
Synthesis of Asebogenin and Balsacone A Precursor by a Novel Synthetic Strategy: Recent Opportunities for and Challenges of Total Synthesis of Balsacone A


Abstract
:
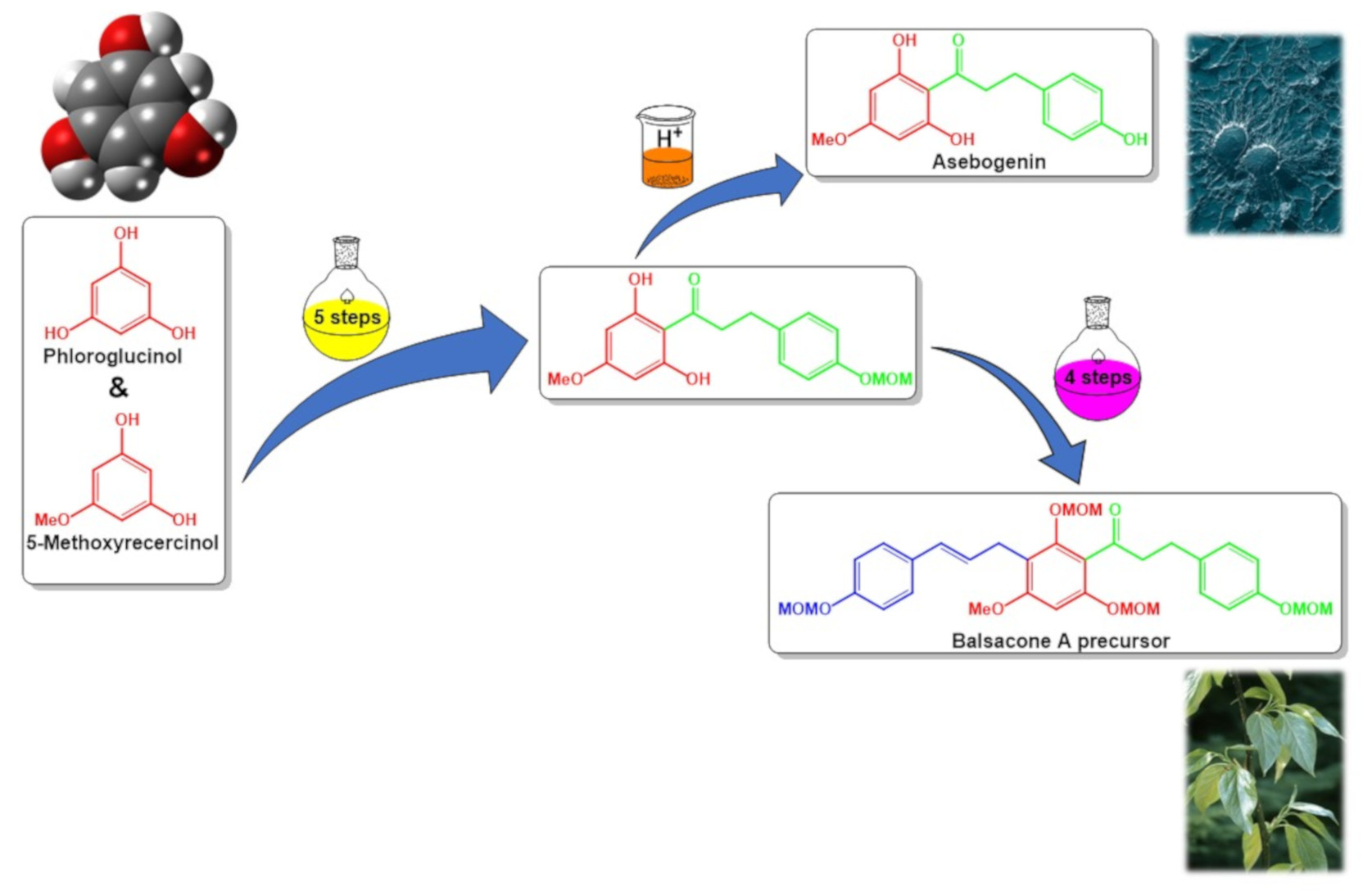{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Result and Discussion
3. Conclusions
4. Experimental
4.1. General
4.2. Chemistry Experimental Procedures
4.2.1. 1-(2,6-Bis(benzyloxy)-4-methoxyphenyl) ethan-1-one (4)
4.2.2. 4-(Methoxymethoxy) benzaldehyde (5)
4.2.3. (E)-1-(2,6-Bis(benzyloxy)-4-methoxyphenyl)-3-(4-(methoxymethoxy)phenyl)prop-2-en-1-one (6a)
4.2.4. 1-(2,6-Dihydroxy-4-methoxyphenyl)-3-(4-(methoxymethoxy)phenyl)propan-1-one (7)
4.2.5. 1-(2,6-Dihydroxy-4-methoxyphenyl)-3-(4-hydroxyphenyl)propan-1-one (8)
4.2.6. 1-(2-(Allyloxy)-6-hydroxy-4-methoxyphenyl)-3-(4-(methoxymethoxy)phenyl)propan-1-one (9a) and 1-(2,6-Dihydroxy-4-methoxyphenyl)-2-(4-(methoxymethoxy)benzyl)pent-4-en-1-one (9b)
4.2.7. 1-(3-Allyl-2,6-dihydroxy-4-methoxyphenyl)-3-(4-(methoxymethoxy)phenyl)propan-1-one (10)
4.2.8. 1-(3-Allyl-4-methoxy-2,6-bis(methoxymethoxy)phenyl)-3-(4-(methoxymethoxy)phenyl)propan-1-one (12)
4.2.9. 1-(Methoxymethoxy)-4-vinylbenzene (13)
4.2.10. (E)-1-(4-Methoxy-2,6-bis(methoxymethoxy)-3-(3-(4-(methoxymethoxy)phenyl)allyl)phenyl)-3-(4-(methoxymethoxy)phenyl)propan-1-one (14)
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dihydrochalcone—An Overview|ScienceDirect Topics. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/topics/chemistry/dihydrochalcone (accessed on 8 February 2022).
- Tomás-Barberán, F.A.; Clifford, M.N. Flavanones, Chalcones and Dihydrochalcones—Nature, Occurrence and Dietary Burden. J. Sci. Food Agric. 2000, 80, 1073–1080. [Google Scholar] [CrossRef]
- Stompor, M.; Broda, D.; Bajek-Bil, A. Dihydrochalcones: Methods of Acquisition and Pharmacological Properties—A First Systematic Review. Molecules 2019, 24, 4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Watanabe, S.; Miyake, N.; Kohno, H.; Osawa, T. Dihydrochalcones: Evaluation as Novel Radical Scavenging Antioxidants. J. Agric. Food Chem. 2003, 51, 3309–3312. [Google Scholar] [CrossRef]
- Funari, C.S.; Gullo, F.P.; Napolitano, A.; Carneiro, R.L.; Mendes-Giannini, M.J.S.; Fusco-Almeida, A.M.; Piacente, S.; Pizza, C.; Silva, D.H.S. Chemical and Antifungal Investigations of Six Lippia Species (Verbenaceae) from Brazil. Food Chem. 2012, 135, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Monge, P.; Solheim, E.; Scheline, R.R. Dihydrochalcone Metabolism in the Rat: Phloretin. Xenobiotica 1984, 14, 917–924. [Google Scholar] [CrossRef]
- Joshi, A.S.; Li, X.-C.; Nimrod, A.C.; ElSohly, H.N.; Walker, L.A.; Clark, A.M. Dihydrochalcones from Piper Longicaudatum. Planta Med. 2001, 67, 186–188. [Google Scholar] [CrossRef]
- Krishnamurty, H.G.; Sathyanarayana, S. Catalytic Transfer Hydrogenation, a Facile Conversion of Hydroxyflavanones into Hydroxydihydrochalcones. Synth. Commun. 1989, 19, 119–123. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, X.; Chen, X.; Shi, S.; Jiang, X.; Liang, X.; Chen, W.; Zhang, S. Analysis and Improved Characterization of Minor Antioxidants from Leaves of Malus Doumeri Using a Combination of Major Constituents’ Knockout with High-Performance Liquid Chromatography–Diode Array Detector–Quadrupole Time-of-Flight Tandem Mass Spectrometry. J. Chromatogr. A 2015, 1398, 57–65. [Google Scholar] [CrossRef]
- Gasull, E.I.; Silber, J.J.; Blanco, S.E.; Tomas, F.; Ferretti, F.H. A Theoretical and Experimental Study of the Formation Mechanism of 4-X-Chalcones by the Claisen–Schmidt Reaction. J. Mol. Struct. THEOCHEM 2000, 503, 131–144. [Google Scholar] [CrossRef]
- Martins, B.T. Synthetic Analogues of Marine Natural Flavonoids as Antifouling Agents: Synthesis and Biological Evaluation. Master’s Thesis, Universidade do Porto, Porto, Portugal, 2017. [Google Scholar]
- Burmaoğlu, S. Total Syntheses of Balsacone B and Balsacone C. J. Turk. Chem. Soc. Sect. A Chem. 2017, 4, 709–720. [Google Scholar] [CrossRef]
- Alsarraf, J.; Bilodeau, J.-F.; Legault, J.; Simard, F.; Pichette, A. Exploring the Biomass-Derived Chemical Space Emerging from Natural Dihydrochalcones through the Single-Step Hemisynthesis of Antibacterial Balsacones. ACS Sustain. Chem. Eng. 2020, 8, 6194–6199. [Google Scholar] [CrossRef]
- Bélanger, A.; Grenier, A.; Simard, F.; Gendreau, I.; Pichette, A.; Legault, J.; Pouliot, R. Dihydrochalcone Derivatives from Populus Balsamifera L. Buds for the Treatment of Psoriasis. Int. J. Mol. Sci. 2020, 21, 256. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, S.; Legault, J.; Simard, F.; Chiasson, É.; Pichette, A. New Antibacterial Dihydrochalcone Derivatives from Buds of Populus Balsamifera. Tetrahedron Lett. 2013, 54, 1631–1633. [Google Scholar] [CrossRef]
- Côté, H.; Pichette, A.; Simard, F.; Ouellette, M.-E.; Ripoll, L.; Mihoub, M.; Grimard, D.; Legault, J. Balsacone C, a New Antibiotic Targeting Bacterial Cell Membranes, Inhibits Clinical Isolates of Methicillin-Resistant Staphylococcus Aureus (MRSA) without Inducing Resistance. Front. Microbiol. 2019, 10, 2341. [Google Scholar] [CrossRef] [Green Version]
- Simard, F.; Legault, J.; Lavoie, S.; Pichette, A. Balsacones DI, Dihydrocinnamoyl Flavans from Populus Balsamifera Buds. Phytochemistry 2014, 100, 141–149. [Google Scholar] [CrossRef]
- Simard, F.; Gauthier, C.; Chiasson, É.; Lavoie, S.; Mshvildadze, V.; Legault, J.; Pichette, A. Antibacterial Balsacones J–M, Hydroxycinnamoylated Dihydrochalcones from Populus Balsamifera Buds. J. Nat. Prod. 2015, 78, 1147–1153. [Google Scholar] [CrossRef]
- Baran, P.S. Natural Product Total Synthesis: As Exciting as Ever and Here to Stay. J. Am. Chem. Soc. 2018, 140, 4751–4755. [Google Scholar] [CrossRef] [Green Version]
- Alonso, F.; Osante, I.; Yus, M. Conjugate Reduction of α, β-Unsaturated Carbonyl Compounds Promoted by Nickel Nanoparticles. Synlett 2006, 2006, 3017–3020. [Google Scholar] [CrossRef]
- Hays, D.S.; Scholl, M.; Fu, G.C. Organotin Hydride-Catalyzed Conjugate Reduction of Alpha, Beta-Unsaturated Ketones. J. Org. Chem. 1996, 61, 6751–6752. [Google Scholar] [CrossRef] [PubMed]
- Nagendiran, A.; Pascanu, V.; Bermejo Gómez, A.; González Miera, G.; Tai, C.-W.; Verho, O.; Martín-Matute, B.; Bäckvall, J.-E. Mild and Selective Catalytic Hydrogenation of the C=C Bond in α, β-Unsaturated Carbonyl Compounds Using Supported Palladium Nanoparticles. Chem. A Eur. J. 2016, 22, 7184–7189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, J.; Fayez, S.; Scheiner, M.; Hoffmann, M.; Oerter, S.; Appelt-Menzel, A.; Maher, P.; Maurice, T.; Bringmann, G.; Decker, M. Sterubin: Enantioresolution and Configurational Stability, Enantiomeric Purity in Nature, and Neuroprotective Activity in Vitro and in Vivo. Chem. A Eur. J. 2020, 26, 7299–7308. [Google Scholar] [CrossRef] [PubMed]
- Fruit, C.; Turck, A.; Plé, N.; Mojovic, L.; Quéguiner, G. A New Route to Septorin via Controlled Metalations of Pyrazines. Diazines XXX. Tetrahedron 2001, 57, 9429–9435. [Google Scholar] [CrossRef]
- PubChem Asebogenin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/442255 (accessed on 13 May 2022).
- Singh, M.; Argade, N.P. Palladium-Catalyzed Routes to Geranylated or Farnesylated Phenolic Stilbenes: Synthesis of Pawhuskin C and Schweinfurthin J. Synthesis 2012, 44, 2895–2902. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polat, M.F. Synthesis of Asebogenin and Balsacone A Precursor by a Novel Synthetic Strategy: Recent Opportunities for and Challenges of Total Synthesis of Balsacone A. Molecules 2022, 27, 3523. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113523
Polat MF. Synthesis of Asebogenin and Balsacone A Precursor by a Novel Synthetic Strategy: Recent Opportunities for and Challenges of Total Synthesis of Balsacone A. Molecules. 2022; 27(11):3523. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113523
Chicago/Turabian StylePolat, M. Fatih. 2022. "Synthesis of Asebogenin and Balsacone A Precursor by a Novel Synthetic Strategy: Recent Opportunities for and Challenges of Total Synthesis of Balsacone A" Molecules 27, no. 11: 3523. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113523