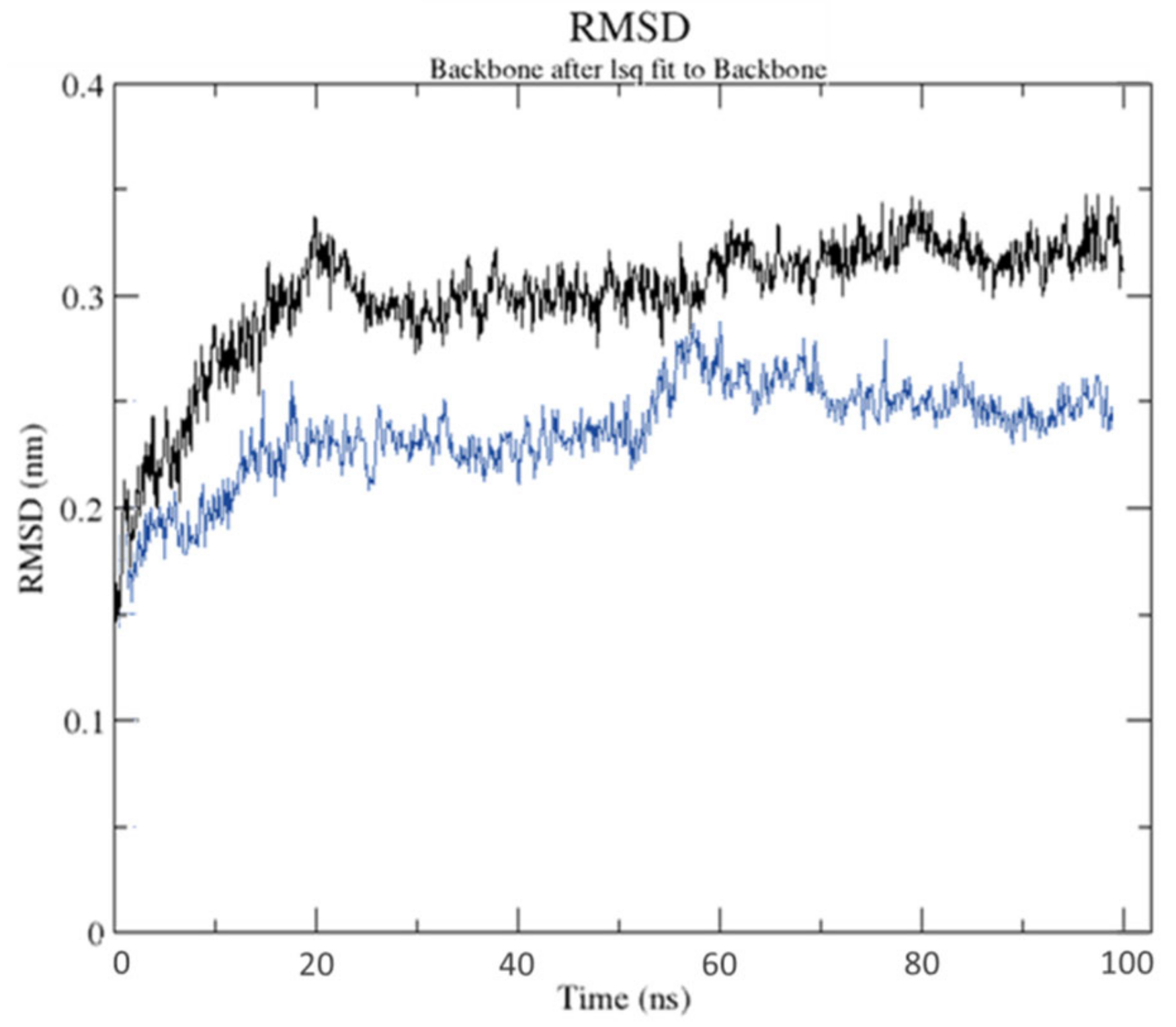
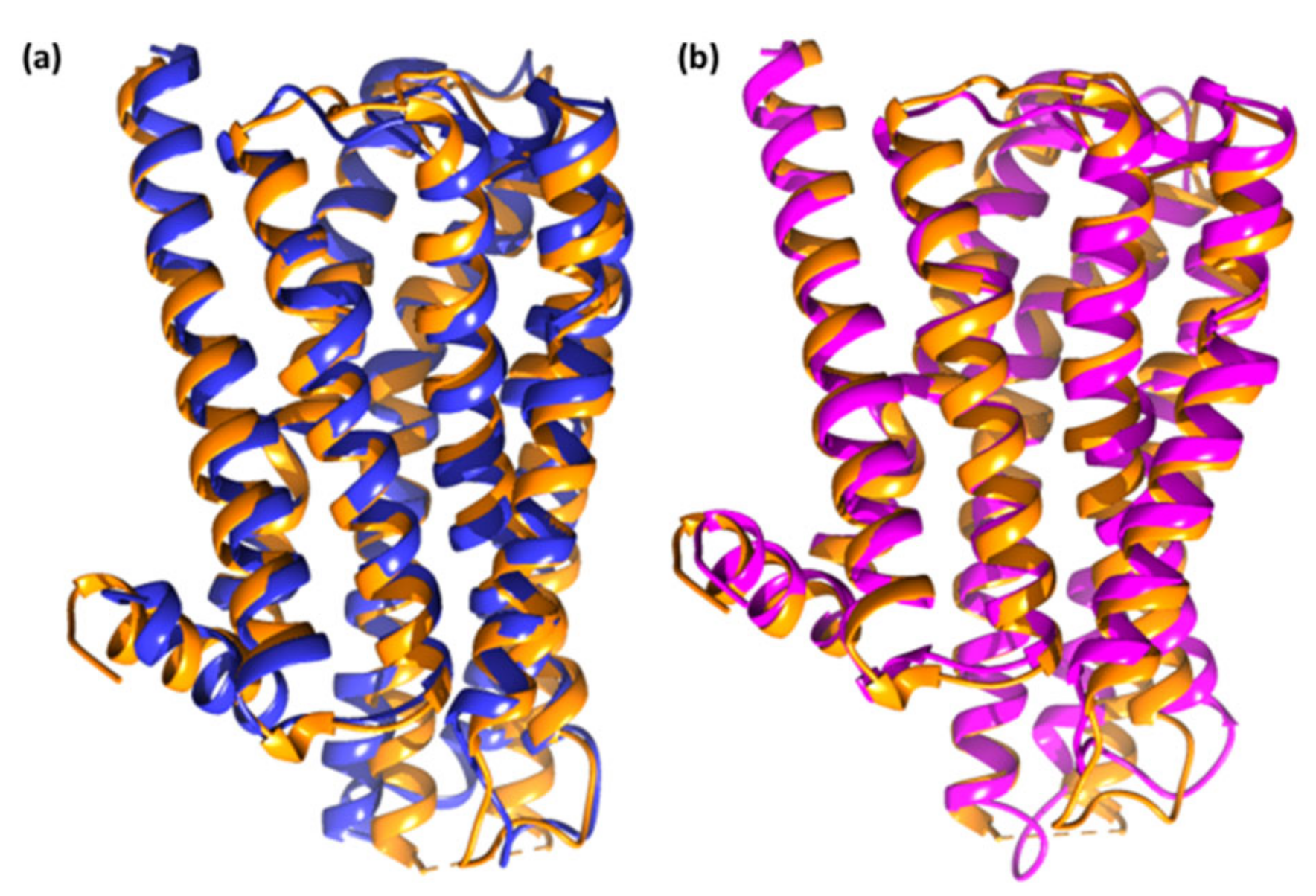
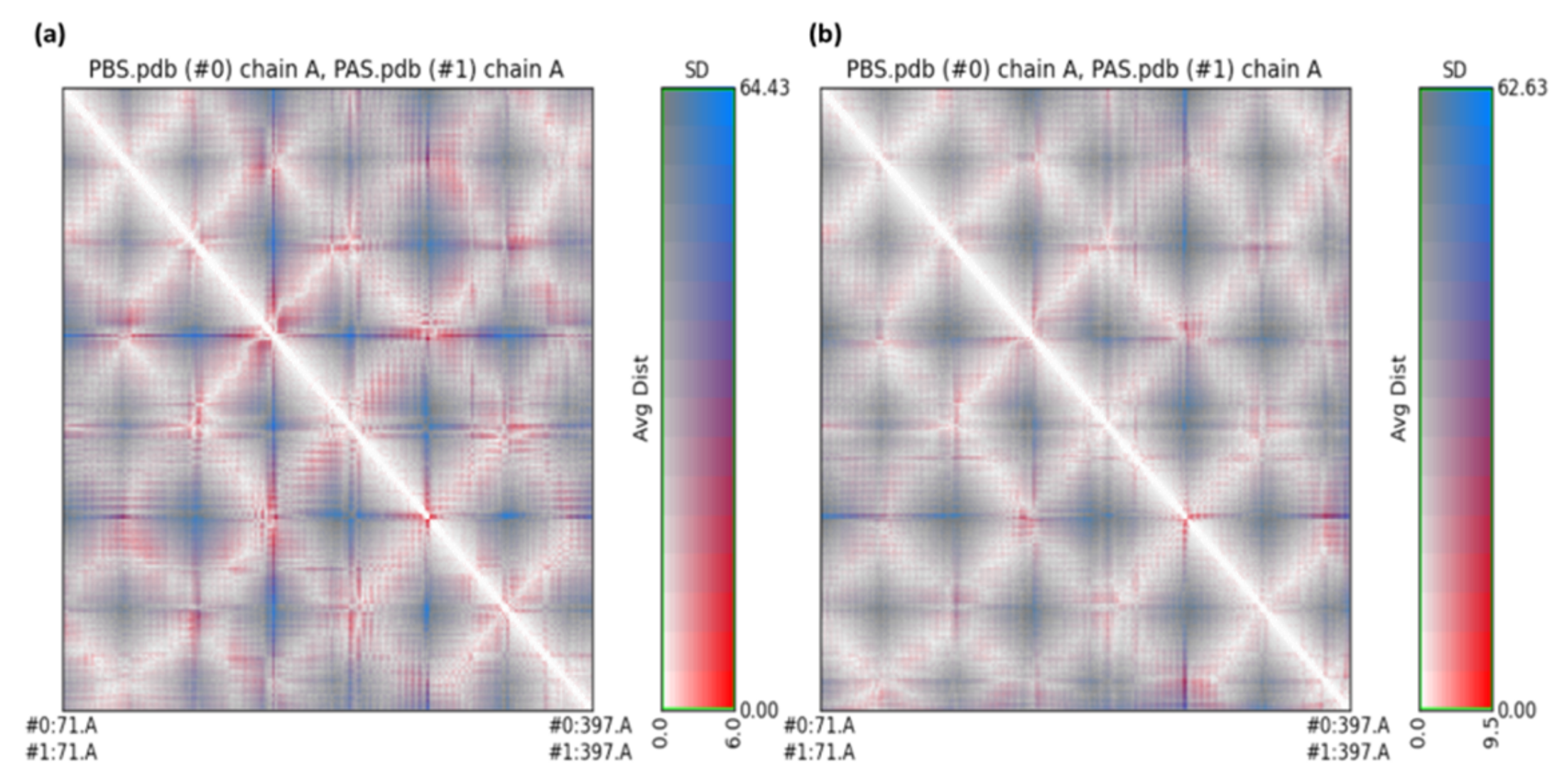
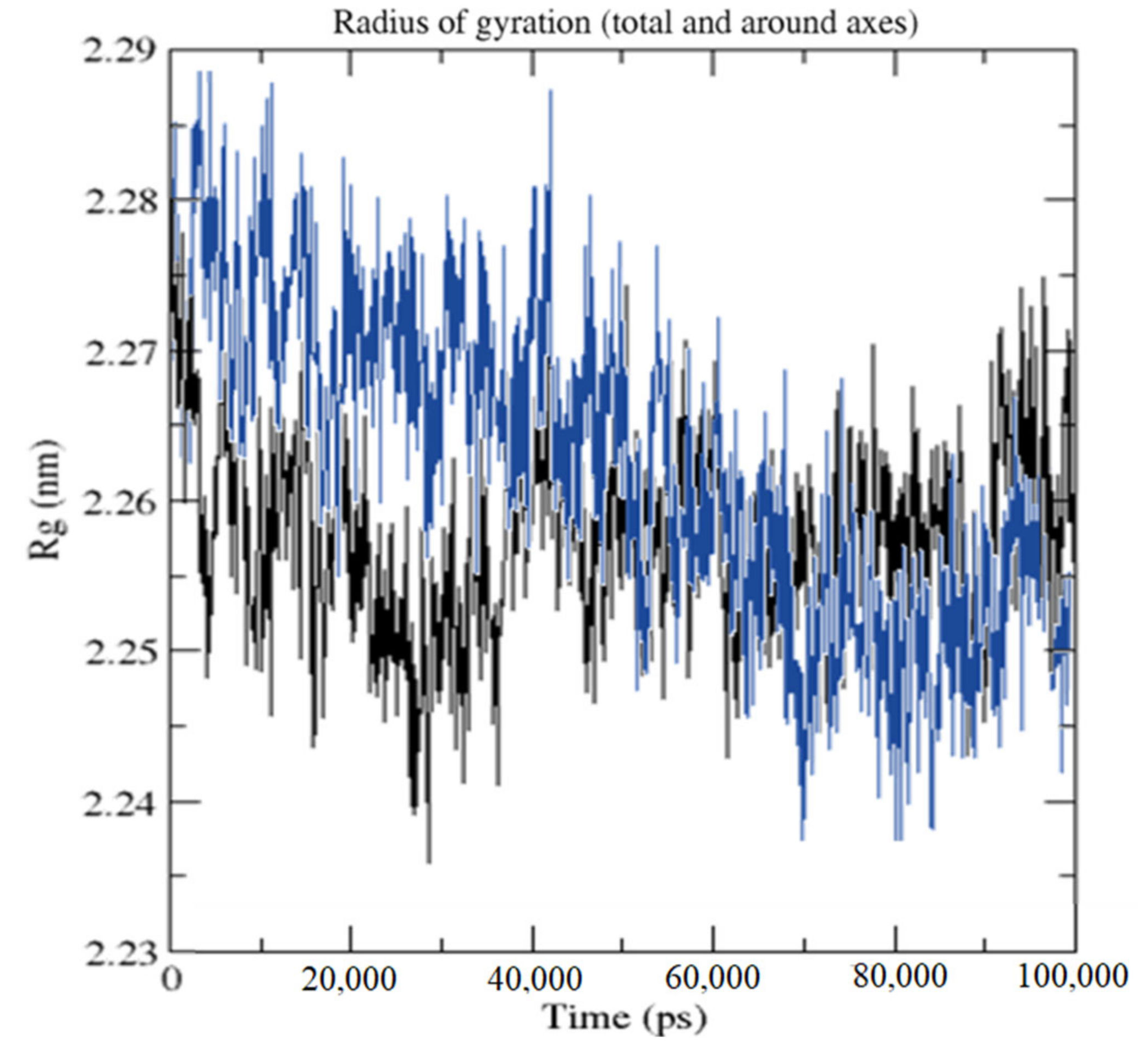
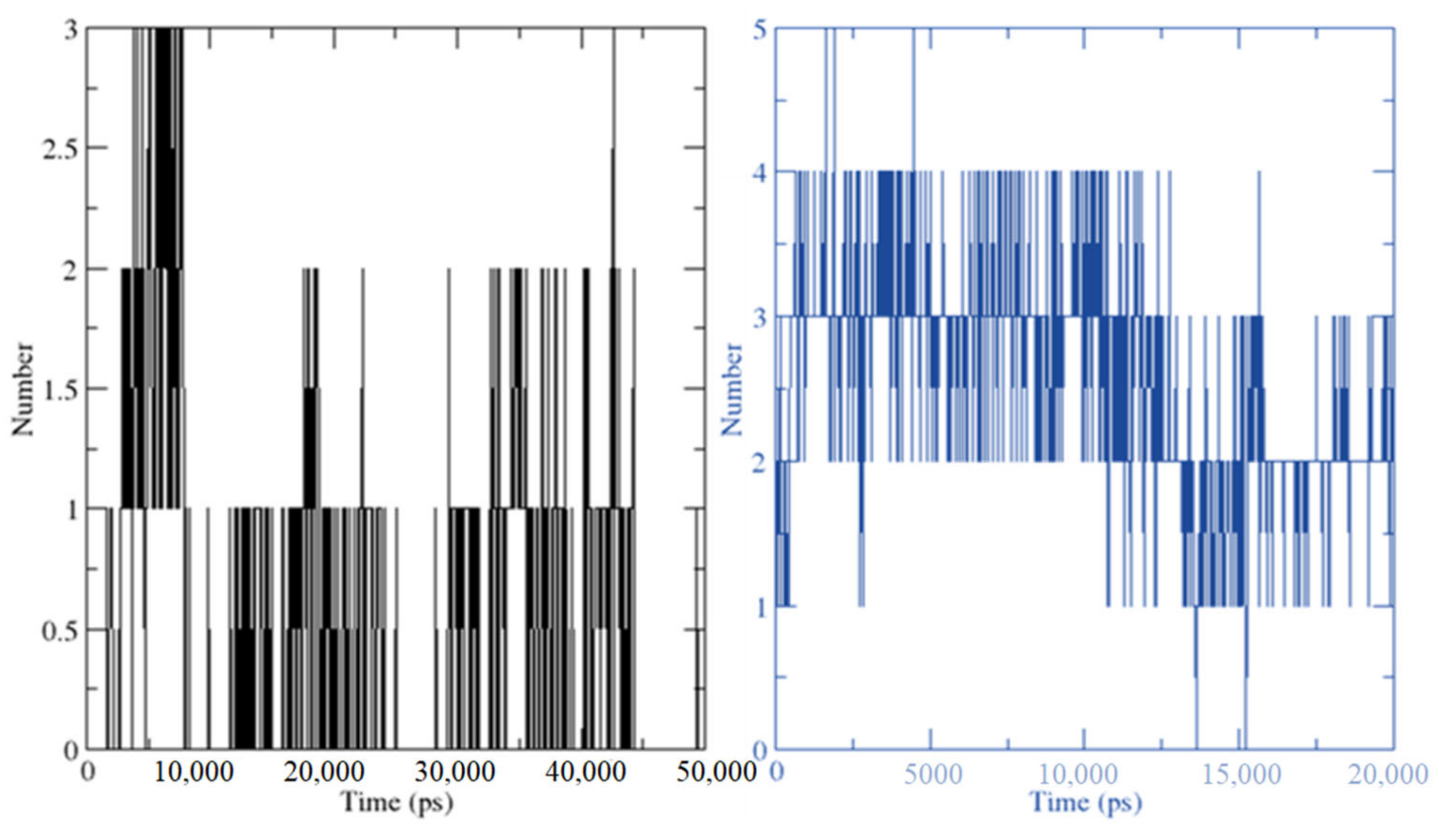
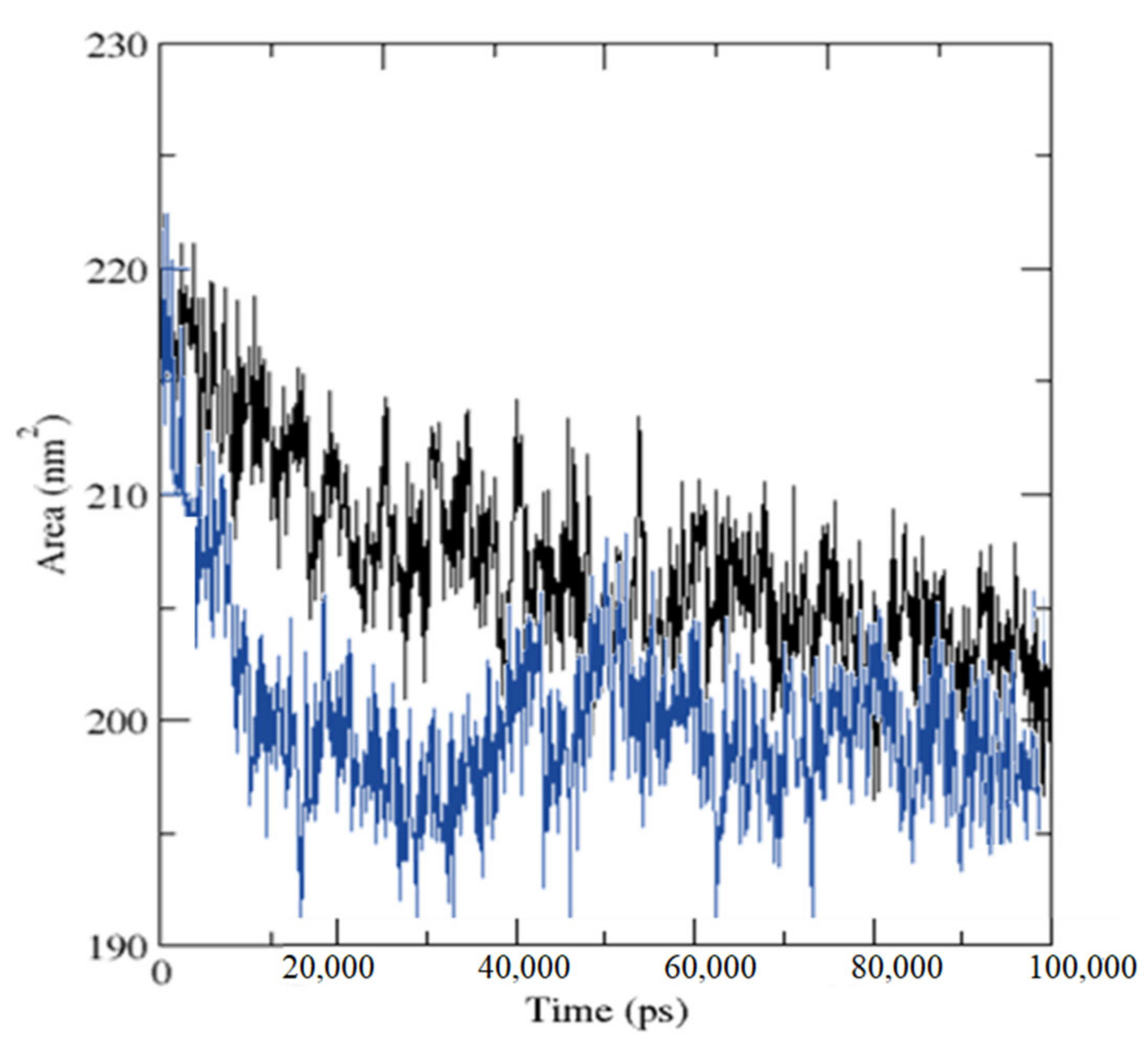
2.3. Molecular Dynamics Simulation
3. Materials and Methods
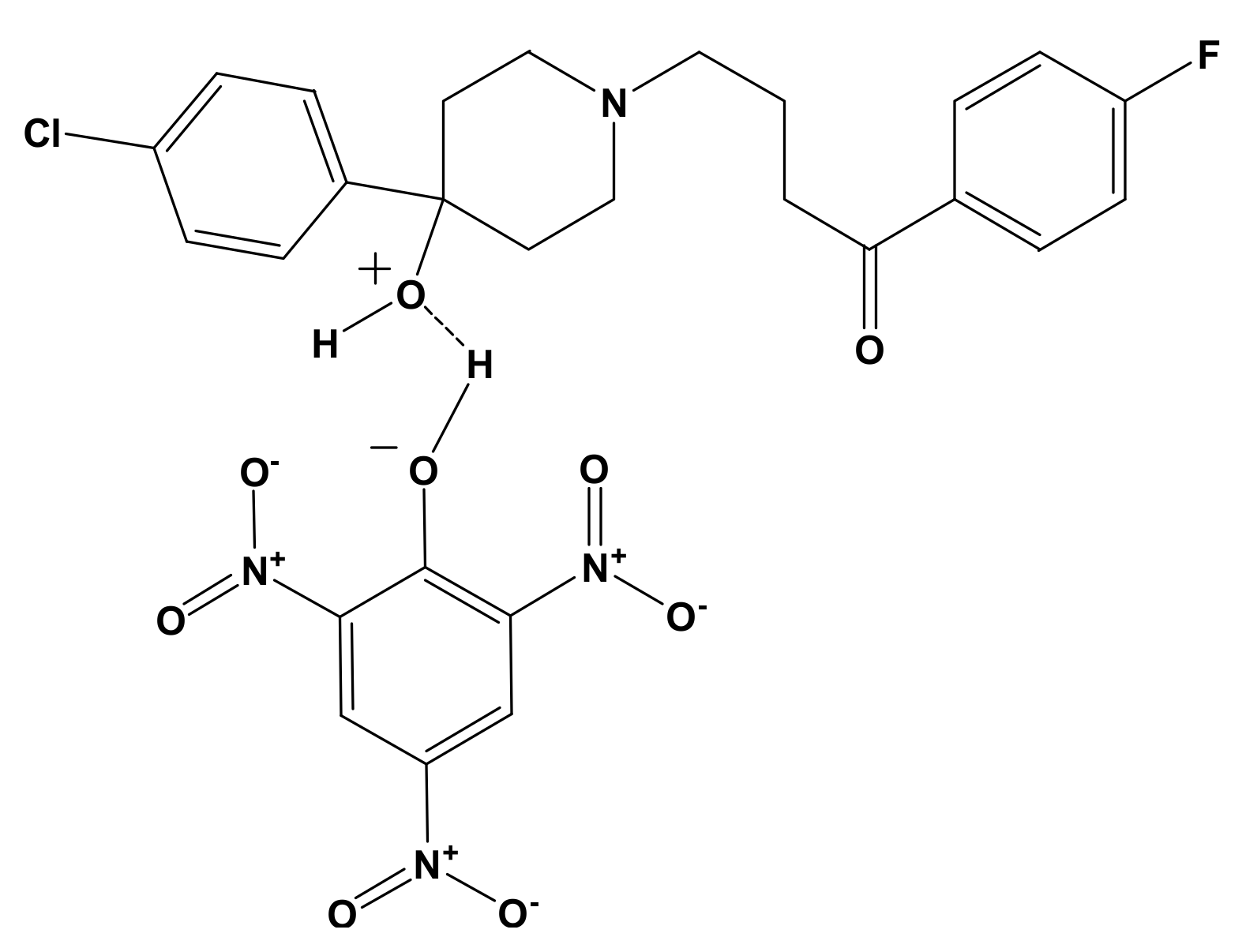
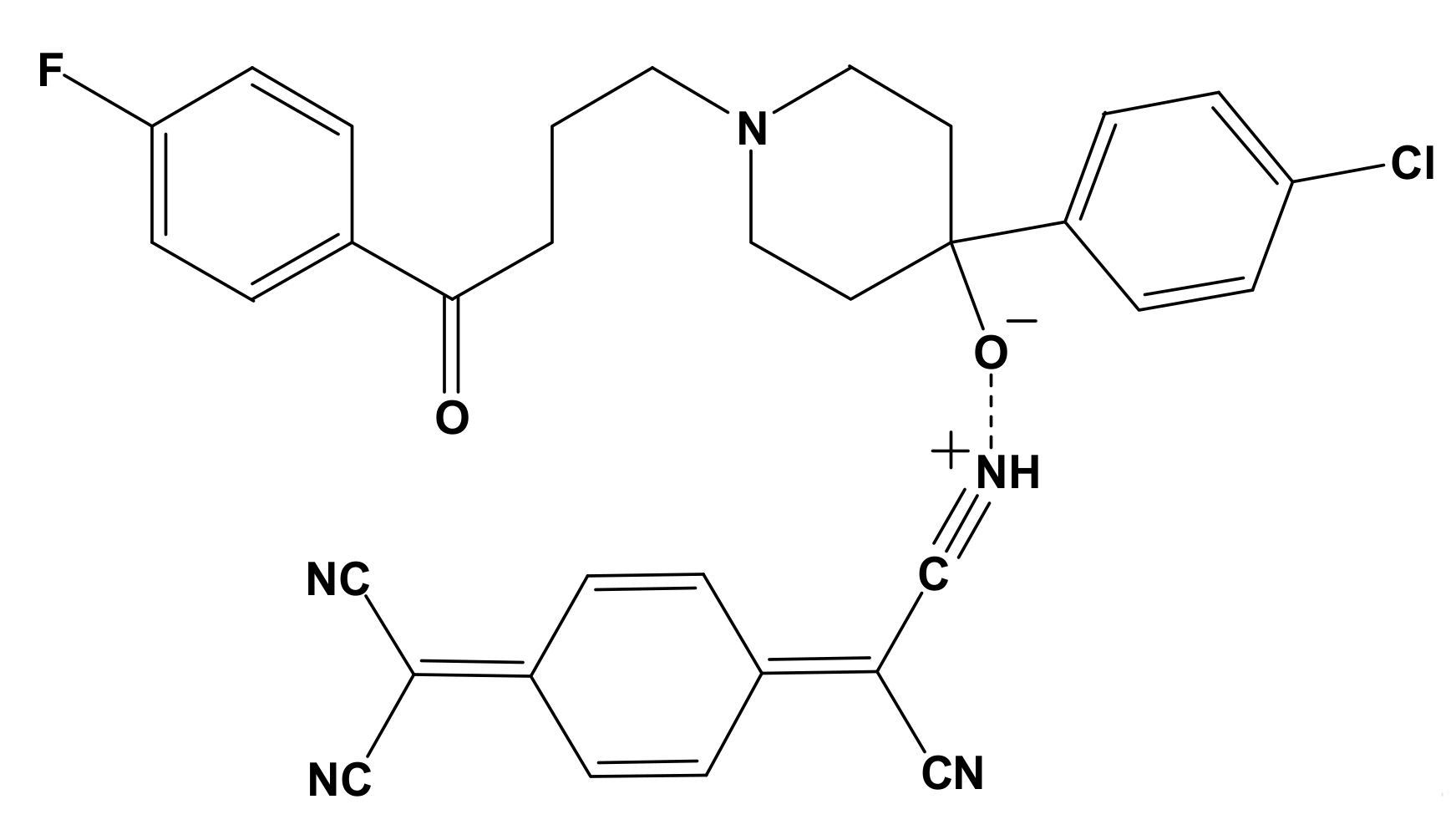
3.1. Synthesis of [(HPL)(PA)] and [(HPL)(TCN)] CT Complexes
3.2. Physical Measurements
3.3. Molecular Docking
3.4. MD Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Irving, C.B.; Adams, C.E.; Lawrie, S.M. Haloperidol versus placebo for schizophrenia. Cochrane Database Syst. Rev. 2004, 4, CD003082. [Google Scholar]
- Mulliken, R.S. Structures of complexes formed by halogen molecules with aromatic and with oxygenated solvents. J. Am. Chem. Soc. 1950, 72, 600–608. [Google Scholar] [CrossRef]
- Vinay, K.B.; Revanasiddappa, H.D.; Raghu, M.S.; Sameer, A.M.A.; Rajendraprasad, N. Spectrophotometric determination of mycophenolate mofetil as its charge-transfer complexes with two π-acceptors. J. Anal. Methods Chem. 2012, 2012, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugesan, V.; Saravanabhavan, M.; Sekar, M. Synthesis, spectroscopic characterization and structural investigation of a new CT complex of 2, 6-diaminopyridine with 4-nitrophenylacetic acid: Antimicrobial, DNA binding/cleavage and antioxidant studies. Spectrochim. Acta A 2015, 147, 99–106. [Google Scholar] [CrossRef]
- Miyan, L.; Ahmad, A.; Alam, M.F.; Younus, H. Synthesis, single-crystal, DNA interaction, spectrophotometric and spectroscopic characterization of the hydrogen-bonded CT complex of 2-aminopyrimidine with π-acceptor chloranilic acid at different temperature in acetonitrile. J. Photochem. Photobiol. B 2017, 174, 195–208. [Google Scholar]
- Singh, N.; Khan, I.M.; Ahmad, A.; Javed, S. Synthesis and dynamics of a novel proton transfer complex containing 3,5-dimethylpyrazole as a donor and 2,4-dinitro-1-naphthol as an acceptor: Crystallographic, UV-visible spectrophotometric, molecular docking and Hirshfeld surface analyses. New J. Chem. 2017, 41, 6810–6821. [Google Scholar] [CrossRef]
- Bai, H.; Wang, Y.; Cheng, P.; Li, Y.; Zhu, D.; Zhan, X. Acceptor-donoracceptor small molecules based on indacenodithiophene for efficient organic solar cells. ACS Appl. Mater. Interfaces 2014, 6, 8426–8433. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System PyMOL DeLano Scientific. 2002. Available online: http://www.pymol.org (accessed on 1 March 2022).
- Horowitz, H.H.; Metzger, G. A new analysis of thermogravimetric traces. Anal. Chem. 1963, 35, 1464–1468. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Mott, N.F.; Davis, E.A. Electronic Process in Non-Crystalline Materials; Calendron Press: Oxford, UK, 1972. [Google Scholar]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Fu, M.L.; Guo, G.C.; Liu, X.; Cai, L.Z.; Huang, J.S. Syntheses, structures, and properties of three selenoarsenates templated by transition metal complexes. Inorg. Chem. Commun. 2005, 8, 18–21. [Google Scholar] [CrossRef]
- Adam, A.M.A.; Refat, M.S.; Hegab, M.S.; Saad, H.A. Spectrophotometric and thermodynamic studies on the 1:1 charge transfer interaction of several clinically important drugs with tetracyanoethylene in solution-state: Part one. J. Mol. Liq. 2016, 224, 311–321. [Google Scholar] [CrossRef]
- Akram, M.; Lal, H.; Shakya, S.; Kabir-ud-Din. Multispectroscopic and computational analysis insight into the interaction of cationic diester-bonded gemini surfactants with serine protease α-chymotrypsin. ACS Omega 2020, 5, 3624–3637. [Google Scholar] [CrossRef]
- Khan, I.M.; Shakya, S.; Islam, M.; Khan, S.; Najnin, H. Synthesis and spectrophotometric studies of CT complex between 1, 2-dimethylimidazole and picric acid in different polar solvents: Exploring antimicrobial activities and molecular (DNA) docking. Phys. Chem. Liq. 2021, 59, 753–769. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar]
- Wu, S.; Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics 2008, 24, 924–931. [Google Scholar] [CrossRef] [Green Version]
- El-Habeeb, A.A.; Al-Saif, F.A.; Refat, M.S. CT complex of some nervous and brain drugs—Part 1: Synthesis, spectroscopic, analytical and biological studies on the reaction between haloperidol antipsychotic drugs with π-acceptors. J. Mol. Struct. 2013, 1034, 1–18. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S. PyRx-Python Prescription, Version 0.8; The Scripps Research Institute: La Jolla, CA, USA, 2008.
- Chu, C.H.; Li, K.M.; Lin, S.W.; Chang, M.D.T.; Jiang, T.Y.; Sun, Y.J. Crystal structures of starch binding domain from Rhizopus oryzae glucoamylase in complex with isomaltooligosaccharide: Insights into polysaccharide binding mechanism of CBM21 family. Proteins 2014, 82, 1079–1085. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additivebiological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to sulfonylcontaining compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulations of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Alhomrani, M.; Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Haynes, J.M.; Shakya, S.; Raafat, B.M.; et al. Enhancing the Antipsychotic Effect of Risperidone by Increasing Its Binding Affinity to Serotonin Receptor via Picric Acid: A Molecular Dynamics Simulation. Pharmaceuticals 2022, 15, 285. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target: PDB: 6A94 | |||
|---|---|---|---|
| Receptor | Binding Free Energy (kcal/Mol) | Interactions | |
| H-Bond | Others | ||
| HPL-PA | −9.4 | Thr160 | Leu229, Phe243, Val366 (π-Alkyl); Trp336, Phe340 (π-π T-Shape) |
| HPL-TCNQ | −10.2 | Asn363 and Tyr139 | Ala321, Val324 (π-Alkyl); Ala360 (π-Alkyl); Val366 (π-Sigma) |
| HPL | −10.0 | Leu229 | Phe332, Phe243, Val366 (π-Alkyl); Phe340, Trp336, Ser159 (π-π T-Shape) |
| Target: PDB: 6CM4 | |||
|---|---|---|---|
| Receptor | Binding Free Energy (kcal/Mol) | Interactions | |
| H-Bond | Others | ||
| HPL-PA | −9.6 | Trp100 and Tro419 | Cys118, Val115, Leu94 (π-Alkyl); Phe390, Trp389, Tyr480 (π-π T-Shape) |
| HPL-TCNQ | −11.8 | His393, Ser193, and Tyr416 | Val91 and Trp413 (π-Alkyl); Tyr408, (π-π T-Shaped); Cys118 (π-Alkyl); Thr412 and Leu94 (π-Sigma); Asp114 (Attractive charge) |
| HPL | −10.9 | Asp114 | Phe198, Phe382, Cys118, Val91 (π-Alkyl); Trp100, Trp386, Phe390 (π-π T-shaped); Lue94, Thr412 (π-Sigma) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alamri, A.S.; Alhomrani, M.; Alsanie, W.F.; Alyami, H.; Shakya, S.; Habeeballah, H.; Alamri, A.; Alzahrani, O.; Alzahrani, A.S.; Alkhatabi, H.A.; et al. Enhancement of Haloperidol Binding Affinity to Dopamine Receptor via Forming a Charge-Transfer Complex with Picric Acid and 7,7,8,8-Tetracyanoquinodimethane for Improvement of the Antipsychotic Efficacy. Molecules 2022, 27, 3295. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103295
Alamri AS, Alhomrani M, Alsanie WF, Alyami H, Shakya S, Habeeballah H, Alamri A, Alzahrani O, Alzahrani AS, Alkhatabi HA, et al. Enhancement of Haloperidol Binding Affinity to Dopamine Receptor via Forming a Charge-Transfer Complex with Picric Acid and 7,7,8,8-Tetracyanoquinodimethane for Improvement of the Antipsychotic Efficacy. Molecules. 2022; 27(10):3295. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103295
Chicago/Turabian StyleAlamri, Abdulhakeem S., Majid Alhomrani, Walaa F. Alsanie, Hussain Alyami, Sonam Shakya, Hamza Habeeballah, Abdulwahab Alamri, Omar Alzahrani, Ahmed S. Alzahrani, Heba A. Alkhatabi, and et al. 2022. "Enhancement of Haloperidol Binding Affinity to Dopamine Receptor via Forming a Charge-Transfer Complex with Picric Acid and 7,7,8,8-Tetracyanoquinodimethane for Improvement of the Antipsychotic Efficacy" Molecules 27, no. 10: 3295. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27103295