
Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Different Molecular Mechanisms Associated with the Same Phenotype
3. Symptomatic Treatments Based on Antioxidant Cocktails
4. Small Molecules, Vitamins and Cofactors Administered as Single Molecules or as a Cocktail to Patients Affected by MD
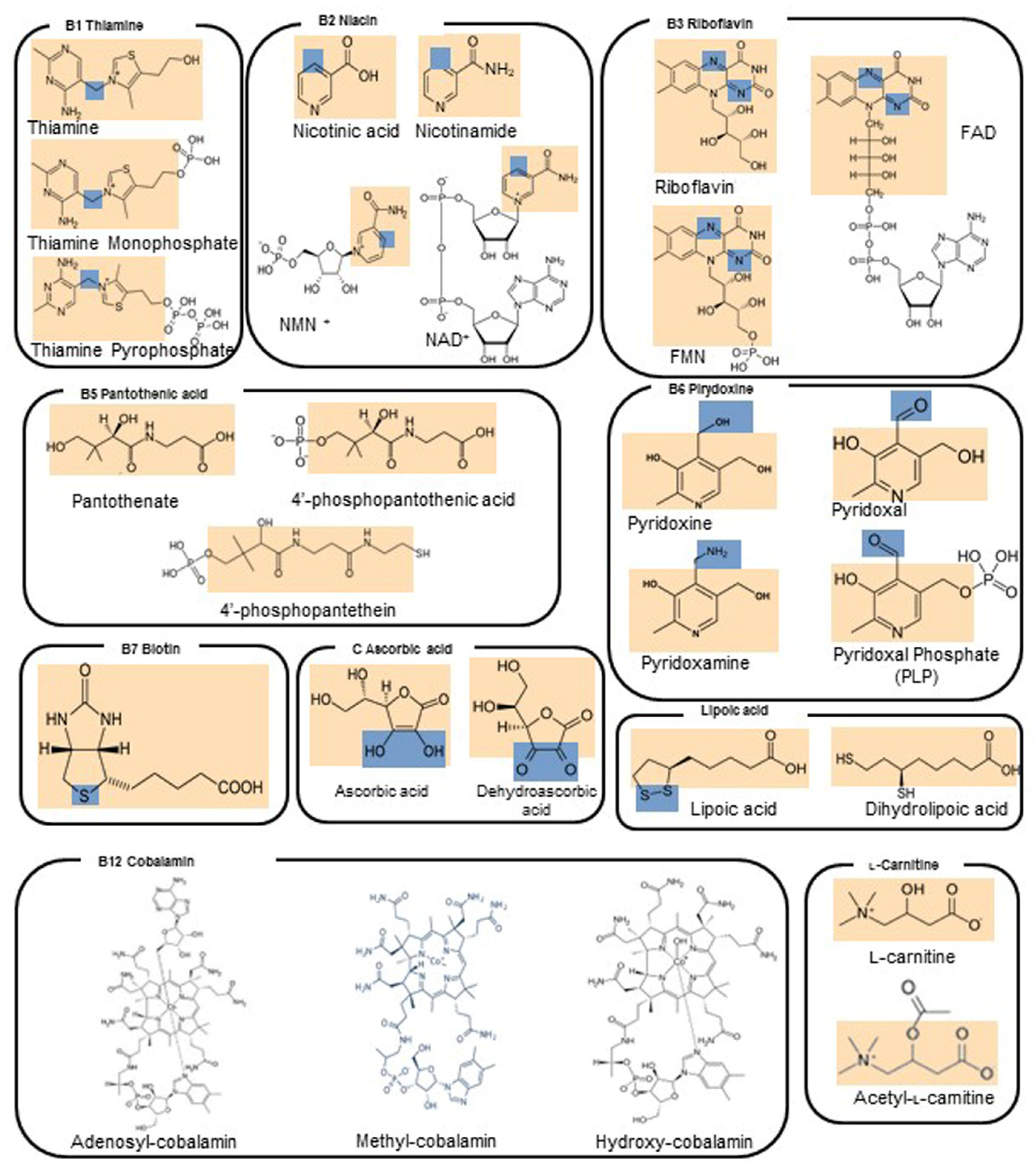
4.1. Vitamin B1—Thiamine
4.2. Vitamin B2—Riboflavin
4.3. Vitamin B3—Niacin
4.4. Vitamin B5—Pantothenic Acid
4.5. Vitamin B6—Pyridoxine
4.6. Vitamin B7—Biotin
4.7. Vitamin B9—Folic Acid
4.8. Vitamin B12—Cobalamin
4.9. Vitamin C—L-Ascorbic Acid
4.10. Lipoic Acid
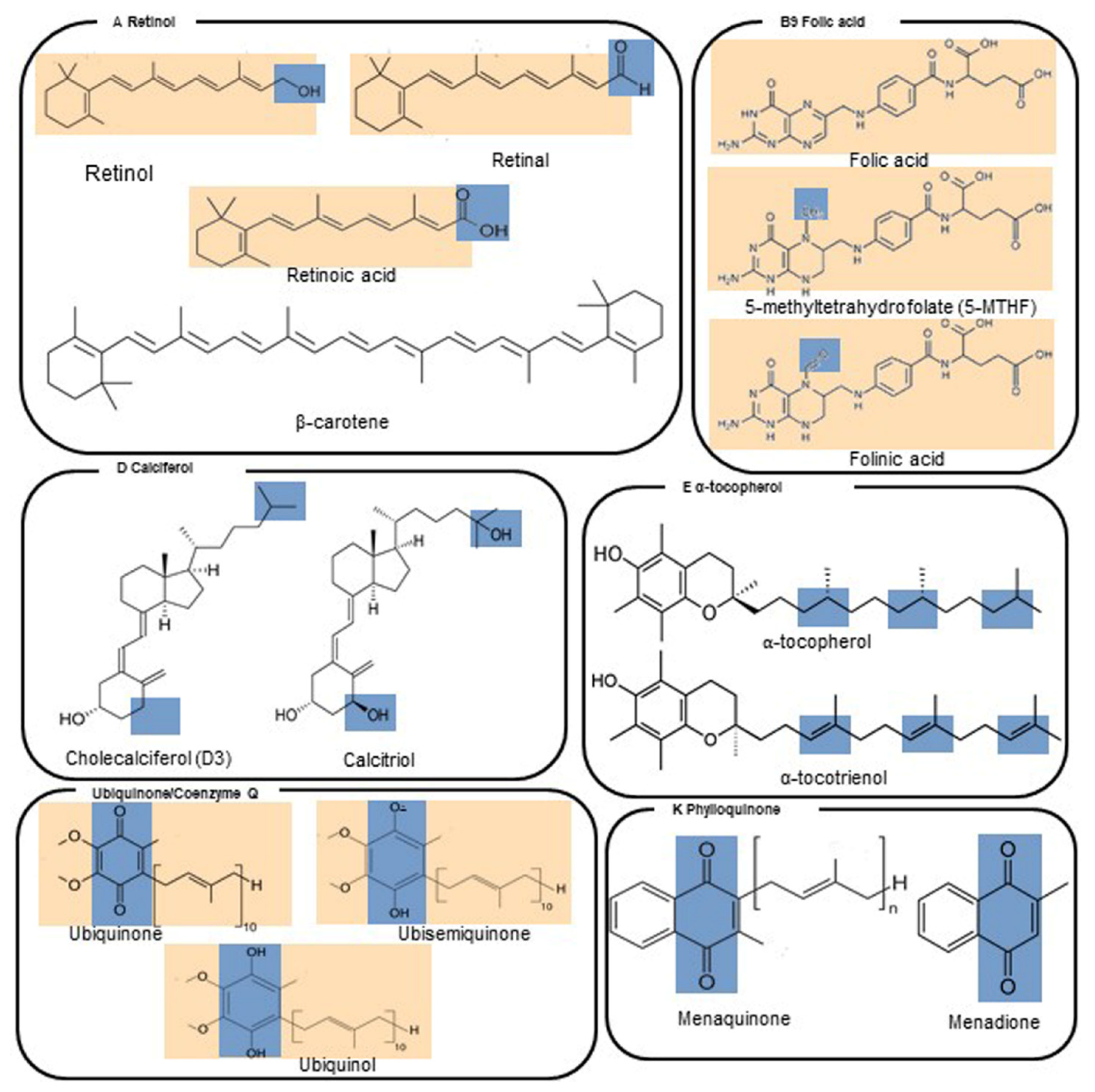
4.11. Vitamin A—Retinol
4.12. Vitamin D—Calciferol
4.13. Vitamin E—α-Tocopherol
4.14. Vitamin K—Phylloquinone
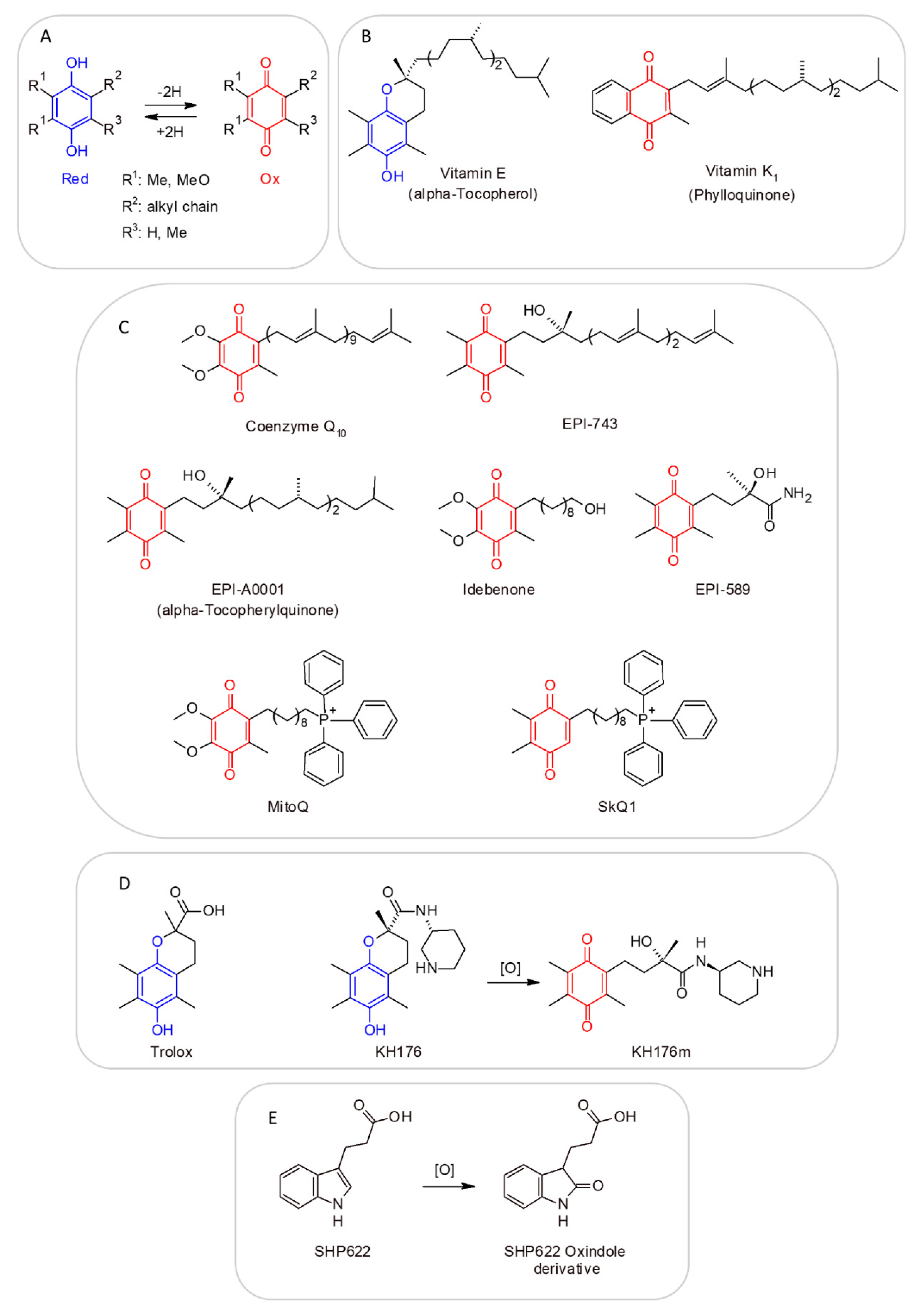
4.15. Coenzyme Q10 (Reduced, as Ubiquinol; Oxidized, as Ubiquinone)
4.16. L-Carnitine/Acetyl-L-Carnitine
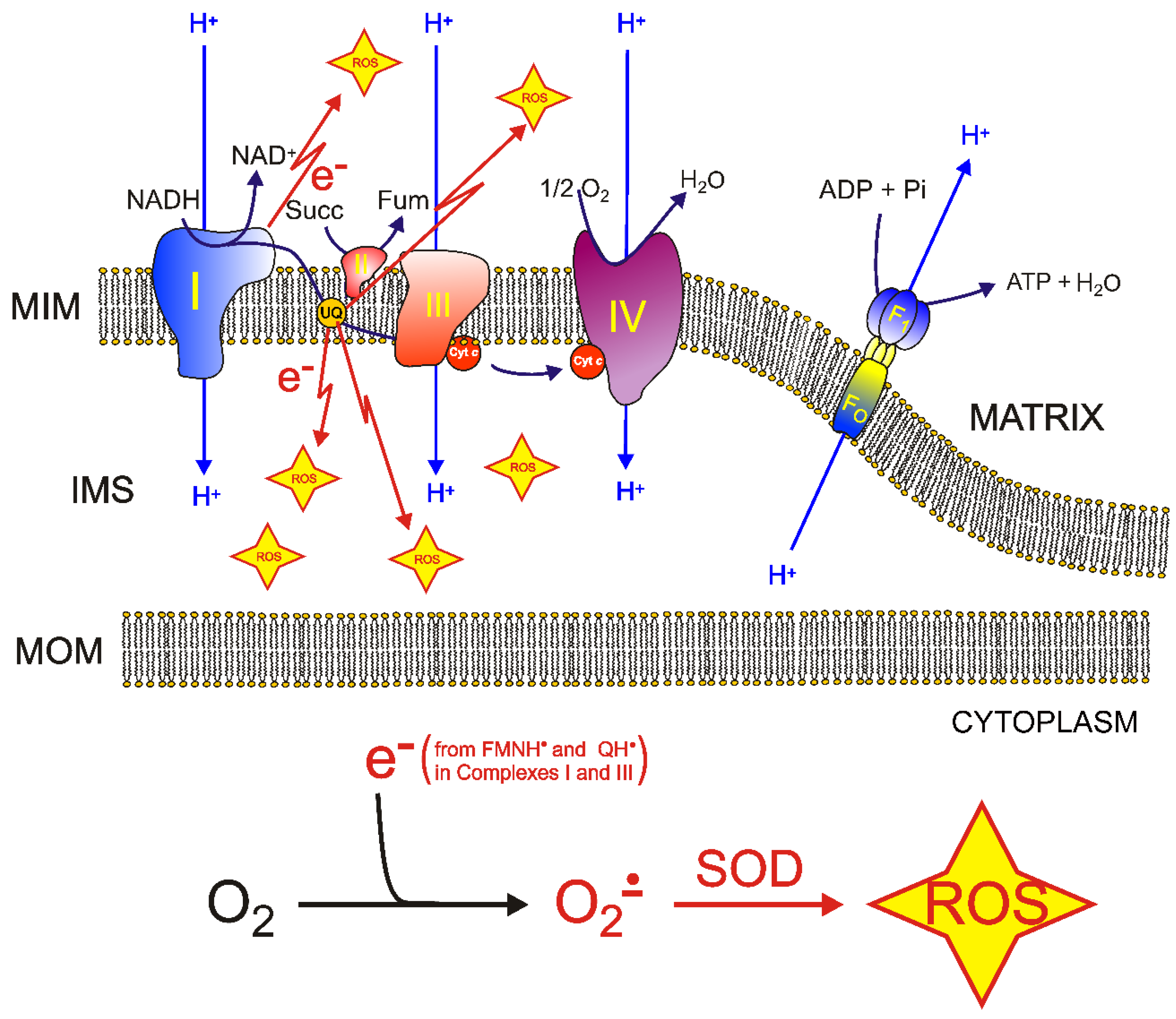
5. Molecular Mechanisms of ROS Production and Mitochondrial Damage Observed in Cells from Mitochondrial Patient Tissues
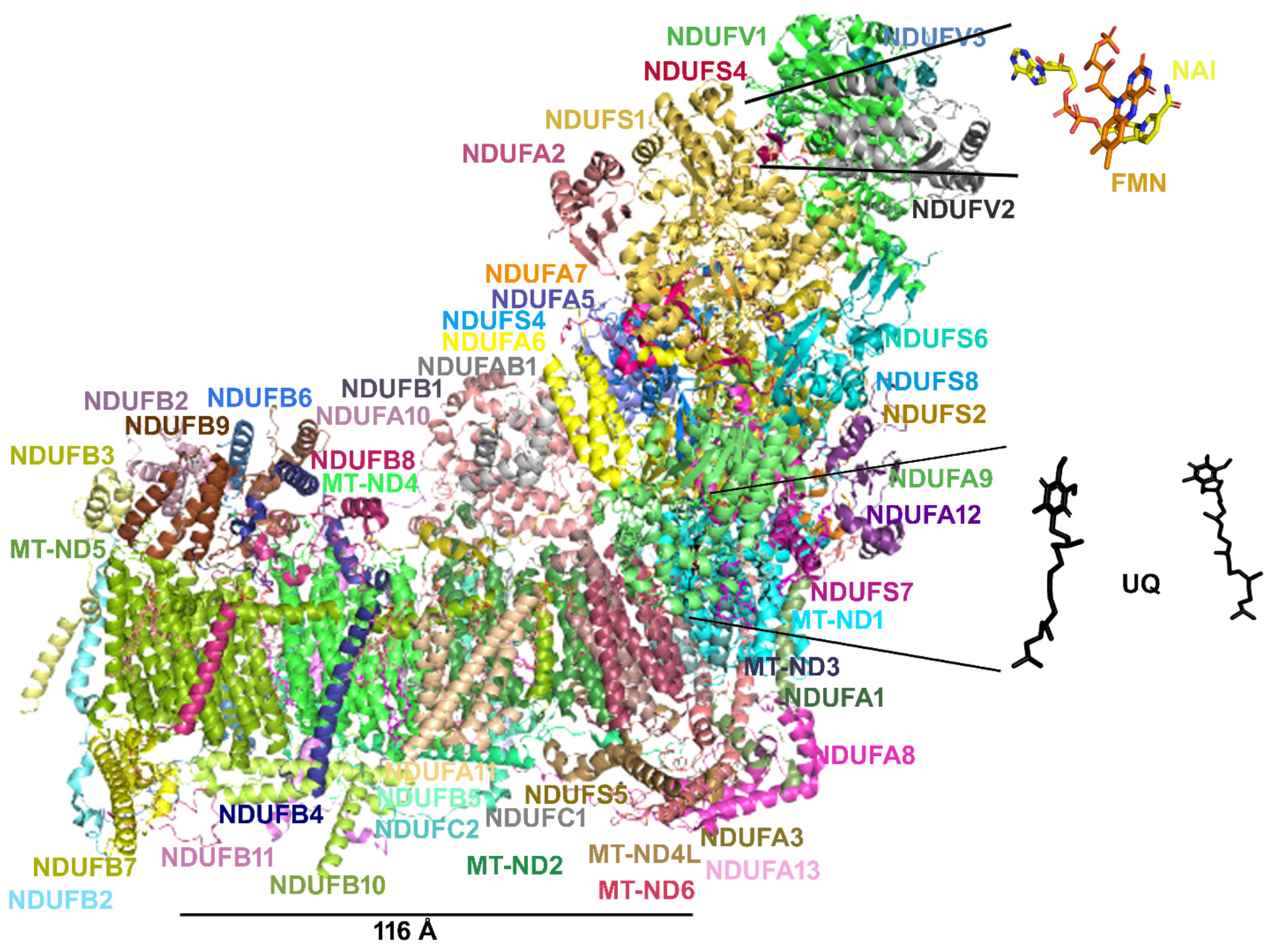
6. The Need for a Structural Analysis for Evaluating the Real Need for Vitamin Supplementation
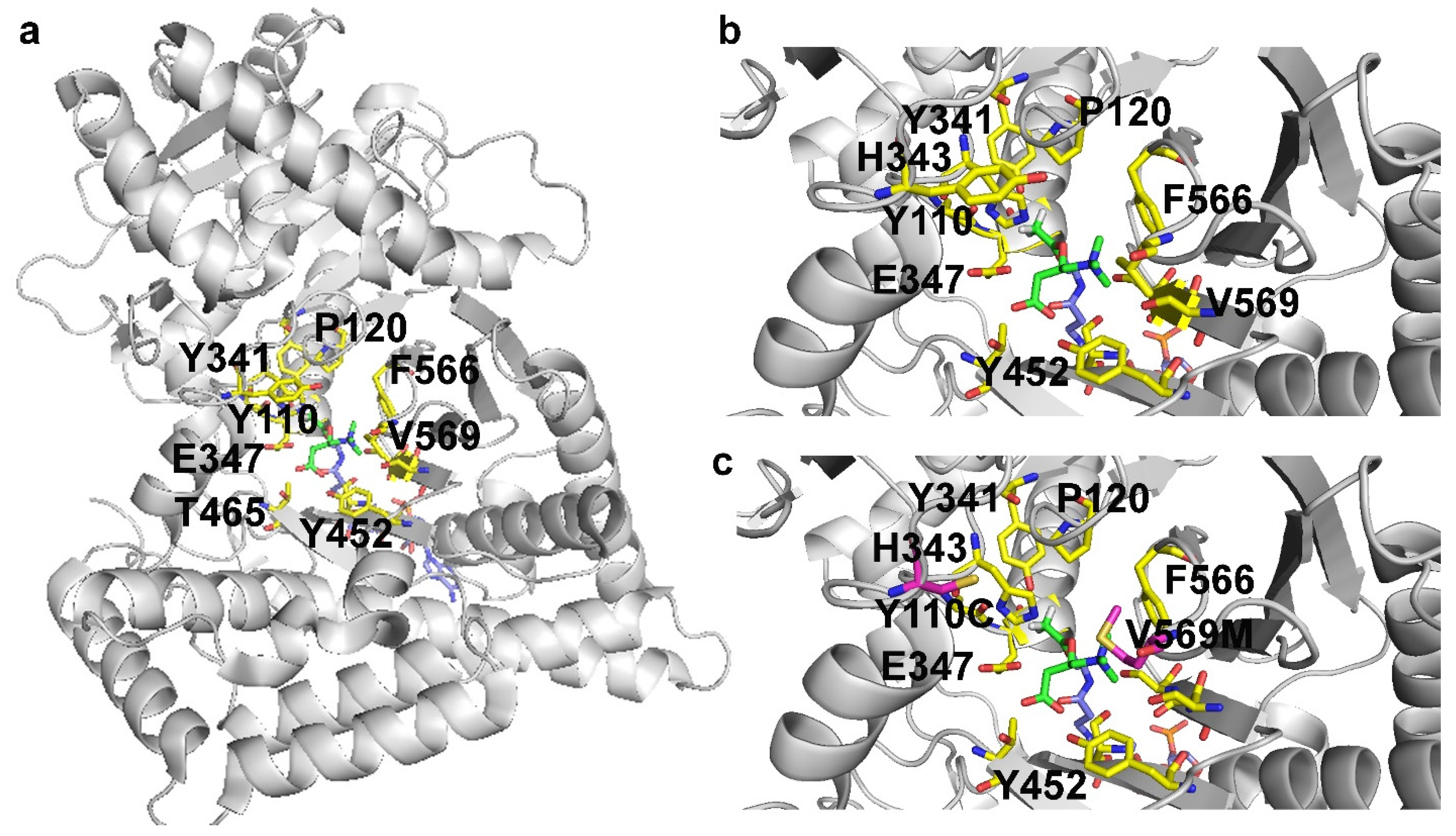
7. Missense Mutations Responsible for Severe Encephalomyopathies: The Case of MT-ND5, NDUFAF6 and SERAC1
8. The Case of Carnitine O-Acetyltransferase (CRAT) Deficiency
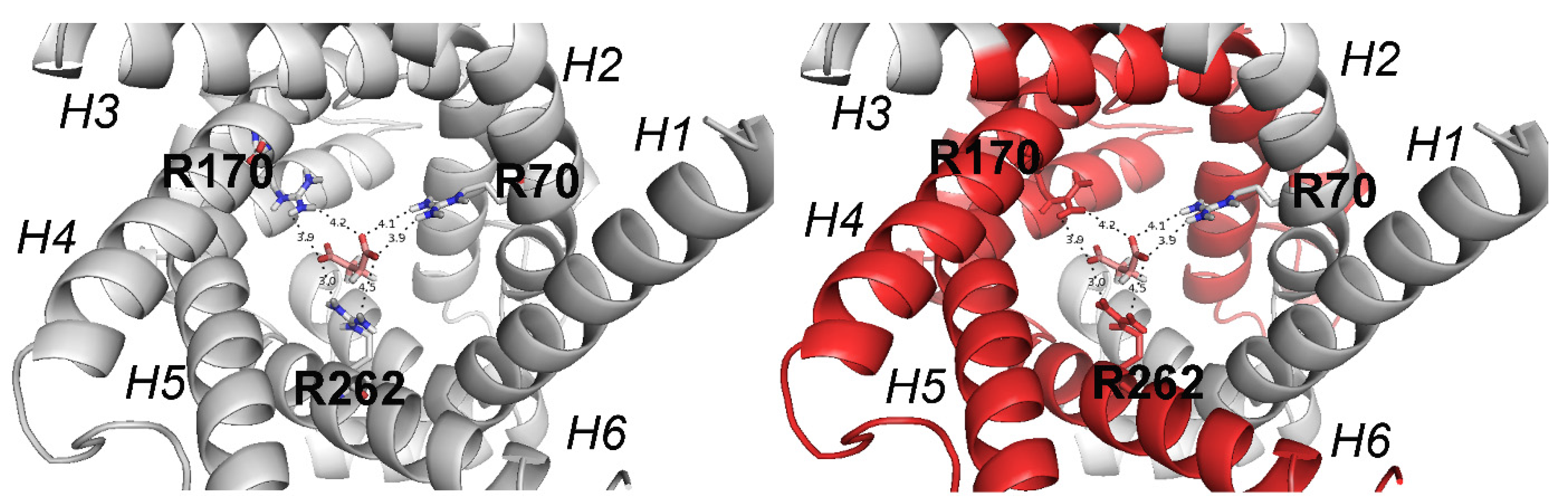
9. SLC25A10 Dicarboxylate Carrier (DIC) Deficiency

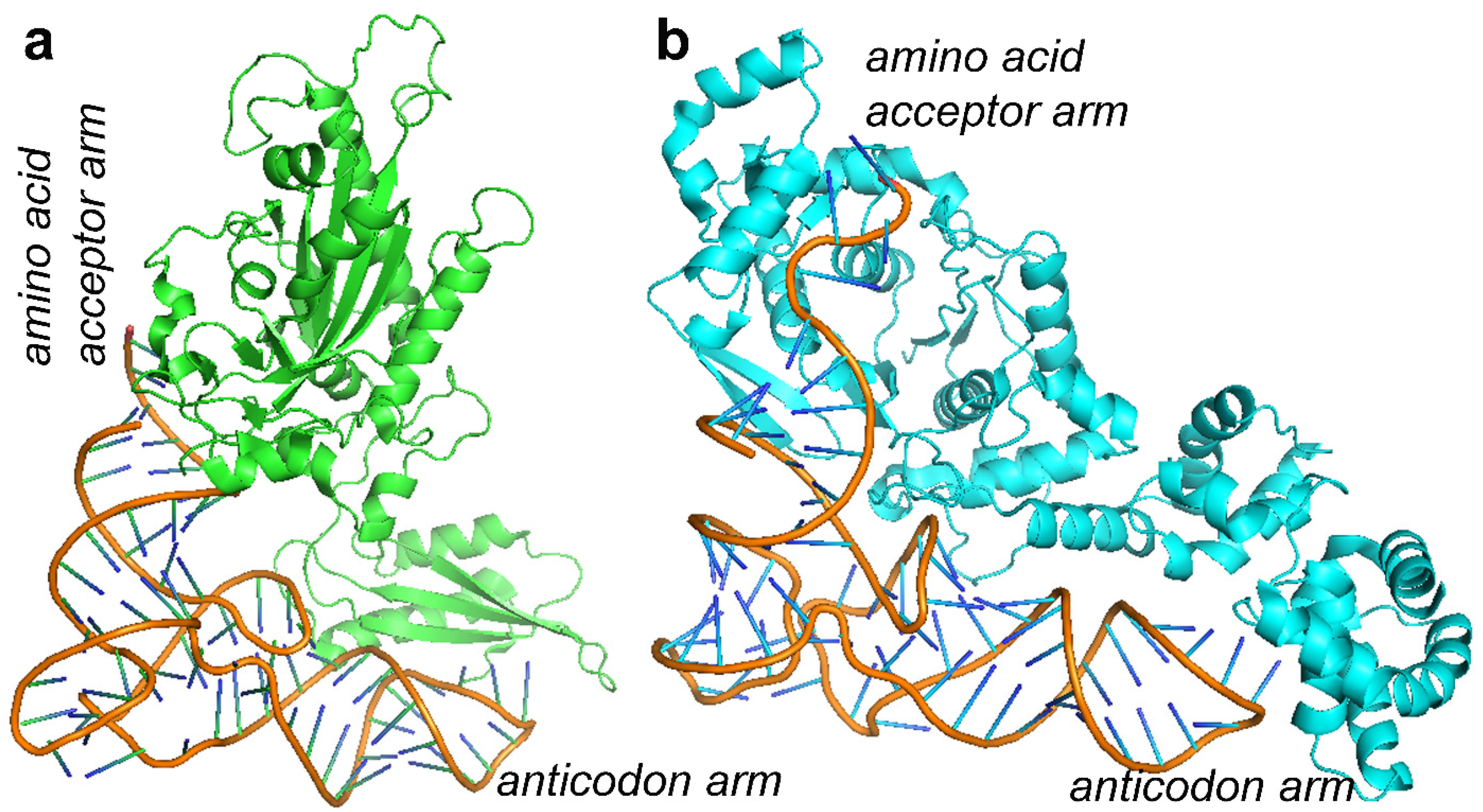
10. MDs Depending on AA-tRNA Mutations or AA-tRNA Synthetase Mutations
11. CoQ Analogs and Mitochondrial Delivery Systems: Organic/Inorganic Chemicals for Stimulating Mitochondrial Activity
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Trisolini, L.; Laera, L.; Favia, M.; Muscella, A.; Castegna, A.; Pesce, V.; Guerra, L.; De Grassi, A.; Volpicella, M.; Pierri, C.L. Differential Expression of ADP/ATP Carriers as a Biomarker of Metabolic Remodeling and Survival in Kidney Cancers. Biomolecules 2020, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccone, C.; Lanave, C.; De Grassi, A. Metazoan OXPHOS gene families: Evolutionary forces at the level of mitochondrial and nuclear genomes. Biochim. Biophys. Acta 2006, 1757, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Grassi, A.; Lanave, C.; Saccone, C. Evolution of ATP synthase subunit c and cytochrome c gene families in selected Metazoan classes. Gene 2006, 371, 224–233. [Google Scholar] [CrossRef]
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef]
- Hughes, K.T. Non-coding. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; p. 83. ISBN 978-0-08-096156-9. [Google Scholar]
- Di Mauro, S. Mitochondrial encephalomyopathies: Where next? Acta Myol. 1996, 19, 489–503. [Google Scholar]
- Di Mauro, S.; Servidei, S.; Zeviani, M.; DiRocco, M.; DeVivo, D.C.; DiDonato, S.; Uziel, G.; Berry, K.; Hoganson, G.; Johnsen, S.D.; et al. Cytochromec oxidase deficiency in leigh syndrome. Ann. Neurol. 1987, 22, 498–506. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 2013, 80, 2049–2054. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef]
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L.; et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum. Mol. Genet. 2017, 27, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Shi, H.; Li, X.; Sun, D.; Li, F.; Li, B.; Ding, Y.; Ma, Y.; Liu, Y.; Zhang, Y.; et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Sci. Rep. 2015, 5, 10480. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Folmes, C.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef]
- Wang, S.-B.; Weng, W.-C.; Lee, N.-C.; Hwu, W.-L.; Fan, P.-C.; Lee, W.-T. Mutation of Mitochondrial DNA G13513A Presenting with Leigh Syndrome, Wolff-Parkinson-White Syndrome and Cardiomyopathy. Pediatr. Neonatol. 2008, 49, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Ardissone, A.; Bruno, C.; Diodato, D.; Donati, A.; Ghezzi, D.; Lamantea, E.; Lamperti, C.; Mancuso, M.; Martinelli, D.; Primiano, G.; et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: A study from the Italian network of mitochondrial diseases. Orphanet J. Rare Dis. 2021, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.Z.; Ng, Y.S.; Blain, A.; Jiminez-Moreno, C.; Alston, C.L.; Nesbitt, V.; Simmons, L.; Santra, S.; Wassmer, E.; Blakely, E.L.; et al. Natural History of Leigh Syndrome: A Study of Disease Burden and Progression. Ann. Neurol. 2021, 91, 117–130. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Liou, C.-W.; Chen, S.-D.; Wang, P.-W.; Chuang, J.-H.; Tiao, M.-M.; Hsu, T.-Y.; Lin, H.-Y.; Lin, T.-K. Mitochondrial Transfer from Wharton’s Jelly Mesenchymal Stem Cell to MERRF Cybrid Reduces Oxidative Stress and Improves Mitochondrial Bioenergetics. Oxidative Med. Cell. Longev. 2017, 2017, 1–22. [Google Scholar] [CrossRef]
- Fornuskova, D.; Brantova, O.; Tesarova, M.; Stiburek, L.; Honzik, T.; Wenchich, L.; Tietzeova, E.; Hansikova, H.; Zeman, J. The impact of mitochondrial tRNA mutations on the amount of ATP synthase differs in the brain compared to other tissues. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2008, 1782, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Laera, L.; Punzi, G.; Porcelli, V.; Gambacorta, N.; Trisolini, L.; Pierri, C.L.; De Grassi, A. CRAT missense variants cause abnormal carnitine acetyltransferase function in an early-onset case of Leigh syndrome. Hum. Mutat. 2019, 41, 110–114. [Google Scholar] [CrossRef]
- Baide-Mairena, H.; Gaudó, P.; Marti-Sánchez, L.; Emperador, S.; Sánchez-Montanez, A.; Alonso-Luengo, O.; Correa, M.; Grau, A.M.; Ortigoza-Escobar, J.D.; Artuch, R.; et al. Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood. Mol. Genet. Metab. 2019, 126, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianciardi, L.; Imperatore, V.; Fernandez-Vizarra, E.; Lopomo, A.; Falabella, M.; Furini, S.; Galluzzi, P.; Grosso, S.; Zeviani, M.; Renieri, A.; et al. Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a Leigh gene. Mol. Genet. Metab. 2016, 119, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Itkis, Y.; Krylova, T.; Pechatnikova, N.L.; De Grassi, A.; Tabakov, V.Y.; Pierri, C.L.; Aleshin, V.; Boyko, A.; Bunik, V.I.; Zakharova, E.Y. A novel variant m.641A>T in the mitochondrial MT-TF gene is associated with epileptic encephalopathy in adolescent. Mitochondrion 2019, 47, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Richter, U.; McFarland, R.; Taylor, R.W.; Pickett, S.J. The molecular pathology of pathogenic mitochondrial tRNA variants. FEBS Lett. 2021, 595, 1003–1024. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A.; Fiorini, A.C.; Scorza, C.A. MEGDEL Syndrome. Pediatr. Neurol. 2020, 110, 25–29. [Google Scholar] [CrossRef]
- Catteruccia, M.; Sauchelli, D.; Della Marca, G.; Primiano, G.; Cuccagna, C.; Bernardo, D.; Leo, M.; Camporeale, A.; Sanna, T.; Cianfoni, A.; et al. “Myo-cardiomyopathy” is commonly associated with the A8344G “MERRF” mutation. J. Neurol. 2015, 262, 701–710. [Google Scholar] [CrossRef]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochim. Biophys. Acta BBA-Bioenerg. 2015, 1847, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Ng, Y.S.; A Bindoff, L.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef]
- Jeppesen, T.D. Aerobic Exercise Training in Patients With mtDNA-Related Mitochondrial Myopathy. Front. Physiol. 2020, 11, 349. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Blain, A.; Jimenez-Moreno, C.; Errington, L.; Ng, Y.S.; McFarland, R.; Turnbull, D.M.; Newman, J.; Gorman, G.S. Measuring the effects of exercise in neuromuscular disorders: A systematic review and meta-analyses. SSRN Electron. J. 2020, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Hamedani, A.G.; Gold, D.R. Eyelid Dysfunction in Neurodegenerative, Neurogenetic, and Neurometabolic Disease. Front. Neurol. 2017, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Primiano, G.; Servidei, S. A severe linezolid-induced rhabdomyolysis and lactic acidosis in Leigh syndrome. J. Inherit. Metab. Dis. 2020, 44, 4–5. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.C.; Brown, D.A.; Allen, M.E.; Bindoff, L.; Gorman, G.S.; Karaa, A.; Keshavan, N.; Lamperti, C.; McFarland, R.; Ng, Y.S.; et al. Safety of drug use in patients with a primary mitochondrial disease: An international Delphi-based consensus. J. Inherit. Metab. Dis. 2020, 43, 800–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2013, 2, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Kuszak, A.; Espey, M.G.; Falk, M.J.; Holmbeck, M.A.; Manfredi, G.; Shadel, G.S.; Vernon, H.J.; Zolkipli-Cunningham, Z. Nutritional Interventions for Mitochondrial OXPHOS Deficiencies: Mechanisms and Model Systems. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 163–191. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2019, 26, 40–57. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Sriram, K.; Manzanares, W.; Joseph, K. Thiamine in nutrition therapy. Nutr. Clin. Pract. 2012, 27, 41–50. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-nucleotide evolution quantifies the importance of each site along the structure of mitochondrial carriers. Cell. Mol. Life Sci. 2013, 71, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic Routes in Inflammation: The Citrate Pathway and its Potential as Therapeutic Target. Curr. Med. Chem. 2020, 26, 7104–7116. [Google Scholar] [CrossRef] [PubMed]
- Santarsiero, A.; Convertini, P.; Todisco, S.; Pierri, C.L.; De Grassi, A.; Williams, N.C.; Iacobazzi, D.; De Stefano, G.; O’Neill, L.A.J.; Infantino, V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells 2021, 10, 2962. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Spera, I.; Menga, A.; Infantino, V.; Porcelli, V.; Iacobazzi, V.; Pierri, C.L.; Hooper, D.C.; Palmieri, F.; Castegna, A. Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim. Et Biophys. Acta 2015, 1847, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardo, A.; DiMauro, S.; Hirano, M. A Diagnostic Algorithm for Metabolic Myopathies. Curr. Neurol. Neurosci. Rep. 2010, 10, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Nakagawa, M.; Higuchi, I.; Osame, M.; Naito, E.; Oizumi, K. Mitochondrial myopathy and familial thiamine deficiency. Muscle Nerve 2000, 23, 1069–1075. [Google Scholar] [CrossRef]
- Mermigkis, C.; Bouloukaki, I.; Mastorodemos, V.; Plaitakis, A.; Alogdianakis, V.; Siafakas, N.; Schiza, S. Medical treatment with thiamine, coenzyme Q, vitamins E and C, and carnitine improved obstructive sleep apnea in an adult case of Leigh disease. Sleep Breath. 2013, 17, 1129–1135. [Google Scholar] [CrossRef]
- Zhang, X.; Li, D.; Lyu, N.; Yang, J.; Yang, C.; Zhang, X.; Ma, W.; Li, D. Clinical and molecular genetic analysis of a case of MEGDEL syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2021, 38, 271–274. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Fiermonte, G.; Song, S.; Struys, E.; De Leonardis, F.; Schwartzberg, P.L.; Chen, A.; Castegna, A.; Verhoeven, N.; Mathews, C.K.; et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc. Natl. Acad. Sci. USA 2006, 103, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Alfadhel, M.; Al-Thihli, K.; Moubayed, H.; Eyaid, W.; Al-Jeraisy, M. Drug treatment of inborn errors of metabolism: A systematic review. Arch. Dis. Child. 2013, 98, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Dueñas, B.; Serrano, M.; Rebollo, M.; Muchart, J.; Gargallo, E.; Dupuits, C.; Artuch, R. Reversible Lactic Acidosis in a Newborn with Thiamine Transporter-2 Deficiency. Pediatrics 2013, 131, e1670–e1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trisolini, L.; Gambacorta, N.; Gorgoglione, R.; Montaruli, M.; Laera, L.; Colella, F.; Volpicella, M.; De Grassi, A.; Pierri, C.L. FAD/NADH Dependent Oxidoreductases: From Different Amino Acid Sequences to Similar Protein Shapes for Playing an Ancient Function. J. Clin. Med. 2019, 8, 2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrozzo, R.; Torraco, A.; Fiermonte, G.; Martinelli, D.; Di Nottia, M.; Rizza, T.; Vozza, A.; Verrigni, D.; Diodato, D.; Parisi, G.; et al. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. The chaperon-like effect of vitamin B2. Mitochondrion 2014, 18, 49–57. [Google Scholar] [CrossRef]
- Leone, P.; Galluccio, M.; Quarta, S.; Anoz-Carbonell, E.; Medina, M.; Indiveri, C.; Barile, M. Mutation of Aspartate 238 in FAD Synthase Isoform 6 Increases the Specific Activity by Weakening the FAD Binding. Int. J. Mol. Sci. 2019, 20, 6203. [Google Scholar] [CrossRef] [Green Version]
- Giancaspero, T.A.; Wait, R.; Boles, E.; Barile, M. Succinate dehydrogenase flavoprotein subunit expression in Saccharomyces cerevisiae- involvement of the mitochondrial FAD transporter, Flx1p. FEBS J. 2008, 275, 1103–1117. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef]
- Russell, A.P.; Schrauwen, P.; Somm, E.; Gastaldi, G.; Hesselink, M.K.C.; Schaart, G.; Kornips, E.; Lo, S.K.; Bufano, D.; Giacobino, J.-P.; et al. Decreased Fatty Acid β-Oxidation in Riboflavin-Responsive, Multiple Acylcoenzyme A Dehydrogenase-Deficient Patients Is Associated with an Increase in Uncoupling Protein-3. J. Clin. Endocrinol. Metab. 2003, 88, 5921–5926. [Google Scholar] [CrossRef] [Green Version]
- Sigoillot, F.D.; Berkowski, J.A.; Sigoillot, S.M.; Kotsis, D.H.; Guy, H.I. Cell Cycle-dependent Regulation of Pyrimidine Biosynthesis. J. Biol. Chem. 2003, 278, 3403–3409. [Google Scholar] [CrossRef] [Green Version]
- Bugiani, M.; Lamantea, E.; Invernizzi, F.; Moroni, I.; Bizzi, A.; Zeviani, M.; Uziel, G. Effects of riboflavin in children with complex II deficiency. Brain Dev. 2006, 28, 576–581. [Google Scholar] [CrossRef]
- Bernsen, P.; Gabreëls, F.; Ruitenbeek, W.; Hamburger, H. Treatment of complex I deficiency with riboflavin. J. Neurol. Sci. 1993, 118, 181–187. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Macchione, F.; Salviati, L.; Bordugo, A.; Vincenzi, M.; Camilot, M.; Teofoli, F.; Pancheri, E.; Zordan, R.; Bertolin, C.; Rossi, S.; et al. Multiple acyl-COA dehydrogenase deficiency in elderly carriers. J. Neurol. 2020, 267, 1414–1419. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 2016, 1863, 2394–2412. [Google Scholar] [CrossRef]
- Wibom, R.; Lasorsa, F.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Parisi, G.; Martinelli, D.; De Leonardis, F.; Torre, G.; Pierri, C.L.; Saccari, A.; Lasorsa, F.M.; Vozza, A.; Palmieri, F.; et al. A new Caucasian case of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD): A clinical, molecular, and functional study. Mol. Genet. Metab. 2011, 104, 501–506. [Google Scholar] [CrossRef]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef]
- Kory, N.; de Bos, J.U.; van der Rijt, S.; Jankovic, N.; Güra, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, eabe5310. [Google Scholar] [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gürtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Eller, J.M.; Lu, M.-J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Di Noia, M.A.; Onofrio, A.; Parisi, G.; Punzi, G.; Redavid, G.; De Grassi, A.; Pierri, C.L. Identification of new highly selective inhibitors of the human ADP/ATP carriers by molecular docking and in vitro transport assays. Biochem. Pharmacol. 2015, 100, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Giudetti, A.M. Dietary long-chain unsaturated fatty acids acutely and differently reduce the activities of lipogenic enzymes and of citrate carrier in rat liver. J. Physiol. Biochem. 2016, 72, 485–494. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Beretti, F.; Carnevale, G.; De Biasi, S.; Pierri, C.L.; Cossarizza, A. Sirtuin 3 interacts with Lon protease and regulates its acetylation state. Biochim. Et Biophys. Acta 2014, 1837, e75. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Kuula, J.; Heinonen, U.; Schmidt, M.S.; et al. Niacin Cures Systemic NAD+ Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 31, 1078–1090.e5. [Google Scholar] [CrossRef]
- Earthman, T.P.; Odom, L.; Mullins, C.A. Lactic Acidosis Associated with High-Dose Niacin Therapy. S. Med. J. 1991, 84, 496–497. [Google Scholar] [CrossRef]
- Traber, M.G.; Vitamin, E. Pantothenic Acid. In Present Knowledge in Nutrition, 10th ed.; Erdman, J.W., Jr., Macdonald, I.A., Zeisel, S.H., Eds.; ILSI Press: Washington, DC, USA, 2012; pp. 214–229. ISBN 978-0-470-95917-6. [Google Scholar]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef]
- Drouin, G.; Godin, J.-R.; Page, B. The Genetics of Vitamin C Loss in Vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef]
- Whittaker, J.W. Intracellular trafficking of the pyridoxal cofactor. Implications for health and metabolic disease. Arch. Biochem. Biophys. 2015, 592, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, M. The mitochondrial cocktail: Rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv. Drug Deliv. Rev. 2008, 60, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.M.; Frost, B.; Scherzer, C.; Feany, M.B. Biotin rescues mitochondrial dysfunction and neurotoxicity in a tauopathy model. Proc. Natl. Acad. Sci. USA 2020, 117, 33608–33618. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Newberry, J.; Erlitzki, R.; Schultz, C.S.; Ames, B.N. Biotin Deficiency Inhibits Heme Synthesis and Impairs Mitochondria in Human Lung Fibroblasts. J. Nutr. 2007, 137, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distelmaier, F.; Haack, T.B.; Wortmann, S.B.; Mayr, J.; Prokisch, H. Treatable mitochondrial diseases: Cofactor metabolism and beyond. Brain 2016, 140, e11. [Google Scholar] [CrossRef]
- Battini, R.; Olivieri, G.; Milone, R.; Mazio, F.; Scalise, R.; Verdolotti, T.; Primiano, G.; Genovese, O.; Mercuri, E.; Servidei, S. Spinal cord demyelination in children: A diagnostic challenge in neuropaediatrics for a good outcome. Brain Dev. 2020, 42, 457–461. [Google Scholar] [CrossRef]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef]
- Florit-Sureda, M.; Conde-Estévez, D.; Vidal, J.; Montagut, C. Hypersensitivity reaction caused by folinic acid administration: A case report and literature review. J. Chemother. 2016, 28, 500–505. [Google Scholar] [CrossRef]
- Green, R.; Miller, J.W. Chapter Fifteen—Vitamin B12 deficiency. In Vitamin B12; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 119, pp. 405–439. ISBN 0083-6729. [Google Scholar]
- Kc, S.; Càrcamo, J.M.; Golde, D.W. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Gluti) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of Vitamin C Transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Jacob, R.A.; Sotoudeh, G. Vitamin C Function and Status in Chronic Disease. Nutr. Clin. Care 2002, 5, 66–74. [Google Scholar] [CrossRef]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 2012, CD004426. [Google Scholar] [CrossRef] [PubMed]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta BBA-Gen. Subj. 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.M.; Pinault, D.; O’Brien, T.J.; Jones, N.C.; Andreasen, N.; Flaum, M.; Angeles, L.; Van Horn, J.D.; Balla, A.; Nattini, M.E.; et al. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Schizophr. Res. 2013, 45, 107–116. [Google Scholar] [CrossRef]
- Chiu, H.; Fischman, D.A.; Hammerling, U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB J. 2008, 22, 3878–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenkin, A. Dietary reference values for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium and zinc. J. Hum. Nutr. Diet. 2003, 16, 199–200. [Google Scholar] [CrossRef]
- de Oliveira, M.R. Vitamin A and Retinoids as Mitochondrial Toxicants. Oxidative Med. Cell. Longev. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Prosser, D.E.; Kaufmann, M. The Activating Enzymes of Vitamin D Metabolism (25- and 1α-Hydroxylases). In Vitamin D, 4th ed.; Feldman, D., Pike, W.J., Bouillon, R., Giovannucci, E., Goltzman, D., Hewison, M., Eds.; Academic Press: Cambridge, MA, USA, 2018; ISBN 9780128099667. [Google Scholar]
- Denlinger, L.C.; King, T.S.; Cardet, J.C.; Craig, T.; Holguin, F.; Jackson, D.J.; Kraft, M.; Peters, S.P.; Ross, K.; Sumino, K.; et al. Vitamin D Supplementation and the Risk of Colds in Patients with Asthma. Am. J. Respir. Crit. Care Med. 2016, 193, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Boğa, A. The Effect Of Vıtamın D Levels On The Frequency Of Upper Respıratory Infectıon In Chıldren. Haydarpasa Numune Train. Res. Hosp. Med. J. 2022, 62, 93–96. [Google Scholar] [CrossRef]
- Lauridsen, C.; Jensen, S.K. α-Tocopherol incorporation in mitochondria and microsomes upon supranutritional vitamin E supplementation. Genes Nutr. 2012, 7, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Majima, H.J.; Indo, H.P.; Suenaga, S.; Matsui, H.; Yen, H.-C.; Ozawa, T. Mitochondria as Possible Pharmaceutical Targets for the Effects of Vitamin E and its Homologues in Oxidative Stress-Related Diseases. Curr. Pharm. Des. 2011, 17, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Weisskopf, M.G.; O’Reilly, E.J.; Jacobs, E.J.; McCullough, M.L.; Calle, E.E.; Cudkowicz, M.; Thun, M.J. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann. Neurol. 2004, 57, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Carnevale, R.; Cangemi, R.; Saliola, M.; Nocella, C.; Bartimoccia, S.; Vicario, T.; Farcomeni, A.; Violi, F.; Pignatelli, P. Vitamin E Serum Levels and Bleeding Risk in Patients Receiving Oral Anticoagulant Therapy: A Retrospective Cohort Study. J. Am. Hear. Assoc. 2013, 2, e000364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, A.; Hennessy, Á.; Walton, J.; A McNulty, B.; Lucey, A.; Kiely, M.; Flynn, A.; Cashman, K.D. Phylloquinone Intakes and Food Sources and Vitamin K Status in a Nationally Representative Sample of Irish Adults. J. Nutr. 2016, 146, 2274–2280. [Google Scholar] [CrossRef]
- Booth, S.L. Vitamin K: Food composition and dietary intakes. Food Nutr. Res. 2012, 56, 5505. [Google Scholar] [CrossRef] [Green Version]
- Mladěnka, P.; Macáková, K.; Kujovská Krčmová, L.; Javorská, L.; Mrštná, K.; Carazo, A.; Protti, M.; Remião, F.; Nováková, L. Vitamin K—Sources, physiological role, kinetics, deficiency, detection, therapeutic use, and toxicity. Nutr. Rev. 2021, 80, 677–698. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Getsov, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018, 16, 352–358. [Google Scholar] [CrossRef]
- Prasuhn, J.; Kasten, M.; Vos, M.; König, I.R.; Schmid, S.M.; Wilms, B.; Klein, C.; Brüggemann, N. The Use of Vitamin K2 in Patients with Parkinson’s Disease and Mitochondrial Dysfunction (PD-K2): A Theranostic Pilot Study in a Placebo-Controlled Parallel Group Design. Front. Neurol. 2021, 11, 592104. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, C.; Algieri, C.; La Mantia, D.; Trombetti, F.; Pagliarani, A.; Forni, M.; Nesci, S. Vitamin K Vitamers Differently Affect Energy Metabolism in IPEC-J2 Cells. Front. Mol. Biosci. 2021, 8, 682191. [Google Scholar] [CrossRef]
- Cerqua, C.; Casarin, A.; Pierrel, F.; Fonseca, L.V.; Viola, G.; Salviati, L.; Trevisson, E. Vitamin K2 cannot substitute Coenzyme Q10 as electron carrier in the mitochondrial respiratory chain of mammalian cells. Sci. Rep. 2019, 9, 6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.H. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion 2007, 7, S136–S145. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Hirano, M.; Schon, E.A. Approaches to the treatment of mitochondrial diseases. Muscle Nerve 2006, 34, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Misevičienė, L.; Anusevičius, Ž.; Šarlauskas, J.; Sevrioukova, I.F.; Čėnas, N. Redox reactions of the FAD-containing apoptosis-inducing factor (AIF) with quinoidal xenobiotics: A mechanistic study. Arch. Biochem. Biophys. 2011, 512, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.S.; Calisto, F.; Langer, J.D.; Mills, D.J.; Refojo, P.N.; Teixeira, M.; Kühlbrandt, W.; Vonck, J.; Pereira, M.M. Structural basis for energy transduction by respiratory alternative complex III. Nat. Commun. 2018, 9, 1728. [Google Scholar] [CrossRef]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Leigh and Leigh-Like Syndrome in Children and Adults. Pediatr. Neurol. 2008, 39, 223–235. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with Nephropathy and CoQ10 Deficiency Due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme q10 analogues: Benefits and challenges for therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- de Barcelos, I.P.; Emmanuele, V.; Hirano, M. Advances in primary mitochondrial myopathies. Curr. Opin. Neurol. 2019, 32, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Marriage, B.; Clandinin, M.; Glerum, D. Nutritional cofactor treatment in mitochondrial disorders. J. Am. Diet. Assoc. 2003, 103, 1029–1038. [Google Scholar] [CrossRef]
- Shults, C.W.; Beal, M.F.; Song, D.; Fontaine, D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson’s disease. Exp. Neurol. 2004, 188, 491–494. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007, 7, S78–S88. [Google Scholar] [CrossRef]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Asp. Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Tein, I. Carnitine transport: Pathophysiology and metabolism of known molecular defects. J. Inherit. Metab. Dis. 2003, 26, 147–169. [Google Scholar] [CrossRef]
- Nicassio, L.; Fracasso, F.; Sirago, G.; Musicco, C.; Picca, A.; Marzetti, E.; Calvani, R.; Cantatore, P.; Gadaleta, M.N.; Pesce, V. Dietary supplementation with acetyl-l-carnitine counteracts age-related alterations of mitochondrial biogenesis, dynamics and antioxidant defenses in brain of old rats. Exp. Gerontol. 2017, 98, 99–109. [Google Scholar] [CrossRef]
- Pesce, V.; Fracasso, F.; Cassano, P.; Lezza, A.M.S.; Cantatore, P.; Gadaleta, M.N. Acetyl-l-Carnitine Supplementation to Old Rats Partially Reverts the Age-Related Mitochondrial Decay of Soleus Muscle by Activating Peroxisome Proliferator-Activated Receptor γ Coactivator-1α–Dependent Mitochondrial Biogenesis. Rejuvenation Res. 2010, 13, 148–151. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, R.; Fukui, F.; Waki, H.; Kawabata, T.; Okazaki, M.; Hasegawa, K.; Ando, S. Acetyl-l-carnitine supplementation restores decreased tissue carnitine levels and impaired lipid metabolism in aged rats. J. Lipid Res. 2004, 45, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnoni, A.; Longo, S.; Gnoni, G.V.; Giudetti, A.M. Carnitine in human muscle bioenergetics: Can carnitine supplementation improve physical exercise? Molecules 2020, 25, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marriage, B.J.; Clandinin, M.; Macdonald, I.M.; Glerum, D. Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol. Genet. Metab. 2004, 81, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cocco, T.M.; Di, M.; Papa, P.; Lorusso, M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic. Biol. Med. 1999, 27, 51–59. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Nohl, H.; Stolze, K. Ubisemiquinones of the Mitochondrial Respiratory Chain Do not Interact with Molecular Oxygen. Free Radic. Res. Commun. 1992, 16, 409–419. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxidative Med. Cell. Longev. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Signorile, A.; Ferretta, A.; Ruggieri, M.; Paolicelli, D.; Lattanzio, P.; Trojano, M.; De Rasmo, D. Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, A Possible Role of Nutraceutics. Antioxidants 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Bobba, A.; Calissano, P.; Passarella, S.; Marra, E. The apoptosis/necrosis transition in cerebellar granule cells depends on the mutual relationship of the antioxidant and the proteolytic systems which regulate ROS production and cytochrome c release en route to death. J. Neurochem. 2003, 84, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, M. Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musatov, A.; Sedlák, E. Role of cardiolipin in stability of integral membrane proteins. Biochimie 2017, 142, 102–111. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef]
- Salehi, B.; Martorell, M.; Arbiser, J.L.; Sureda, A.; Martins, N.; Maurya, P.K.; Sharifi-Rad, M.; Kumar, P.; Sharifi-Rad, J. Antioxidants: Positive or negative actors? Biomolecules 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Liu, T.; Guo, R.; Zhang, L.; Yang, M. The coupling mechanism of mammalian mitochondrial complex I. Nat. Struct. Mol. Biol. 2022, 29, 172–182. [Google Scholar] [CrossRef]
- Corona, P.; Antozzi, C.; Carrara, F.; D’Incerti, L.; Lamantea, E.; Tiranti, V.; Zeviani, M. A novel mtDNA mutation in the ND5 subunit of complex I in two MELAS patients. Ann. Neurol. 2001, 49, 106–110. [Google Scholar] [CrossRef]
- Papa, S.; Petruzzella, V.; Scacco, S.; Sardanelli, A.M.; Iuso, A.; Panelli, D.; Vitale, R.; Trentadue, R.; De Rasmo, D.; Capitanio, N.; et al. Pathogenetic mechanisms in hereditary dysfunctions of complex I of the respiratory chain in neurological diseases. Biochim. Biophys. Acta 2009, 1787, 502–517. [Google Scholar] [CrossRef] [Green Version]
- Naini, A.B.; Lu, J.; Kaufmann, P.; Bernstein, R.A.; Mancuso, M.; Bonilla, E.; Hirano, M.; DiMauro, S. Novel Mitochondrial DNA ND5 Mutation in a Patient with Clinical Features of MELAS and MERRF. Arch. Neurol. 2005, 62, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Pierri, C.L.; De Grassi, A.; Turi, A. Lattices for ab initio protein structure prediction. Proteins: Struct. Funct. Bioinform. 2008, 73, 351–361. [Google Scholar] [CrossRef]
- Onofrio, A.; Parisi, G.; Punzi, G.; Todisco, S.; Di Noia, M.A.; Bossis, F.; Turi, A.; De Grassi, A.; Pierri, C.L. Distance-dependent hydrophobic–hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys. 2014, 16, 18907–18917. [Google Scholar] [CrossRef]
- Devi, A.R.R.; Lingappa, L. Novel mutations in SERAC1 gene in two Indian patients presenting with dystonia and intellectual disability. Eur. J. Med Genet. 2018, 61, 100–103. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Vaz, F.M.; Gardeitchik, T.; Vissers, L.E.L.M.; Renkema, G.H.; Schuurs-Hoeijmakers, J.H.M.; Kulik, W.; Lammens, M.; Christin, C.; Kluijtmans, L.A.J.; et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012, 44, 797–802. [Google Scholar] [CrossRef]
- Catania, A.; Ardissone, A.; Verrigni, D.; Legati, A.; Reyes, A.; Lamantea, E.; Diodato, D.; Tonduti, D.; Imperatore, V.; Pinto, A.M.; et al. Compound heterozygous missense and deep intronic variants in NDUFAF6 unraveled by exome sequencing and mRNA analysis. J. Hum. Genet. 2018, 63, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassone, E.; Taanman, J.-W.; Hargreaves, I.P.; Sebire, N.J.; Cleary, M.A.; Burch, M.; Rahman, S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J. Med. Genet. 2011, 48, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lu, G.; Qiao, L.; Li, Y. A Leigh syndrome caused by compound heterozygous mutations on NDUFAF5 induce early infant death: A case report. Mol. Genet. Genom. Med. 2021, 10, e1852. [Google Scholar] [CrossRef]
- Chen, W.-W.; Chao, Y.-J.; Chang, W.-H.; Chan, J.-F.; Hsu, Y.-H.H. Phosphatidylglycerol Incorporates into Cardiolipin to Improve Mitochondrial Activity and Inhibits Inflammation. Sci. Rep. 2018, 8, 4919. [Google Scholar] [CrossRef]
- Khalifat, N.; Rahimi, M.; Bitbol, A.-F.; Seigneuret, M.; Fournier, J.-B.; Puff, N.; Arroyo, M.; Angelova, M.I. Interplay of Packing and Flip-flop in Local Bilayer Deformation. How Phosphatidylglycerol Could Rescue Mitochondrial Function in a Cardiolipin-deficient Yeast Mutant. Biophys. J. 2014, 107, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Dabner, L.; E Pieles, G.; Steward, C.G.; Hamilton-Shield, J.P.; Ness, A.R.; A Rogers, C.; Bucciarelli-Ducci, C.; Greenwood, R.; Ellis, L.; Sheehan, K.; et al. Treatment of Barth Syndrome by Cardiolipin Manipulation (CARDIOMAN) with Bezafibrate: Protocol for a Randomized Placebo-Controlled Pilot Trial Conducted in the Nationally Commissioned Barth Syndrome Service. JMIR Res. Protoc. 2021, 10, e22533. [Google Scholar] [CrossRef]
- DiDonato, S.; Rimoldi, M.; Moise, A.; Bertagnoglio, B.; Uziel, G. Fatal ataxic encephalopathy and carnitine acetyltransferase deficiency: A functional defect of pyruvate oxidation? Neurology 1979, 29, 1578–1583. [Google Scholar] [CrossRef]
- Przyrembel, H. Therapy of mitochondrial disorders. J. Inherit. Metab. Dis. 1987, 10, 129–146. [Google Scholar] [CrossRef]
- Seiler, S.E.; Martin, O.J.; Noland, R.C.; Slentz, D.H.; DeBalsi, K.L.; Ilkayeva, O.R.; An, J.; Newgard, C.B.; Koves, T.; Muoio, D.M. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J. Lipid Res. 2014, 55, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Ehinger, J.K.; Piel, S.; Ford, R.; Karlsson, M.; Sjövall, F.; Frostner, E.; Morota, S.; Taylor, R.W.; Turnbull, D.; Cornell, C.; et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016, 7, 12317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunetti, P.; Cappello, A.R.; Marsano, R.M.; Pierri, C.L.; Carrisi, C.; Martello, E.; Caggese, C.; Dolce, V.; Capobianco, L. Mitochondrial glutamate carriers from Drosophila melanogaster: Biochemical, evolutionary and modeling studies. Biochim. Biophys. Acta 2013, 1827, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozza, A.; De Leonardis, F.; Paradies, E.; De Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical characterization of a new mitochondrial transporter of dephosphocoenzyme A in Drosophila melanogaster. Biochim. Biophys. Acta 2016, 1858, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, A.; Pierri, C.L.; Palmieri, F.; Klingenberg, M. The switching mechanism of the mitochondrial ADP/ATP carrier explored by free-energy landscapes. Biochim. Biophys. Acta 2016, 1857, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Tragni, V.; Cotugno, P.; De Grassi, A.; Massari, F.; Di Ronzo, F.; Aresta, A.M.; Zambonin, C.; Sanzani, S.M.; Ippolito, A.; Pierri, C.L. Targeting mitochondrial metabolite transporters in Penicillium expansum for reducing patulin production. Plant Physiol. Biochem. 2020, 158, 158–181. [Google Scholar] [CrossRef]
- Curcio, R.; Muto, L.; Pierri, C.L.; Montalto, A.; Lauria, G.; Onofrio, A.; Fiorillo, M.; Fiermonte, G.; Lunetti, P.; Vozza, A.; et al. New insights about the structural rearrangements required for substrate translocation in the bovine mitochondrial oxoglutarate carrier. Biochim. Et Biophys. Acta BBA Proteins Proteom. 2016, 1864, 1473–1480. [Google Scholar] [CrossRef]
- Biancheri, R.; Lamantea, E.; Severino, M.; Diodato, D.; Pedemonte, M.; Cassandrini, D.; Ploederl, A.; Trucco, F.; Fiorillo, C.; Minetti, C.; et al. Expanding the Clinical and Magnetic Resonance Spectrum of Leukoencephalopathy with Thalamus and Brainstem Involvement and High Lactate (LTBL) in a Patient Harboring a Novel EARS2 Mutation. JIMD Rep. 2015, 23, 85–89. [Google Scholar] [CrossRef]
- Goto, Y. MERRF. Nippon rinsho. Jpn. J. Clin. Med. 2002, 60, 296–297. [Google Scholar]
- Klipcan, L.; Moor, N.; Finarov, I.; Kessler, N.; Sukhanova, M.; Safro, M.G. Crystal Structure of Human Mitochondrial PheRS Complexed with tRNAPhe in the Active “Open” State. J. Mol. Biol. 2012, 415, 527–537. [Google Scholar] [CrossRef]
- Guo, M.; Ignatov, M.; Musier-Forsyth, K.; Schimmel, P.; Yang, X.-L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Hao, Z.; Tian, Q.; Zhang, Z.; Zhou, C.; Xie, W. Cocrystal Structures of Glycyl-tRNA Synthetase in Complex with tRNA Suggest Multiple Conformational States in Glycylation. J. Biol. Chem. 2014, 289, 20359–20369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.-I.; Nureki, O.; Shimada, A.; Vassylyev, D.G.; Yokoyama, S. Structural basis for anticodon recognition by discriminating glutamyl-tRNA synthetase. Nat. Genet. 2001, 8, 203–206. [Google Scholar] [CrossRef]
- Tian, Q.; Wang, C.; Liu, Y.; Xie, W. Structural basis for recognition of G-1-containing tRNA by histidyl-tRNA synthetase. Nucleic Acids Res. 2015, 43, 2980–2990. [Google Scholar] [CrossRef]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II Aminoacyl Transfer Rna Synthetases: Crystal Structure of Yeast Aspartyl-trna Synthetase Complexed with tRNA Asp. Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nureki, O.; Ishitani, R.; Yaremchuk, A.; Tukalo, M.; Cusack, S.; Sakamoto, K.; Yokoyama, S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Mol. Biol. 2003, 10, 425–432. [Google Scholar] [CrossRef]
- Yaremchuk, A.; Kriklivyi, I.; Tukalo, M.; Cusack, S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002, 21, 3829–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, K.M.; Wu, J.; Ling, J.; Simonović, M. The crystal structure of yeast mitochondrial ThrRS in complex with the canonical threonine tRNA. Nucleic Acids Res. 2016, 44, 1428–1439. [Google Scholar] [CrossRef]
- Shen, N.; Guo, L.; Yang, B.; Jin, Y.; Ding, J. Structure of human tryptophanyl-tRNA synthetase in complex with tRNATrp reveals the molecular basis of tRNA recognition and specificity. Nucleic Acids Res. 2006, 34, 3246–3258. [Google Scholar] [CrossRef]
- Wang, C.; Guo, Y.; Tian, Q.; Jia, Q.; Gao, Y.; Zhang, Q.; Zhou, C.; Xie, W. SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res. 2015, 43, 10534–10545. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yoon, I.; Son, J.; Park, J.; Kim, K.; Lee, J.-H.; Park, S.-Y.; Kang, B.S.; Han, J.M.; Hwang, K.Y.; et al. Leucine-sensing mechanism of leucyl-tRNA synthetase 1 for mTORC1 activation. Cell Rep. 2021, 35, 109031. [Google Scholar] [CrossRef]
- Fukai, S.; Nureki, O.; Sekine, S.-I.; Shimada, A.; Tao, J.; Vassylyev, D.G.; Yokoyama, S. Structural Basis for Double-Sieve Discrimination of L-Valine from L-Isoleucine and L-Threonine by the Complex of tRNAVal and Valyl-tRNA Synthetase. Cell 2000, 103, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Yaremchuk, A.; Tukalo, M.; Grøtli, M.; Cusack, S. A succession of substrate induced conformational changes ensures the amino acid specificity of Thermus thermophilus prolyl-tRNA synthetase: Comparison with histidyl-tRNA synthetase. J. Mol. Biol. 2001, 309, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Biou, V.; Yaremchuk, A.; Tukalo, M.; Cusack, S. The 2.9 Å Crystal Structure of T. thermophilus Seryl-tRNA Synthetase Complexed with tRNA Ser. Science 1994, 263, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Dock-Bregeon, A.-C.; Romby, P.; Caillet, J.; Springer, M.; Rees, B.; Ehresmann, C.; Ehresmann, B.; Moras, D. The Structure of Threonyl-tRNA Synthetase-tRNAThr Complex Enlightens Its Repressor Activity and Reveals an Essential Zinc Ion in the Active Site. Cell 1999, 97, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Rould, M.A.; Perona, J.J.; Steitz, T.A. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature 1991, 352, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Cohen, B.H. A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. J. Cachex Sarcopenia Muscle 2020, 11, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Satrialdi; Hibino, M.; Sasaki, D.; Abe, J.; Harashima, H. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv. Drug Deliv. Rev. 2020, 154–155, 187–209. [Google Scholar] [CrossRef]
- Colella, F.; Scillitani, G.; Pierri, C.L. Sweet as honey, bitter as bile: Mitochondriotoxic peptides and other therapeutic proteins isolated from animal tissues, for dealing with mitochondrial apoptosis. Toxicology 2020, 447, 152612. [Google Scholar] [CrossRef]
- Chitwood, P.; Hegde, R.S. An intramembrane chaperone complex facilitates membrane protein biogenesis. Nature 2020, 584, 630–634. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 Peptide Reverses the Mitochondrial Fragmentation Present in Fibroblasts from Patients with DCMA, a Mitochondrial Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Mirabella, M.; Di Giovanni, S.; Silvestri, G.; Tonali, P.; Servidei, S. Apoptosis in mitochondrial encephalomyopathies with mitochondrial DNA mutations: A potential pathogenic mechanism. Brain 2000, 123, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giovanni, S.; Mirabella, M.; D’Amico, A.; Tonali, P.; Servidei, S. Apoptotic features accompany acute quadriplegic myopathy. Neurology 2000, 55, 854–858. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S.; Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [Green Version]
- Chiaretti, A.; Eftimiadi, G.; Buonsenso, D.; Rendeli, C.; Staccioli, S.; Conti, G. Intranasal nerve growth factor administration improves neurological outcome after GBS meningitis. Child’s Nerv. Syst. 2020, 36, 2083–2088. [Google Scholar] [CrossRef]
- Chiaretti, A.; Conti, G.; Falsini, B.; Buonsenso, D.; Crasti, M.; Manni, L.; Soligo, M.; Fantacci, C.; Genovese, O.; Calcagni, M.L.; et al. Intranasal Nerve Growth Factor administration improves cerebral functions in a child with severe traumatic brain injury: A case report. Brain Inj. 2017, 31, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [Green Version]
- Mahata, B.; Mukherjee, S.; Mishra, S.; Bandyopadhyay, A.; Adhya, S. Functional Delivery of a Cytosolic tRNA into Mutant Mitochondria of Human Cells. Science 2006, 314, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. tRNA therapeutics burst onto startup scene. Nat. Biotechnol. 2022, 40, 283–286. [Google Scholar] [CrossRef]
- Templeton, N.S. Liposomal Delivery of Nucleic AcidsIn Vivo. DNA Cell Biol. 2002, 21, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid Delivery Systems for Nucleic-Acid-Based-Drugs: From Production to Clinical Applications. Pharmaceutics 2019, 11, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissig, V.; Boddapati, S.V.; Cheng, S.-M.; D’Souza, G.G.M. Liposomes and Liposome-like Vesicles for Drug and DNA Delivery to Mitochondria. J. Liposome Res. 2006, 16, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Cheng, S.-M.; D’Souza, G.G. Mitochondrial pharmaceutics. Mitochondrion 2004, 3, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Mitochondrial Delivery of Biologically Active Molecules. Pharm. Res. 2011, 28, 2633–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Schneider, A. Mitochondrial tRNA Import and Its Consequences for Mitochondrial Translation. Annu. Rev. Biochem. 2011, 80, 1033–1053. [Google Scholar] [CrossRef] [Green Version]
- Baleva, M.; Gowher, A.; Kamenski, P.; Tarassov, I.; Entelis, N.; Masquida, B. A Moonlighting Human Protein Is Involved in Mitochondrial Import of tRNA. Int. J. Mol. Sci. 2015, 16, 9354–9367. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yashiro, Y.; Kikuchi, I.; Ishigami, Y.; Saito, H.; Matsuzawa, I.; Okada, S.; Mito, M.; Iwasaki, S.; Ma, D.; et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020, 11, 4269. [Google Scholar] [CrossRef]
- Kamenski, P.A.; Krasheninnikov, I.A.; Tarassov, I. 40 Years of Studying RNA Import into Mitochondria: From Basic Mechanisms to Gene Therapy Strategies. Mol. Biol. 2019, 53, 813–819. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Beyrath, J.; Pellegrini, M.; Renkema, H.; Houben, L.; Pecheritsyna, S.; Van Zandvoort, P.; Broek, P.V.D.; Bekel, A.; Eftekhari, P.; Smeitink, J.A.M. KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery. Sci. Rep. 2018, 8, 6577. [Google Scholar] [CrossRef] [PubMed]
- Tiet, M.Y.; Lin, Z.; Gao, F.; Jennings, M.J.; Horvath, R. Targeted Therapies for Leigh Syndrome: Systematic Review and Steps Towards a ‘Treatabolome’. J. Neuromuscul. Dis. 2021, 8, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Diodato, D.; Ricci, D.; Falsini, B.; Iarossi, G.; Olivieri, G.; Ceravolo, F.; Gambardella, M.L.; Ferrantini, G.; Mercuri, E.; et al. Effect of epi-743 on the clinical course of visual damage in Cobalamin c patients: A prospective randomized double-blind phase ii clinical trial. J. Inborn Errors Metab. Screen. In Proceedings of the III Congress Annual SIMMESN, Roma, Italy, 29 November–1 December 2017. [Google Scholar]
- Zesiewicz, T.; Salemi, J.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Cohen, B.H. Clinical Trials in Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef]
- Ronis, M.J.J.; Pedersen, K.B.; Watt, J. Adverse Effects of Nutraceuticals and Dietary Supplements. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 583–601. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Lichtenstein, A.H.; Howard, B.V.; Steinberg, D.; Witztum, J.L. Antioxidant Vitamin Supplements and Cardiovascular Disease. Circulation 2004, 110, 637–641. [Google Scholar] [CrossRef]
- Angebault, C.; Gueguen, N.; Desquiret-Dumas, V.; Chevrollier, A.; Guillet, V.; Verny, C.; Cassereau, J.; Ferre, M.; Milea, D.; Amati-Bonneau, P.; et al. Idebenone increases mitochondrial complex I activity in fibroblasts from LHON patients while producing contradictory effects on respiration. BMC Res. Notes 2011, 4, 557. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Sharpley, M.S.; Manas, A.-R.B.; Frerman, F.E.; Hirst, J.; Smith, R.A.J.; Murphy, M.P. Interaction of the Mitochondria-targeted Antioxidant MitoQ with Phospholipid Bilayers and Ubiquinone Oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Slone, J.; Huang, T. The special considerations of gene therapy for mitochondrial diseases. NPJ Genom. Med. 2020, 5, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, M.; Salman, L.; Sabag, O.; Patel, M.S.; Lorberboum-Galski, H. Successful TAT-mediated enzyme replacement therapy in a mouse model of mitochondrial E3 deficiency. J. Mol. Med. 2010, 89, 161–170. [Google Scholar] [CrossRef]
- van Konijnenburg, E.M.M.H.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D.M. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J. Rare Dis. 2021, 16, 1–35. [Google Scholar] [CrossRef]
- Gustafson, M.A.; Perera, L.; Shi, M.; Copeland, W.C. Mechanisms of SSBP1 variants in mitochondrial disease: Molecular dynamics simulations reveal stable tetramers with altered DNA binding surfaces. DNA Repair 2021, 107, 103212. [Google Scholar] [CrossRef]
- Euro, L.; Haapanen, O.; Róg, T.; Vattulainen, I.; Suomalainen-Wartiovaara, A.; Sharma, V. Atomistic Molecular Dynamics Simulations of Mitochondrial DNA Polymerase γ: Novel Mechanisms of Function and Pathogenesis. Biochemistry 2017, 56, 1227–1238. [Google Scholar] [CrossRef]
- Kubo, S.; Niina, T.; Takada, S. Molecular dynamics simulation of proton-transfer coupled rotations in ATP synthase FO motor. Sci. Rep. 2020, 10, 8225. [Google Scholar] [CrossRef]
- Pierri, C.L.; Parisi, G.; Porcelli, V. Computational approaches for protein function prediction: A combined strategy from multiple sequence alignment to molecular docking-based virtual screening. Biochim. Biophys. Acta BBA Proteins Proteom. 2010, 1804, 1695–1712. [Google Scholar] [CrossRef]
- Palese, L.L. Random Matrix Theory in molecular dynamics analysis. Biophys. Chem. 2015, 196, 1–9. [Google Scholar] [CrossRef]
- Bossis, F.; De Grassi, A.; Palese, L.L.; Pierri, C.L. Prediction of high- and low-affinity quinol-analogue-binding sites in the aa3 and bo3 terminal oxidases from Bacillus subtilis and Escherichia coli. Biochem. J. 2014, 461, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Amoedo, N.D.; Dard, L.; Sarlak, S.; Mahfouf, W.; Blanchard, W.; Rousseau, B.; Izotte, J.; Claverol, S.; Lacombe, D.; Rezvani, H.R.; et al. Targeting Human Lung Adenocarcinoma with a Suppressor of Mitochondrial Superoxide Production. Antioxid. Redox Signal. 2020, 33, 883–902. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Molecule | Vitamin and Coenzyme | DRI (mg/d) | Dosage for Children | Dosage for Adults | Food Sources (mg/serving; %Daily Value) | Web Reference |
|---|---|---|---|---|---|---|
| Thiamine | B1 | 0.4–1.3 | 10 mg/kg/day | 100–1000 mg/day | Rice, white, long grain, enriched, parboiled, ½ cup (1.4; 117); Pork chop, bone-in, broiled, 3 ounces (0.4; 33); Trout, cooked, dry heat, 3 ounces (0.4; 33); Black beans, boiled, ½ cup (0.4; 33); Mussels, blue, cooked, moist heat, 3 ounces (0.3; 25); Tuna, Bluefin, cooked, dry heat, 3 ounces (0.2; 17); Macaroni, whole wheat, cooked, 1 cup (0.2; 17); Acorn squash, cubed, baked, ½ cup (0.2; 17); Rice, brown, long grain, not enriched, cooked, ½ cup (0.1; 8); Bread, whole wheat, 1 slice (0.1; 8); Orange juice, prepared from concentrate, 1 cup (0.1; 8); Sunflower seeds, toasted, 1 ounce (0.1; 8); Beef steak, bottom round, trimmed of fat, braised, 3 ounces (0.1; 8); Yogurt, plain, low fat, 1 cup (0.1; 8); Oatmeal, regular and quick, unenriched, cooked with water, ½ cup (0.1; 8); Corn, yellow, boiled, 1 medium ear (0.1; 8); Milk, 2%, 1 cup (0.1; 8); Barley, pearled, cooked, 1 cup (0.1; 8). | https://ods.od.nih.gov/factsheets/Thiamin-HealthProfessional/ |
| Riboflavin | B2 | 50–400 mg po daily | 50–400 mg po daily | 50–400 mg/day (divided in 2–3 doses) | Beef liver, pan fried, 3 ounces (2.9; 223); Yogurt, plain, fat free, 1 cup (0.6; 46); Milk, 2% fat, 1 cup (0.5; 38); Beef, tenderloin steak, boneless, trimmed of fat, grilled, 3 ounces (0.4; 31); Clams, mixed species, cooked, moist heat, 3 ounces (0.4; 31); Mushrooms, portabella, sliced, grilled, ½ cup (0.3; 23); Almonds, dry roasted, 1 ounce (0.3; 23); Cheese, Swiss, 3 ounces (0.3; 23); Rotisserie chicken, breast meat only, 3 ounces (0.2; 15); Egg, whole, scrambled, 1 large (0.2; 15); Quinoa, cooked, 1 cup (0.2; 15); Salmon, pink, canned, 3 ounces (0.2; 15); Spinach, raw, 1 cup (0.1; 8); Apple, with skin, 1 large (0.1; 8); Kidney beans, canned, 1 cup (0.1; 8); Macaroni, elbow shaped, whole wheat, cooked, 1 cup (0.1; 8); Bread, whole wheat, 1 slice (0.1; 8); Cod, Atlantic, cooked, dry heat, 3 ounces (0.1; 8); Sunflower seeds, toasted, 1 ounce (0.1; 8); Tomatoes, crushed, canned, ½ cup (0.1; 8). | https://ods.od.nih.gov/factsheets/Riboflavin-HealthProfessional/ |
| Niacin | B3 (PP) | 5–14 | 25–250 mg/day | 250 mg/day up to 1 g/day | Beef liver, pan fried, 3 ounces (14.9; 93); Chicken breast, meat only, grilled, 3 ounces (10.3; 64); Marinara (spaghetti) sauce, ready to serve, 1 cup (10.3; 64); Turkey breast, meat only, roasted, 3 ounces (10;0; 63); Salmon, sockeye, cooked, 3 ounces (8.6; 54); Tuna, light, canned in water, drained, 3 ounces (8.6; 54); Pork, tenderloin, roasted, 3 ounces (6.3; 39); Beef, ground, 90% lean, pan-browned, 3 ounces (5.8; 36); Rice, brown, cooked, 1 cup (5.2; 33); Peanuts, dry roasted, 1 ounce (4.2; 26); Potato (russet), baked, 1 medium (2.3; 14); Sunflower seeds, dry roasted, 1 ounce (2.0; 13); Bread, whole wheat, 1 slice (1.4; 9); Pumpkin seeds, dry roasted, 1 ounce (1.3; 8); Soymilk, unfortified, 1 cup (1.3; 8); Lentils, boiled and drained, ½ cup (1.0; 6); Bulgur, cooked, 1 cup (0.9; 6); Banana, 1 medium (0.8; 5); Edamame, frozen, prepared, ½ cup (0.7; 4); Raisins, ½ cup (0.6; 4); Tomatoes, cherry, ½ cup (0.5; 3); Broccoli, boiled, drained, chopped, ½ cup (0.4; 3); Cashews, dry roasted, 1 ounce (0.4; 3); Yogurt, plain, low fat, 1 cup (0.3; 2); Apple, 1 medium (0.2; 1); Chickpeas, canned, drained, 1 cup (0.2; 1); Milk, 1% milkfat, 1 cup (0.2; 1); Spinach, frozen, chopped, boiled, ½ cup (0.2; 1); Tofu, raw, firm, ½ cup (0.2; 1); Onions, chopped, ½ cup (0.1; 1). | https://ods.od.nih.gov/factsheets/Niacin-HealthProfessional/ |
| Coenzyme Q10 (reduced, i.e., as ubiquinol, or oxidized, i.e., as ubiquinone) | 3–6 | CoQ10 as ubiquinol (preferred) 2–8 mg/kg po daily divided in two doses CoQ10 as ubiquinone 10–30 mg/kg po daily divided in 2 doses | CoQ10 as ubiquinol (preferred) 50–600 mg po daily CoQ10 as ubiquinone 300–2400 mg po daily divided 2–3 times a day | Sardines, salmon, trout, mackerel, 3 ounces (2.3–4/up to 50)). Chicken, beef, pork, 3 ounces (1.4–2.6/up to 50). Spinach, broccoli, cauliflower, ½ cup, chopped (0.4–0.5/up to 20). Fruits: strawberries, oranges, ½ cup (0.1–0.3/up to 20). Soybean and canola oils, 1 tablespoon (1.0–1.3/up to 30). Soybeans, lentils, peanuts, 1 ounce (0.6/up to 20). Pistachio, sesame seeds, 1 ounce (3.6, up to 80). Egg boiled, 1 medium (0.1/up to 3) | https://lpi.oregonstate.edu/mic/dietary-factors/coenzyme-Q10 | |
| Carnitine | NE | 20–100 mg/kg/day divided into two or three doses | 300–990 mg/dose two or three times per day | Beef steak, cooked, 4 ounces (56–162 mg); Ground beef, cooked, 4 ounces (87–99 mg); Milk, whole, 1 cup (8 mg); Codfish, cooked, 4 ounces (4–7 mg); Chicken breast, cooked, 4 ounces (3–5 mg); Ice cream, ½ cup (3 mg); Cheese, cheddar, 2 ounces (2 mg); Whole–wheat bread, 2 slices (0.2 mg); Asparagus, cooked, ½ cup (0.1 mg) | https://ods.od.nih.gov/factsheets/Carnitine-HealthProfessional/ | |
| Pantothenic acid | B5 | 2–7 | B vitamin complexes 1 tab po given; one to two times per day | 25 mg per day | Beef liver, boiled, 3 ounces (8.3; 166); Shitake mushrooms, cooked, ½ cup pieces (2.6; 52); Sunflower seeds, ¼ cup (2.4; 48); Chicken, breast meat, skinless, roasted, 3 ounces (1.3; 26); Tuna, fresh, bluefin, cooked, 3 ounces (1.2; 24); Avocados, raw, ½ avocado (1.0; 20); Milk, 2% milkfat, 1 cup (0.9; 18); Mushrooms, white, stir fried, ½ cup sliced (0.8; 16); Potatoes, russet, flesh and skin, baked, 1 medium (0.7; 14); Egg, hard boiled, 1 large (0.7; 14); Greek yogurt, vanilla, nonfat, 5.3-ounce container (0.6; 12); Ground beef, 85% lean meat, broiled, 3 ounces (0.6; 12); Peanuts, roasted in oil, ¼ cup (0.5; 10); Broccoli, boiled, ½ cup (0.5; 10); Whole-wheat pita, 1 large (0.5; 10); Chickpeas, canned, ½ cup (0.4; 8); Rice, brown, medium grain, cooked, ½ cup (0.4; 8); Oats, regular and quick, cooked with water, ½ cup (0.4; 8); Cheese, cheddar, 1.5 ounces (0.2; 4); Carrots, chopped, raw, ½ cup (0.2; 4); Cabbage, boiled, ½ cup (0.1; 2); Clementine, raw, 1 clementine (0.1; 2); Tomatoes, raw, chopped or sliced, ½ cup (0.1; 2). | https://ods.od.nih.gov/factsheets/PantothenicAcid-HealthProfessional/ |
| Biotin | B7 (H) | 5–30 | 5–10 mg/kg/ day | 25 mg per day | Beef liver, cooked, 3 ounces (30.8; 103); Egg, whole, cooked (10.0; 33); Salmon, pink, canned in water, 3 ounces (5.0; 17); Pork chop, cooked, 3 ounces (3.8; 13); Hamburger patty, cooked, 3 ounces (3.8; 13); Sunflower seeds, roasted, ¼ cup (2.6; 9); Sweet potato, cooked, ½ cup (2.4; 8); Almonds, roasted, ¼ cup (1.5; 5); Tuna, canned in water, 3 ounces (0.6; 2); Spinach, boiled, ½ cup (0.5; 2); Broccoli, fresh, ½ cup (0.4; 1); Cheddar cheese, mild, 1 ounce (0.4; 1); Milk, 2%, 1 cup (0.3; 1); Plain yogurt, 1 cup (0.2; 1); Oatmeal, 1 cup (0.2; 1); Banana, ½ cup (0.2; 1). | https://ods.od.nih.gov/factsheets/Biotin-HealthProfessional/ |
| Pyridoxine | B6 | 0.4–1.7 | 25 mg/day (generally in multivitamin complexes) | 25 mg/day (generally in multivitamin complexes) | Chickpeas, canned, 1 cup (1.1; 65); Beef liver, pan fried, 3 ounces (0.9; 53); Tuna, yellowfin, fresh, cooked, 3 ounces (0.9; 53); Salmon, sockeye, cooked, 3 ounces (0.6; 35); Chicken breast, roasted, 3 ounces (0.5; 29); Potatoes, boiled, 1 cup (0.4; 25); Turkey, meat only, roasted, 3 ounces (0.4; 25); Banana, 1 medium (0.4; 25); Marinara (spaghetti) sauce, ready to serve, 1 cup (0.4; 25); Ground beef, patty, 85% lean, broiled, 3 ounces (0.3; 18); Waffles, plain, ready to heat, toasted, 1 waffle (0.3; 18); Bulgur, cooked, 1 cup (0.2; 12); Cottage cheese, 1% low-fat, 1 cup (0.2; 12); Squash, winter, baked, ½ cup (0.2; 12); Nuts, mixed, dry-roasted, 1 ounce (0.1; 6); Raisins, seedless, ½ cup (0.1; 6); Onions, chopped, ½ cup (0.1; 6); Spinach, frozen, chopped, boiled, ½ cup (0.1; 6); Tofu, raw, firm, prepared with calcium sulfate, ½ cup (0.1; 6); Watermelon, raw, 1 cup (0.1; 6). | https://ods.od.nih.gov/factsheets/VitaminB6-HealthProfessional/ |
| Folic acid | B9 | 0.1–0.5 | 1 mg/day | 1 mg/day | Beef liver, braised, 3 ounces (215 mcg; 54); Spinach, boiled, ½ cup (131 mcg; 33); Black-eyed peas (cowpeas), boiled, ½ cup (105 mcg; 26); Rice, white, medium-grain, cooked, ½ cup (90 mcg; 22); Asparagus, boiled, 4 spears (89 mcg; 22); Brussels sprouts, frozen, boiled, ½ cup (78 mcg; 20); Lettuce, romaine, shredded, 1 cup (64 mcg; 16); Avocado, raw, sliced, ½ cup (59 mcg; 15); Spinach, raw, 1 cup (58 mcg; 15); Broccoli, chopped, frozen, cooked, ½ cup (52 mcg; 13); Mustard greens, chopped, frozen, boiled, ½ cup (52 mcg; 13); Bread, white, 1 slice (50 mcg; 13); Green peas, frozen, boiled, ½ cup (47 mcg; 12); Kidney beans, canned, ½ cup (46 mcg; 12); Wheat germ, 2 tablespoons (40 mcg; 10); Tomato juice, canned, ¾ cup (36 mcg; 9); Crab, Dungeness, 3 ounces (36 mcg; 9); Orange juice, ¾ cup (35 mcg; 9); Turnip greens, frozen, boiled, ½ cup (32 mcg; 8); Peanuts, dry roasted, 1 ounce (27 mcg; 7); Orange, fresh, 1 small (29 mcg; 7) Papaya, raw, cubed, ½ cup (27 mcg; 7); Banana, 1 medium (24 mcg; 6); Yeast, baker’s, ¼ teaspoon (23 mcg; 6); Egg, whole, hard-boiled, 1 large (22 mcg; 6); Cantaloupe, raw, cubed, ½ cup (17 mcg; 4); Vegetarian baked beans, canned, ½ cup (15 mcg; 4); Fish, halibut, cooked, 3 ounces (12 mcg; 3); Milk, 1% fat, 1 cup (12 mcg; 3); Ground beef, 85% lean, cooked, 3 ounces (7 mcg; 2); Chicken breast, roasted, 3 ounces (3 mcg; 1). | https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/ |
| Cobalamine | B12 | 0.0007–0.0024 | 25 mcg/day (generally in multivitamin complexes) | 25 mcg/day (generally in multivitamin complexes) | Beef liver, cooked, pan-fried, 3 ounces (70.7 mcg; 2944); Clams (without shells), cooked, 3 ounces (17 mcg; 708); Tuna, bluefin, cooked, dry heat, 3 ounces (9.3 mcg; 385); Salmon, Atlantic, cooked, 3 ounces (2.6 mcg; 108); Beef, ground, 85% lean meat/15% fat, pan-browned, 3 ounces (2.4 mcg; 100); Milk, 2% milkfat, 1 cup (1.3 mcg; 54); Yogurt, plain, fat free, 6-ounce container (1.0 mcg; 43); Cheese, cheddar, 1½ ounces (0.5 mcg; 19); Egg, whole, cooked, 1 large (0.5 mcg; 19); Turkey, breast meat, roasted, 3 ounces (0.3 mcg; 14); Tempeh, 1/2 cup (0.1 mcg; 3). | https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/ |
| Ascorbic acid | C | 25–90 | 5 mg/kg/day (or 10 mg qds) | 50–200 mg po daily | Red pepper, sweet, raw, ½ cup (95; 106); Orange juice, ¾ cup (93; 103); Orange, 1 medium (70; 78); Grapefruit juice, ¾ cup (70; 78); Kiwifruit, 1 medium (64; 71); Green pepper, sweet, raw, ½ cup (60; 67); Broccoli, cooked, ½ cup (51; 57); Strawberries, fresh, sliced, ½ cup (49; 54); Brussels sprouts, cooked, ½ cup (48; 53); Grapefruit, ½ medium (39; 43); Broccoli, raw, ½ cup (39; 43); Tomato juice, ¾ cup (33; 37); Cantaloupe, ½ cup (29; 32); Cabbage, cooked, ½ cup (28; 31); Cauliflower, raw, ½ cup (26; 29); Potato, baked, 1 medium (17; 19); Tomato, raw, 1 medium (17; 19); Spinach, cooked, ½ cup (9; 10); Green peas, frozen, cooked, ½ cup (8; 9). | https://ods.od.nih.gov/factsheets/VitaminC-HealthProfessional/ |
| Small Molecule | Vitamin and Coenzyme | DRI (mg/d) | Dosage for Children | Dosage for Adults | Food Sources (mcg/serving; % Daily Value) | Web Reference |
|---|---|---|---|---|---|---|
| Lipoic acid | 50–600 | 50–200 mg/day | 50–200 mg/day | Beef kidney, heart and liver (1–3 mcg/g dry wt) as lipoyllysine); Spinach and broccoli (1–3 mcg/g dry wt, as lipoyllysine); Tomatoes, peas and brussels sprouts (0.5 mcg/g dry wt, as lip). | https://lpi.oregonstate.edu/mic/dietary-factors/lipoic-acid | |
| Retinol | A | 0.7–1.3 | 0.3–0.9 mg per day (depending on the age) | Up to 3 mg per day | Beef liver, pan fried, 3 ounces (6582 mcg; 731); Sweet potato, baked in skin, 1 whole (1403 mcg; 156); Spinach, frozen, boiled, ½ cup (573 mcg; 64); Pumpkin pie, commercially prepared, 1 piece (488 mcg; 54); Carrots, raw, ½ cup (459 mcg; 51); Ice cream, French vanilla, soft serve, 1 cup (278 mcg; 31); Cheese, ricotta, part skim, 1 cup (263 mcg; 29); Herring, Atlantic, pickled, 3 ounces (219 mcg; 24); Cantaloupe, raw, ½ cup (135 mcg; 15); Peppers, sweet, red, raw, ½ cup (117 mcg; 13); Mangos, raw, 1 whole (112 mcg; 12); Egg, hard boiled, 1 large (75 mcg; 8); Black-eyed peas (cowpeas), boiled, 1 cup (66 mcg; 7); Apricots, dried, sulfured, 5 apricots (63 mcg; 7); Broccoli, boiled, ½ cup (60 mcg; 7); Salmon, sockeye, cooked, 3 ounces (59 mcg; 7); Tomato juice, canned, ¾ cup (42 mcg; 5); Yogurt, plain, low fat, 1 cup (32 mcg; 4); Tuna, light, canned in oil, drained solids, 3 ounces (20 mcg; 2); Baked beans, canned, plain or vegetarian, 1 cup (13 mcg; 1); Summer squash, all varieties, boiled, ½ cup (10 mcg; 1); Chicken, breast meat and skin, roasted, ½ breast (5 mcg; 1); Pistachio nuts, dry roasted, 1 ounce (4 mcg; 0). | https://ods.od.nih.gov/factsheets/VitaminA-HealthProfessional/ |
| Calciferol | D | 0.1–0.2 | 0.25–1 mg | 1 mg | Cod liver oil, 1 tablespoon (34.0; 170); Trout (rainbow), farmed, cooked, 3 ounces (16.2 mcg; 81); Salmon (sockeye), cooked, 3 ounces (14.2 mcg; 71); Mushrooms, white, raw, sliced, exposed to UV light, ½ cup (9.2 mcg; 46); Sardines (Atlantic), canned in oil, drained, 2 sardines (1.2 mcg; 6); Egg, 1 large, scrambled (1.1 mcg; 6); Liver, beef, braised, 3 ounces (1.0 mcg; 5); Tuna fish (light), canned in water, drained, 3 ounces (1.0 mcg; 5); Cheese, cheddar, 1.5 ounce (0.4 mcg; 2); Mushrooms, portabella, raw, diced, ½ cup (0.1 mcg; 1); Chicken breast, roasted, 3 ounces (0.1 mcg; 1). | https://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional/ |
| α-Tocopherol | E | 4–19 | 1–2 IU/kg po daily | 100–200 IU po daily | Wheat germ oil, 1 tablespoon (20.3; 135); Sunflower seeds, dry roasted, 1 ounce (7.4; 49); Almonds, dry roasted, 1 ounce (6.8; 45); Sunflower oil, 1 tablespoon (5.6; 37); Safflower oil, 1 tablespoon (4.6; 31); Hazelnuts, dry roasted, 1 ounce (4.3; 29); Peanut butter, 2 tablespoons (2.9;19); Peanuts, dry roasted, 1 ounce (2.2; 15); Corn oil, 1 tablespoon (1.9; 13); Spinach, boiled, ½ cup (1.9; 13); Broccoli, chopped, boiled, ½ cup (1.2; 8); Soybean oil, 1 tablespoon (1.1; 7); Kiwifruit, 1 medium (1.1; 7); Mango, sliced, ½ cup (0.7; 5); Tomato, raw, 1 medium (0.7; 5); Spinach, raw, 1 cup (0.6; 4). | https://ods.od.nih.gov/factsheets/VitaminE-HealthProfessional/ |
| Phylloquinone | K | 0.09–0.12 | 2–75 mcg | 90–120 mcg | Natto, 3 ounces (as MK-7; 850 mcg; 708); Collards, frozen, boiled, ½ cup (530 mcg; 442); Turnip greens, frozen, boiled ½ cup (426 mcg; 355); Spinach, raw, 1 cup (145 mcg; 121); Kale, raw, 1 cup (113 mcg; 94); Broccoli, chopped, boiled, ½ cup (110 mcg; 92); Soybeans, roasted, ½ cup (43 mcg; 36); Carrot juice, ¾ cup (28 mcg; 23); Soybean oil, 1 tablespoon (25 mcg; 21); Edamame, frozen, prepared, ½ cup (21 mcg; 18); Pumpkin, canned, ½ cup (20 mcg; 17); Pomegranate juice, ¾ cup (19 mcg; 16); Okra, raw, ½ cup (16 mcg; 13); Salad dressing, Caesar, 1 tablespoon (15 mcg; 13); Pine nuts, dried, 1 ounce (15 mcg; 13); Blueberries, raw, ½ cup (14 mcg; 12); Iceberg lettuce, raw, 1 cup (14 mcg; 12); Chicken, breast, rotisserie, 3 ounces (as MK-4; 13 mcg; 11); Grapes, ½ cup (11 mcg; 9); Vegetable juice cocktail, ¾ cup (10 mcg; 8); Canola oil, 1 tablespoon cup (10 mcg; 8); Cashews, dry roasted, 1 ounce cup (10 mcg; 8); Carrots, raw, 1 medium (8 mcg; 7); Olive oil, 1 tablespoon (8 mcg; 7); Ground beef, broiled, 3 ounces (as MK-4; 6 mcg; 5); Figs, dried, ¼ cup (6 mcg; 5); Chicken liver, braised, 3 ounces (as MK-4; 6 mcg; 5); Ham, roasted or pan-broiled, 3 ounces (as MK-4; 4 mcg; 3); Cheddar cheese, 1½ ounces (as MK-4; 4 mcg; 3); Mixed nuts, dry roasted, 1 ounce (4 mcg; 3); Egg, hard boiled, 1 large (as MK-4; 4 mcg; 3); Mozzarella cheese, 1½ ounces (as MK-4; 2 mcg; 2); Milk, 2%, 1 cup (as MK-4; 1 mcg; 1); Salmon, sockeye, cooked, 3 ounces (as MK-4; 0.3 mcg; 0); Shrimp, cooked, 3 ounces (as MK-4; 0.3 mcg; 0). | https://ods.od.nih.gov/factsheets/vitaminK-HealthProfessional/ |
| Small Molecule | Related Cofactor | Cofactor Protein Associations | Gene Name | Protein Targets from HMDB |
|---|---|---|---|---|
| Thiamine—B1 | Thiamine monophosphate | Cancer-related nucleoside-triphosphatase; membrane | NTPCR | https://hmdb.ca/metabolites/HMDB0002666/metabolite_protein_links |
| 14 kDa phosphohistidine phosphatase; cytosol | PHPT1 | |||
| Thiamine pyrophosphate | Pyruvate dehydrogenase E1 | PDH | https://hmdb.ca/metabolites/HMDB0001372/metabolite_protein_links | |
| 2-Oxoisovalerate dehydrogenase | BCKDH | |||
| 2-Oxoglutarate dehydrogenase | OGDH | |||
| 2-Oxoglutarate dehydrogenase-like | OGDHL | |||
| Thiamine pyrophosphate carrier | SLC25A19 | |||
| Riboflavin—B2 | FMN | Dihydroorotate dehydrogenase | DHODH | https://hmdb.ca/metabolites/HMDB0001520/metabolite_protein_links |
| NADH dehydrogenase UQ flavoprotein 1 | NDUFV1 | |||
| Sarcosine dehydrogenase | SARDH | |||
| FAD | FAD (and/or folate) carrier | SLC25A32 | https://hmdb.ca/metabolites/HMDB0001248/metabolite_protein_links | |
| Dihydrolipoyl dehydrogenase | DLD | |||
| Acyl carrier protein | NDUFAB1 | |||
| Long-chain specific acyl-CoA dehydrogenase | ACADL | |||
| Short-chain specific acyl-CoA dehydrogenase | ACADS | |||
| Medium-chain specific acyl-CoA dehydrogenase | ACADM | |||
| NADH-ubiquinone oxidoreductase | MT-ND | |||
| Succinate dehydrogenase | SDHD | |||
| Electron-transport flavoprotein dehydrogenase | IVD | |||
| Thioredoxin reductase 2 | ETFDH | |||
| Isovaleryl-CoA dehydrogenase | TXNRD2 | |||
| Dihydroorotate dehydrogenase | DHODH | |||
| Glutathione reductase | GSR | |||
| Glycerol-3-phosphate dehydrogenase | GPD2 | |||
| Choline dehydrogenase | CHDH | |||
| Glutaryl-CoA dehydrogenase | GCDH | |||
| Dimethylglycine dehydrogenase, | DMGDH | |||
| Sarcosine dehydrogenase | SARDH | |||
| Proline dehydrogenase 1 | PRODH | |||
| Hydroxyglutarate dehydrogenase | HGDH | |||
| Apoptosis-inducing factor 1 | AIFM1 | |||
| Probable D-lactate dehydrogenase | LDHD | |||
| Isobutyryl-CoA dehydrogenase | ACAD8 | |||
| Protein MTO1 homolog | MTO1 | |||
| (Pyruvate dehydrogenase (acetyl-transferring))-phosphatase 2 | PDP2 | |||
| Sulfide:quinone oxidoreductase | SQRDL | |||
| Niacin—B3 | NMN | 5′(3′)-deoxyribonucleotidase | NT5M | https://hmdb.ca/metabolites/HMDB0000229/metabolite_protein_links |
| NADH | Pyruvate dehydrogenase | PDH | https://hmdb.ca/metabolites/HMDB0000902/metabolite_protein_links | |
| Methylmalonate-semialdehyde dehydrogenase | ALDH6A1 | |||
| Dihydrolipoyl dehydrogenase | DLD | |||
| Acyl-CoA dehydrogenase | ACAD | |||
| NADH-ubiquinone oxidoreductase | MT-ND | |||
| Delta-1-pyrroline-5-carboxylate dehydrogenase | ALDH4A1 | |||
| Acyl carrier protein | NDUFAB1 | |||
| Glutathione reductase | GSR | |||
| Succinate-semialdehyde dehydrogenase | ALDH5A1 | |||
| Aldehyde dehydrogenase | ALDH2 | |||
| Pyrroline-5-carboxylate reductase 1 | PYCR1 | |||
| Trifunctional enzyme | HADH | |||
| 3-Hydroxyisobutyrate dehydrogenase | HIBADH | |||
| 2-Oxoglutarate dehydrogenase | OGDH | |||
| D-beta-hydroxybutyrate dehydrogenase | BDH1 | |||
| Glutamate dehydrogenase | GLUD | |||
| Aminomethyltransferase | AMT | |||
| Alpha-aminoadipic semialdehyde synthase | AASS | |||
| Malate dehydrogenase | MDH | |||
| NAD-dependent malic enzyme | ME | |||
| Isocitrate dehydrogenase | IDH3 | |||
| NAD(P) transhydrogenase | NNT | |||
| Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase | MTHFD2 | |||
| Tricarboxylate transport protein | SLC25A1 | |||
| Proline dehydrogenase 1 | PRODH | |||
| Glycine cleavage system H protein | GCSH | |||
| NAD-dependent protein deacetylase sirtuin-3 | SIRT3 | |||
| NAD-dependent protein deacetylase sirtuin-5 | SIRT5 | |||
| 2-Oxoglutarate dehydrogenase-like | OGDHL | |||
| Dihydrofolate reductase | DHFRL1 | |||
| NAD kinase domain-containing protein 1 | NADKD1 | |||
| Ubiquinone/coenzyme Q10 (reduced, i.e., as ubiquinol, or oxidized, i.e., as ubiquinone) | UQ/CoQ10 (reduced, i.e., UQH2) | NADH dehydrogenase (ubiquinone) | NDFU | https://hmdb.ca/metabolites/HMDB0001072/metabolite_protein_links and https://hmdb.ca/metabolites/HMDB0001304/metabolite_protein_links |
| NADH-ubiquinone oxidoreductase | MT-ND | |||
| Succinate dehydrogenase | SDH | |||
| Acyl carrier protein | NDUFA5 | |||
| Cytochrome b-c1 complex | UQCR | |||
| Electron transfer flavoprotein-ubiquinone oxidoreductase | ETFDH | |||
| Cytochrome c1, heme protein | CYC1 | |||
| Carnitine, acetyl-L-carnitine | Carnitine O-acetyltransferase | CRAT | https://hmdb.ca/metabolites/HMDB0000201/metabolite_protein_links and https://hmdb.ca/metabolites/HMDB0000062/metabolite_protein_links | |
| Carnitine O-palmitoyltransferase 2 | CPT2 | |||
| Carnitine/acylcarnitine carrier | SLC25A20 | |||
| Pantothenic acid—B5 | Pantothenate kinase 2 | PANK2 | https://hmdb.ca/metabolites/HMDB0000210/metabolite_protein_links | |
| Pyridoxine—B6 | Alpha-aminoadipic semialdehyde dehydrogenase | ALDH7A1 | https://hmdb.ca/metabolites/HMDB0000239/metabolite_protein_links | |
| Biotin—B7 | Pyruvate carboxylase | PC | https://hmdb.ca/metabolites/HMDB0000030/metabolite_protein_links | |
| Propionyl-CoA carboxylase | PCC | |||
| Methylcrotonoyl-CoA carboxylase | MCCC | |||
| Folic acid—B9 | Folate (and/or FAD) carrier | SLC25A32 | https://hmdb.ca/metabolites/HMDB0000121/metabolite_protein_links | |
| Dihydrofolate reductase | DHFRL1 | |||
| Cobalamine—B12 | Methylmalonyl-CoA mutase | MUT | https://hmdb.ca/metabolites/HMDB0002174/metabolite_protein_links | |
| Methylmalonic aciduria type A protein | MMAA | |||
| Ascorbic acid—C | Trimethyllysine dioxygenase | TMLHE | https://hmdb.ca/metabolites/HMDB0000044/metabolite_protein_links |
| Small Molecule/Cofactor | Cofactor Protein Associations | Gene Name | Protein Targets from HMDB |
|---|---|---|---|
| Lipoic acid | Lipoyl synthase | LIAS | https://hmdb.ca/metabolites/HMDB0014312/metabolite_protein_links |
| Acyl-coenzyme A synthetase ACSM1 | ACSM1 | ||
| Lipoyltransferase 1 | LIPT1 | ||
| Retinol—A | Diacylglycerol O-acyltransferase 1; endoplasmic reticulum | DGAT1 | https://hmdb.ca/metabolites/HMDB0000305/metabolite_protein_links |
| Retinol dehydrogenase; endoplasmic reticulum | RDH | ||
| Alcohol dehydrogenase; cytoplasm | ADH | ||
| Lecithin retinol acyltransferase; endoplasmic reticulum and other locations | LRAT | ||
| 17-Beta-hydroxysteroid dehydrogenase type 6; endosome and endoplasmic reticulum | HSD17B6 | ||
| Acyl-CoA wax alcohol acyltransferase; endoplasmic reticulum | AWAT | ||
| All-trans-retinol 13,14-reductase; endoplasmic reticulum | RETSAT | ||
| Short-chain dehydrogenase/reductase 3; membrane | DHRS3 | ||
| Epidermal retinol dehydrogenase 2; endoplasmic reticulum | SDR16C5 | ||
| Calciferol—D | Cholesterol side-chain cleavage enzyme | CYP11A1 | https://hmdb.ca/metabolites/HMDB0000876/metabolite_protein_links |
| Sterol 26-hydroxylase | CYP27A1 | ||
| 25-Hydroxyvitamin D-1 alpha hydroxylase | CYP27B1 | ||
| 1,25-Dihydroxyvitamin D(3) 24-hydroxylase | CYP24A1 | ||
| UMP-CMP kinase 2 | CMPK2 | ||
| α-Tocopherol—E | Superoxide dismutase (Cu-Zn) | SOD1 | https://hmdb.ca/metabolites/HMDB0001893/metabolite_protein_links |
| Phylloquinone—K | NAD(P)H dehydrogenase (quinone) 1; cytoplasm | NQO1 | https://hmdb.ca/metabolites/HMDB0003555/metabolite_protein_links |
| Vitamin K-dependent gamma-carboxylase; endoplasmic reticulum | GCCX | ||
| Osteocalcin; secreted | BGLAP | ||
| Vitamin K epoxide reductase complex subunit 1; endoplasmic reticulum | VKORC1 |
| Gene | NT Mutation | AA Mutation | Syndromes |
|---|---|---|---|
| MT-ND5 | m.13513G>A | D393N | LS, MELAS/LS overlap syndrome |
| MT-ND5 | m.13042G>A | A236T | LS, MERRF/MELAS syndrome, LHON-like syndrome |
| NDUFAF6 | compound heterozygosity, NM_152416.4: c.532G>C; p.A178P; deep intronic c.420 + 784C>T NM_152416.4: c.371T>C; p.I124T | A178P + intron I124T | LS |
| MT-TK | m.8344A>G | na | MERRF, LS |
| MT-TK | m.8363G>A | na | MERRF, Cardiomiopathy and deafness, LS |
| MT-TL1 | m.3243A>G | na | MELAS, MIDD, PEO |
| MT-TF | m.641A>T | na | Mitochondrial encephalomyopathy |
| CRAT | compound heterozygosity, NM_000755.4: c.329A>G, p.Y110C c.1705G>A, p.V569M | Y110C; V569M | Mitochondrial encephalomyopathy |
| SLC25A10 | compound heterozygosity, NM_001270888.1: c.304AZT, p.K102 *; c.684C>T, p.P228P; intronic c.790-37G>A | K102 *; P228P + intronic | Mitochondrial encephalomyopathy |
| EARS2 | compound heterozygosity, NM_001083614.2 c.322C>T, p.R108W; c.502A>G, p.R168G; hexon 7 insertion c.1278_1279insCTC, p.Leu427ins | R108W; R168G + L427ins | Leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) |
| SERAC1 | Homozygous, NM_03286.1 c.1709 G>A; p.G526E; c. 1643_1646 dup ATCT, p.Leu550-SerfsX19; c.1593T>G, p.S531R | G526E L550-SfsX19; S531R | MEGDEL |
| PDB_ID | Aminoacyl tRNA Synthetase Name | Number of Residues | Aminoacyl tRNA | Number of Nucleotides | Organism | Cell Compartment | Resolution |
|---|---|---|---|---|---|---|---|
| 1f7u (1f7v) | Arginyl-tRNA synthetase | 608 (607) | Arg tRNA | 76 | S. cerevisiae | cytoplasmic/mitochondrion | 2.20 Å (2.90 Å) |
| 2zue (2zuf) | Arginyl-tRNA synthetase | 629 | Arg tRNA | 78 | P. horikoshii | cytoplasmic | 2.00 Å (2.30 Å) |
| 3kfu | Glutamyl-tRNA amidotransferase subunit A, aspartyl/Glutamyl-tRNA amidotrasferase subunit B, Glutamyl amidotrasferase subunit C-tRNA | 471, 466, 92 | Asn tRNA | 76 | T. thermophilus HB8 | cytoplasmic | 3.00 Å |
| 1c0a | Aspartyl-tRNA synthetase | 585 | Asp tRNA | 77 | E. coli | cytoplasmic | 2.40 Å |
| 1efw | Aspartyl-tRNA synthetase | 580 | Asp tRNA | 73 | T. thermpohilus/E. coli | cytoplasmic | 3.00 Å |
| 1il2 | Aspartyl-tRNA synthetase | 590 | Asp tRNA | 75 | E. coli/S. cerevisiae | cytoplasmic/mitochondrion | 2.60 Å |
| 1u0b | Cysteinyl-tRNA synthetase | 461 | Cys tRNA | 74 | E. coli | cytoplasmic | 2.30 Å |
| 1exd | Glutaminyl-tRNA synthetase | 548 | Gln Aptamer tRNA | 73 | E. coli | cytoplasmic | 2.70 Å |
| 3aL0 | Glutamyl-tRNA amidotransferase subunit A | 475 | Gln tRNA | 74 | T. maritima MSB8 | cytoplasmic | 3.37 Å |
| 1euy (1euq) | Glutaminyl-tRNA synthetase | 548 | Gln tRNA | 74 (72) | E. coli | cytoplasmic | 2.60 Å (3.10 Å) |
| 1gsg | Glutaminyl-tRNA synthetase | 553 | Gln tRNA | 75 | E. coli | cytoplasmic | 2.80 Å |
| 1gts | Glutaminyl-tRNA (E.C.6.1.1.18) synthetase | 553 | Gln tRNA | 74 | E. coli | cytoplasmic | 2.80 Å |
| 1o0b (1o0c) | Glutaminyl-tRNA synthetase | 554 | Gln tRNA | 75 | E. coli | cytoplasmic | 2.70 Å (2.70 Å) |
| 1zjw | Glutaminyl-tRNA synthetase | 553 | Gln tRNA | 75 | E. coli | cytoplasmic | 2.50 Å |
| 2rd2 | Glutaminyl-tRNA synthetase | 556 | Gln tRNA | 75 | E. coli K-12 (E. coli) | cytoplasmic | 2.60 Å |
| 3akz | Glutamyl-tRNA synthetase2 | 487 | Gln tRNA | T. maritima MSB8 | cytoplasmic | 2.90 Å | |
| 1qrs (1qrt, 1qru) | Glutaminyl-tRNA (E.C.6.1.1.18) synthetase | 553 | Gln2 tRNA | 75 | E. coli | cytoplasmic | 2.60 Å (2.70 Å, 3.00 Å |
| 1qtq | Glutaminyl-tRNA synthetase | 553 | Gln2 tRNA | 75 | E. coli | cytoplasmic | 2.25 Å |
| 1g59 | Glutamyl-tRNA synthetase | 648 | Glu tRNA | 75 | T. thermophilus | cytoplasmic | 2.40 Å |
| 1n78 (1n77, 1j09, 1n78) | Glutamyl-tRNA synthetase | 468 | Glu tRNA (Glu tRNA, /, /) | 75 (75, /, /) | T. thermophilus | cytoplasmic | 2.10 Å (2.40 Å, 1.80 Å, 1.90 Å) |
| 4kr2 (4kr3) | Glycine-tRNA synthetase | 637 | Gly tRNA-ccc | 74 | H. sapiens | cytoplasmic/mitochondria | 3.29 Å (3.31 Å) |
| 5e6m | Glycine-tRNA synthetase | 693 | Gly tRNA-ccc | 74 | H. sapiens | cytoplasmic/mitochondria | 2.93 Å |
| 4qei (4kqe) | Glycine-tRNA synthetase | 630 | Gly tRNA-ccc-2-2 (/) | 69 (/) | H. sapiens | cytoplasmic/mitochondria | 2.88 Å |
| 4rdx | Histidine-tRNA synthetase | 423 | His tRNA | 78 | T. thermophilus HB27 | cytoplasmic | 2.55 Å |
| 1ffy (1qu2, 1qu3) | Isoleucyl-tRNA synthetase | 917 | Ile tRNA | 75 | S. aureus | cytoplasmic | 2.20 Å (2.20 Å, 2.90 Å) |
| 1wz2 | Leucyl-tRNA synthetase | 967 | Leu tRNA | 88 | P. horikoshii OT3 | cytoplasmic | 3.21 Å |
| 3zjt (3zjv, 3zju) | Leucyl-tRNA synthetase | 880 | Leu5 tRNA UAA isoacceptor | 88 (87, 87) | E. coli/E. coli K-12 | cytoplasmic | 2.20 Å (2.30 Å, 2.40 Å) |
| 4arc (4asi, 4ari, 4aq7) | Leucyl-tRNA synthetase | 880 | Leu5 tRNA UAA isoacceptor | 87 | E. coli K-12 | cytoplasmic/mitochondria | 2.00 Å (2.02 Å, 2.08 Å, 2.50 Å) |
| 5ah5 | Leucyl-tRNA synthetase | 822 | Leu tRNA UAA isoacceptor | 84 | A. radiobacter K84 | cytoplasmic | 2.10 Å |
| 3zgz | Leucyl-tRNA synthetase | 880 | Leu5 tRNA UAA isoacceptor | 88 | E. coliK-12 | cytoplasmic | 2.40 Å |
| 4cqn | Leucyl-tRNA synthetase | 880 | Leu5 tRNA UAA isoacceptor | 82 | E. coli K-12 | cytoplasmic | 2.50 Å |
| 7k98 (7kao, 7k9m, 7kab) | Phenylalanyl-tRNA synthetase | 344 | Phe tRNA | 77 | M. tubercolosis H37Rv | cytoplasmic | 2.19 Å (2.40 Å, 2.50 Å, 2.50 Å) |
| 3tup | Phenylalanyl-tRNA synthetase | 415 | Phe tRNA | 76 | H. sapiens | cytoplasmic/mitochondria | 3.05 Å |
| 1eiy | Phenylalanyl-tRNA synthetase | 350 | Phe tRNA | 76 | T. thermophilus HB8 | cytoplasmic | 3.30 Å |
| 3w3s | Seryl-tRNA type-2 synthetase | 527 | Selenocysteine tRNA | 99 | M. kandleri AV19 | cytoplasmic | 3.10 Å |
| 4rqf (4rqe) | Seryl-tRNA synthetase | 522 | Selenocysteine tRNA | 90 | H. sapiens | cytoplasmic | 3.50 Å (4.00 Å) |
| 1ser | Seryl-tRNA synthetase E.C.6.1.1.11 | 421 | Ser tRNA | 94 | T. thermophilus | cytoplasmic | 2.90 Å |
| 1qf6 | Threonyl-tRNA synthetase | 642 | Thr tRNA | 76 | E. coli | cytoplasmic | 2.90 Å |
| 1kog | Threonyl-tRNA synthetase | 401 | Thr tRNA synthetase mRNA | 37 | E. coli | cytoplasmic | 3.50 Å |
| 2ake (2dr2) | Tryptophanyl-tRNA synthetase | 384 | Trp transfer tRNA | 72 (75) | H. sapiens/B. taurus | cytoplasmic | 3.10 Å (2.40) |
| 1j1u | Tyrosyl-tRNA synthetase | 306 | Tyr tRNA | 77 | M. jannaschii | cytoplasmic | 1.95 Å |
| 1ivs (1iyw) | Valyl-tRNA synthetase | 862 | Val tRNA (/) | 75 (/) | T. thermophilus | cytoplasmic | 2.90 Å (4.00 Å) |
| 1gax | Valyl-tRNA synthetase | 862 | Val tRNA | 75 | T. thermophilus | cytoplasmic | 2.90 Å |
| 2cv2 | Glutamyl-tRNA synthetase | 468 | tRNA | 75 | T. thermophilus | cytoplasmic | 2.69 Å |
| 2dxi (2cv0, 2cv2, 2cuz) | Glutamyl-tRNA synthetase | 468 | tRNA (tRNA, tRNA, /) | 75 (75, 75, /) | T. thermophilus | cytoplasmic | 2.20 Å (2.40 Å, 2.40 Å, 1.98 Å) |
| 2d6f | Glutamyl-tRNA(Gln) amidotransferase subunit D | 435 | tRNA | 74 | M. thermautrophicus | cytoplasmic | 3.15Å |
| 2du3 (2du4, 2du5, 2du6) | O-Phosphoseryl-tRNA synthetase | 534 | tRNA | 71 | A. fulgidus | / | 2.60 Å (2.80Å, 3.20 Å, 3.20 Å) |
| 2du7 | O-Phosphoseryl-tRNA synthetase | 549 | / | / | M. jannaschii | cytoplasmic | 3.60 Å |
| 4yye | Threonyl-tRNA synthetase | 460 | tRNA | 76 | S. cerevisiae S288c | cytoplasmic/mitochondria | 2.30 Å |
| 5yyn | Arginyl-tRNA synthetase | 586 | tRNA | 77 | E. coli K-12 | cytoplasmic | 3.00Å |
| 2zni | Pyrrolysyl-tRNA synthetase | 308 | Bacterial tRNA | 72 | D. hefniense | cytoplasmic | 3.10 Å |
| 3a2k | Lysidine-tRNA synthase | 464 | Bacterial tRNA | G. kaustophilus | cytoplasmic | 3.65 Å | |
| 2hrk (2hsn, 2hsm) | Glutamyl-tRNA synthetase | 207 | / | / | S. cerevisiae | cytoplasmic/mitochondria | 2.05 Å (2.20 Å, 3.00 Å) |
| 2rkj | Tyrosyl-tRNA synthetase | 392 | 238-MER RNA | 246 | N. crassa | cytoplasmic/mitochondria | 4.50 Å |
| 5ud5 (5v6x) | Pyrrolysyl-tRNA synthetase | 109 | 70-MER RNA | 72 | M. mazei Go1 | cytoplasmic | 2.35 Å (2.76 Å) |
| 4jxx (4jxz, 4jyzx) | Glutaminyl-tRNA synthetase | 553 | 71-MER RNA (71-MER RNA, 72-MER RNA) | 75 | E. coli K-12 | cytoplasmic | 2.30 Å (2.40Å, 2.50 Å) |
| 2azx | Tryptophanyl-tRNA synthetase | 477 | 72-MER | 75 | H. sapiens | cytoplasmic | 2.80 Å |
| 2csx (2ct8) | Methionyl-tRNA synthetase | 497 | 75-MER RNA (74-MER RNA) | 75 (74) | A. aeolicus | cytoplasmic | 2.70 Å (2.70 Å) |
| 3wqy (3wqz) | Alenine-tRNA synthetase | 906 | 75-MER RNA | 75 | A. filgidus DSM 4304 | cytoplasmic | 3.30 Å (3.49 Å) |
| 4wj3 (4wj4) | Glutamyl-tRNA(Gln) amidotransferase subunit A | 484 (599) | 76-MER tRNA | 76 | P. aeruginosa PAO1 | cytoplasmic | 3.71 Å (3.29 Å) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tragni, V.; Primiano, G.; Tummolo, A.; Cafferati Beltrame, L.; La Piana, G.; Sgobba, M.N.; Cavalluzzi, M.M.; Paterno, G.; Gorgoglione, R.; Volpicella, M.; et al. Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs. Molecules 2022, 27, 3494. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113494
Tragni V, Primiano G, Tummolo A, Cafferati Beltrame L, La Piana G, Sgobba MN, Cavalluzzi MM, Paterno G, Gorgoglione R, Volpicella M, et al. Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs. Molecules. 2022; 27(11):3494. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113494
Chicago/Turabian StyleTragni, Vincenzo, Guido Primiano, Albina Tummolo, Lucas Cafferati Beltrame, Gianluigi La Piana, Maria Noemi Sgobba, Maria Maddalena Cavalluzzi, Giulia Paterno, Ruggiero Gorgoglione, Mariateresa Volpicella, and et al. 2022. "Personalized Medicine in Mitochondrial Health and Disease: Molecular Basis of Therapeutic Approaches Based on Nutritional Supplements and Their Analogs" Molecules 27, no. 11: 3494. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113494