
Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex
Department of Chemistry, Tokyo Institute of Technology, Ookayama, Meguro-ku, Tokyo 152-8551, Japan
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(17), 5553; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175553
Submission received: 10 August 2022
/
Revised: 25 August 2022
/
Accepted: 26 August 2022
/
Published: 29 August 2022
(This article belongs to the Special Issue Design Strategies for Metal Complexes that Activate Bio-Related Small Molecules)
Abstract
:The side-on end-on dinitrogen hydride complex [{Na(dme)}2{(O3)Nb}2(μ-η1:η2-N2)(μ-H)2] (3-Na, [O3]3− = [(3,5-tBu2-2-O-C6H2)3CH]3−) was observed to undergo facile elimination of H2 and cleavage of the N–N bond in the presence of 9-borabicyclo[3.3.1]nonane (9-BBN), AlMe3, and ZnMe2. Treatment of 3-Na with 9-BBN and ZnMe2 afforded the nitride complex [{K(dme)2}2{(O3)Nb}2(μ-N)2] (2-Na). The reaction of 3-Na with AlMe3 afforded [{Na(dme)}2{(O3)AlMe}2(NbMe2)2(μ-N)2] (5). The nitride complex 2-Na was treated with 9-BBN and AlMe3 to form [{Na(dme)}2{(O3)Nb}(μ-NH)(μ-NBC8H14){Nb(O3C)}] (4) and 5, respectively. Complex 2-Na, 4, and 5 were structurally characterized.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Activation of dinitrogen under mild conditions has been of long-standing interest in chemistry owing to its industrial and biological significance. In this context, since the discovery of the first dinitrogen complex [Ru(N2)(NH3)5]2+ in 1965 [1], a number of dinitrogen complexes have been synthesized and characterized [2,3,4]. A variety of coordination modes have been found in dinitrogen complexes to date, and dinitrogen complexes show diverse reactivity patterns [5,6,7,8,9,10]. With dinuclear complexes, the side-on end-on coordination mode produces substantial polarization of the coordinated dinitrogen [10,11,12,13], and seminal works from Fryzuk have demonstrated that complexes of this type display a rich functionalization chemistry [14]. However, only a limited number of side-on end-on dinitrogen complexes are known [11,15,16,17]. Therefore, this coordination mode becomes one of synthetic targets in dinitrogen chemistry.
The reactions of Lewis acids with dinitrogen complexes provide suitable reference points to evaluate the reactivity of coordinated N2 toward electrophiles. The reactions of mononuclear dinitrogen complexes with group 13 Lewis acids are known and frequently proceed at the β-N atom to form stable adducts [18,19,20,21,22]. Addition of group 13 Lewis acids to side-on end-on dinitrogen hydride complexes has also been reported to occur at the β-N atom. For example, Fryzuk described the reaction of [{(NPN)Ta}2(μ-η1:η2-N2)(μ-H)2] ([NPN] = [PhP(CH2SiMe2NPh)2]2−) with EX3 (B(C6F5)3, AlMe3, and GaMe3) to give the corresponding adducts [{(NPN)Ta}2(μ-η1:η2-N2EX3)(μ-H)2] [23]. The analogous titanium complex [{(PNP)Ti}2(μ-η1:η2-N2)(μ-H)2] ([PNP] = 4,5-bis(diisopropylphosphino)-2,7,9,9-tetramethyl-9H-acridin-10-ide) was also reported to initially form the Lewis acid–base adducts with B(C6F5)3 and AlMe3 [16].
We have previously reported the synthesis of a series of anionic dinuclear Nb(IV) hydride complexes containing alkali metal ions [{M(dme)}2{(O3)Nb}2(μ-H)4] (1-M, M = Li, Na, K, [O3]3− = [(3,5-tBu2-2-O-C6H2)3CH]3−) and the counterion dependence of their reactivity toward N2 [24]. The potassium derivative 1-K readily reacted with N2 via the reductive elimination of two equivalents of H2 to yield the nitride bridged complex [{K(thf)2}2{(O3)Nb}2(μ-N)2] (2-K) [25]. For the lithium and sodium derivatives, the reactions with N2 did not result in the cleavage of the N2 triple bond but led to the elimination of only one equivalent of H2 and the formation of the corresponding side-on end-on dinitrogen hydride complexes. The lithium salt of the side-on end-on complex was unstable and converted to the end-on bridging dinitrogen complex with concomitant loss of the birding hydride ligands, whereas the sodium salt [{Na(dme)}2{(O3)Nb}2(μ-η1:η2-N2)(μ-H)2] (3-Na) was stable enough to use as a starting material for further reactivity studies.
The complex 3-Na is the first example of an anionic side-on end-on dinitrogen complex, and the anionic charge is expected to enhance the nucleophilicity of the β-N atom. We preliminary demonstrated that the reaction of 3-Na with Me3SiCl occurred at the β-N atom of the coordinated N2 unit [25]. Therefore, we were interested in the reactions with other Lewis acids to investigate the nucleophilicity of the N2 unit in the anionic complex 3-Na. Here we report the reactions of the side-on end-on dinitrogen hydride complex 3-Na with ZnMe2, AlMe3, and 9-borabicyclo[3.3.1]nonane (9-BBN).
2. Results
2.1. Reaction with 9-BBN
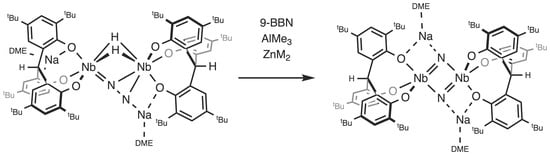
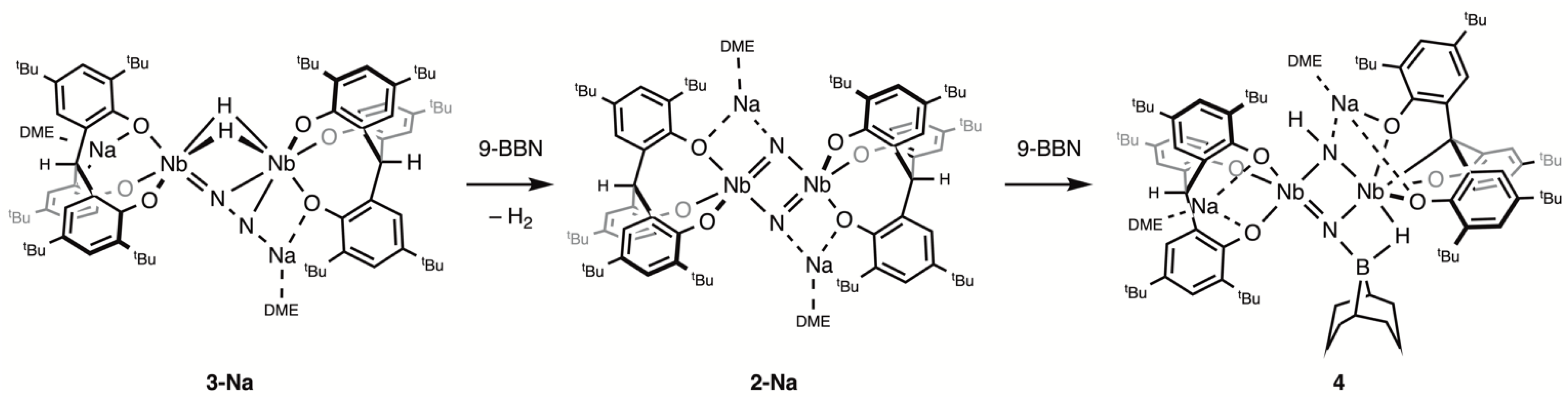
Addition of 9-BBN to a green solution of 3-Na in toluene at room temperature gradually gave a brown solution with concomitant liberation of H2, from which [{Na(dme)}2{(O3)Nb}2(μ-N)2] (2-Na) was isolated as a brown powder in a 67% yield upon workup (Scheme 1). Monitoring the reaction by 1H NMR spectroscopy in C6D6 indicated that 9-BBN was left unreacted. To probe the origin of the eliminated H2, the deuterated complex [{Na(dme)}2{(O3)Nb}2(μ-η1:η2-N2)(μ-D)2] (3-Na-D2) was allowed to react with 9-BBN, which resulted in the formation of 2-Na and D2. The complex 3-Na is stable in toluene in the absence of 9-BBN. These findings indicate that 9-BBN promotes the reductive elimination of H2, providing two electrons for N–N bond cleavage to form the nitride complex 2-Na.
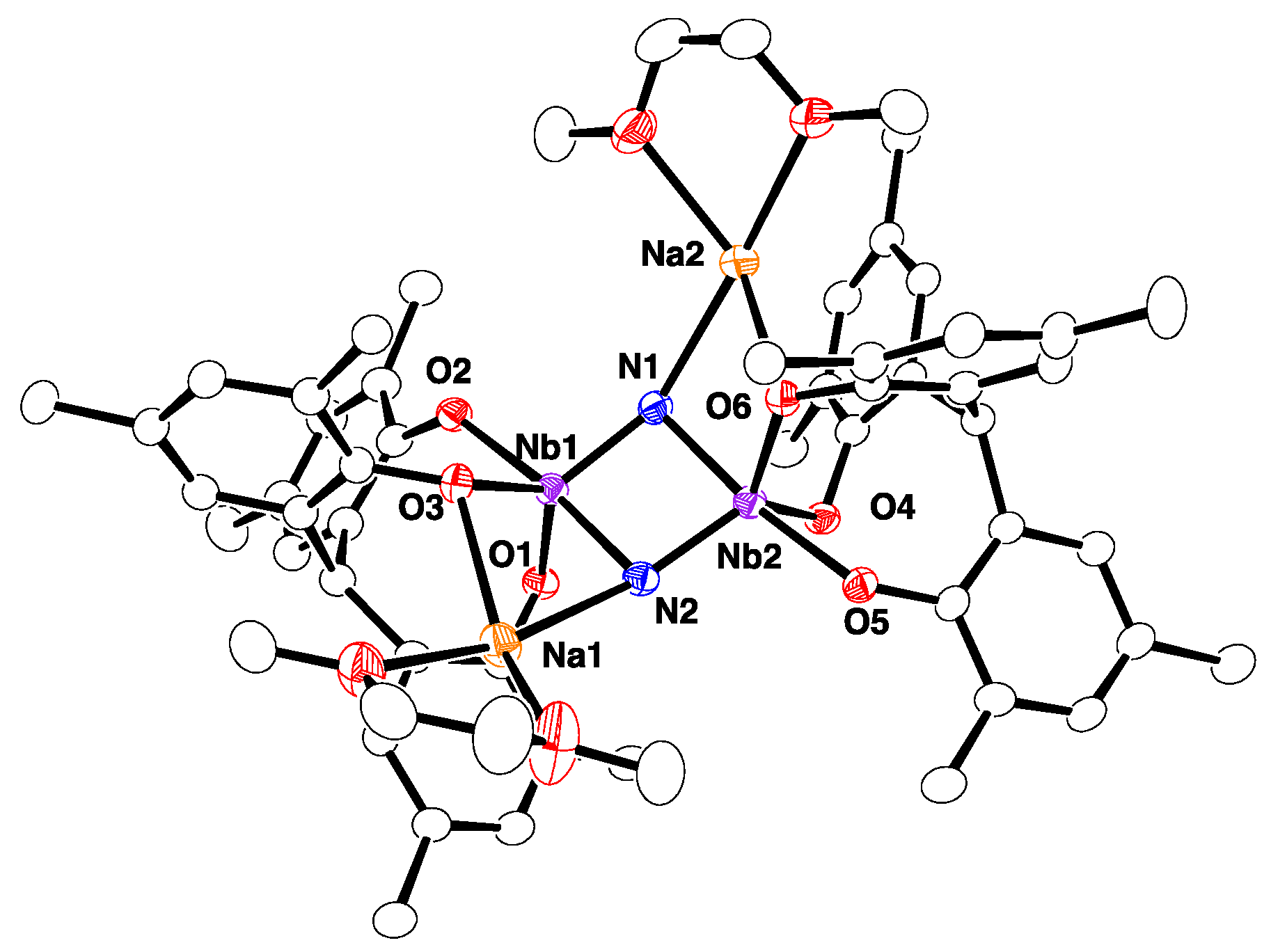
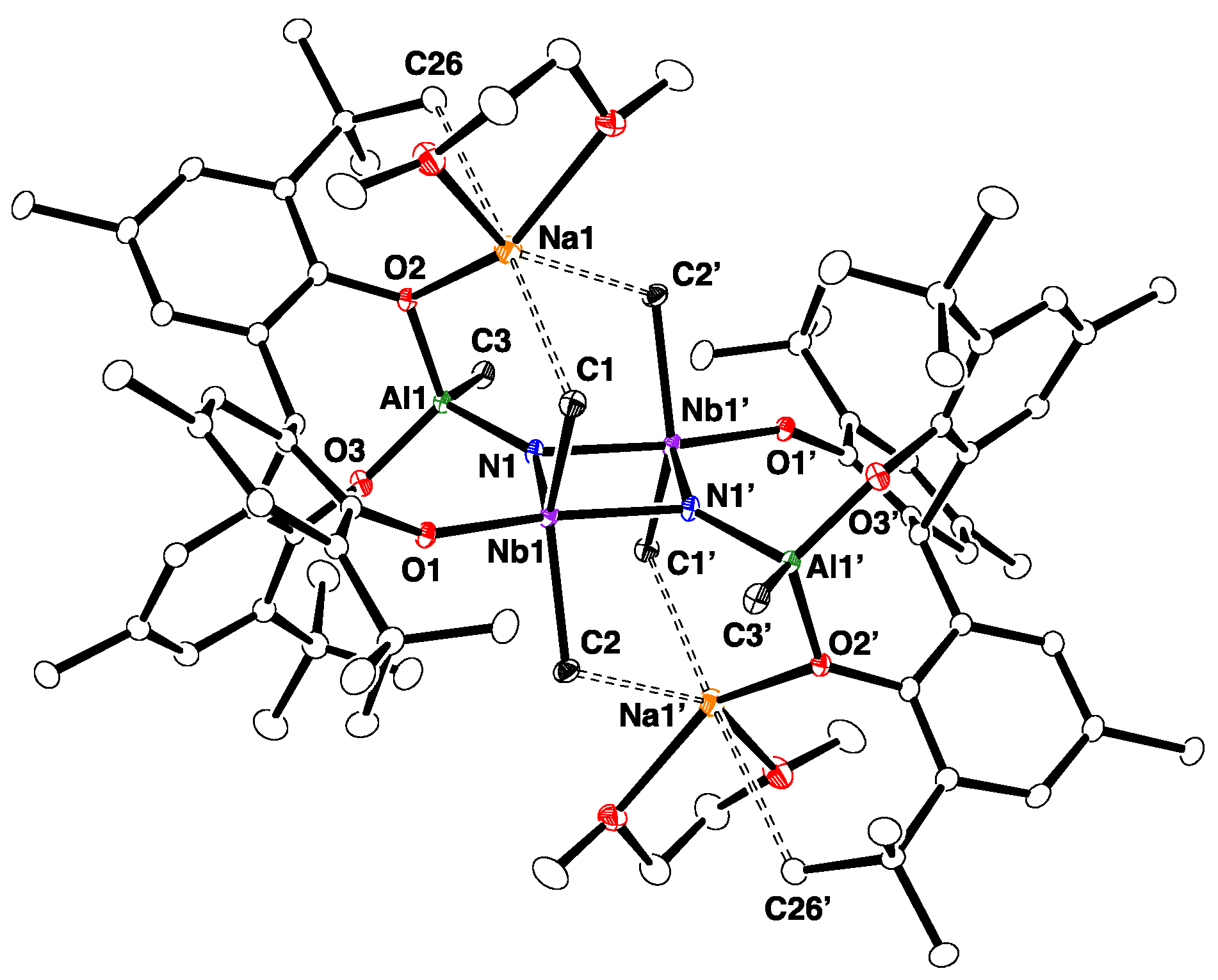
The molecular structure of 2-Na, which was determined by single-crystal X-ray diffraction (Figure 1), revealed the dimer to be bridged by two nitride ligands. The structure of 2-Na closely resembles that of the potassium salt 2-K [25]. As with the potassium salt 2-K, the complex occurs as an ion pair, wherein each DME-solvated sodium cation is coordinated by a nitride group and a phenoxide group. The planar Nb2N2 core of 2-Na is asymmetric, such that the Nb–N bond lengths are different (Nb1-N1 = 1.842(2) Å, Nb2–N2 = 1.838(2) Å, and Nb1-N2 = 2.025(2) Å, Nb2–N1 = 2027(2) Å). The average Nb–N bond length of 1.933(2) Å is similar to that of [{Na(dme)}2{[p-tBu-calix[4]-(O)4]Nb}2(μ-N)2] (average 1.910(8) Å) [26].
Reactions of mono- and dinuclear dinitrogen complexes with hydroboranes were reported to result in B–N bond formation due to the considerable strength of a B–N bond [27,28,29,30]. In the reaction with 2-Na, 9-BBN was observed to induce H2 elimination and N–N bond cleavage rather than undergo N-borylation. This is in contrast with what Fryzuk reported for the reaction of [{(NPN)Ta}2(μ-η1:η2-N2)(μ-H)2] with 9-BBN, which initially formed [{(NPN)TaH}(μ-N2BC8H14)(μ-H)2{Ta(NPN)}] via hydroboration of the Ta–N2 unit [31,32]. The resulting boryl–N2 complex subsequently underwent H2 reductive elimination, N–N bond cleavage, and ligand arrangement, yielding the borylimide–nitride complex. This difference is presumably due to the great tendency of 2-Na to reductively eliminate H2.
The 9-BBN-induced formation of 2-Na from 3-Na is closely related to the reaction of [{(PNP)Ti}2(μ-η1:η2-N2)(η-H)2] with pinacol borane (HBpin), wherein the binding of the borane center to the coordinated N2 unit was proposed to trigger reductive elimination of H2 and N–N bond cleavage [16]. In addition, the nitride bridged titanium dimer was shown to react with HBpin, yielding the borylimide complex. Mézailles also reported that the N2-derived nitride complex [(PPP)Mo(N)I] (PPP = PhP(CH2CH2PCy2)2) reacted with HBpin to afford borylamines via stepwise functionalization at the nitride group [33]. Therefore, it was of interest to investigate the possibility of hydroboration of the bridging nitride ligands in 2-Na.
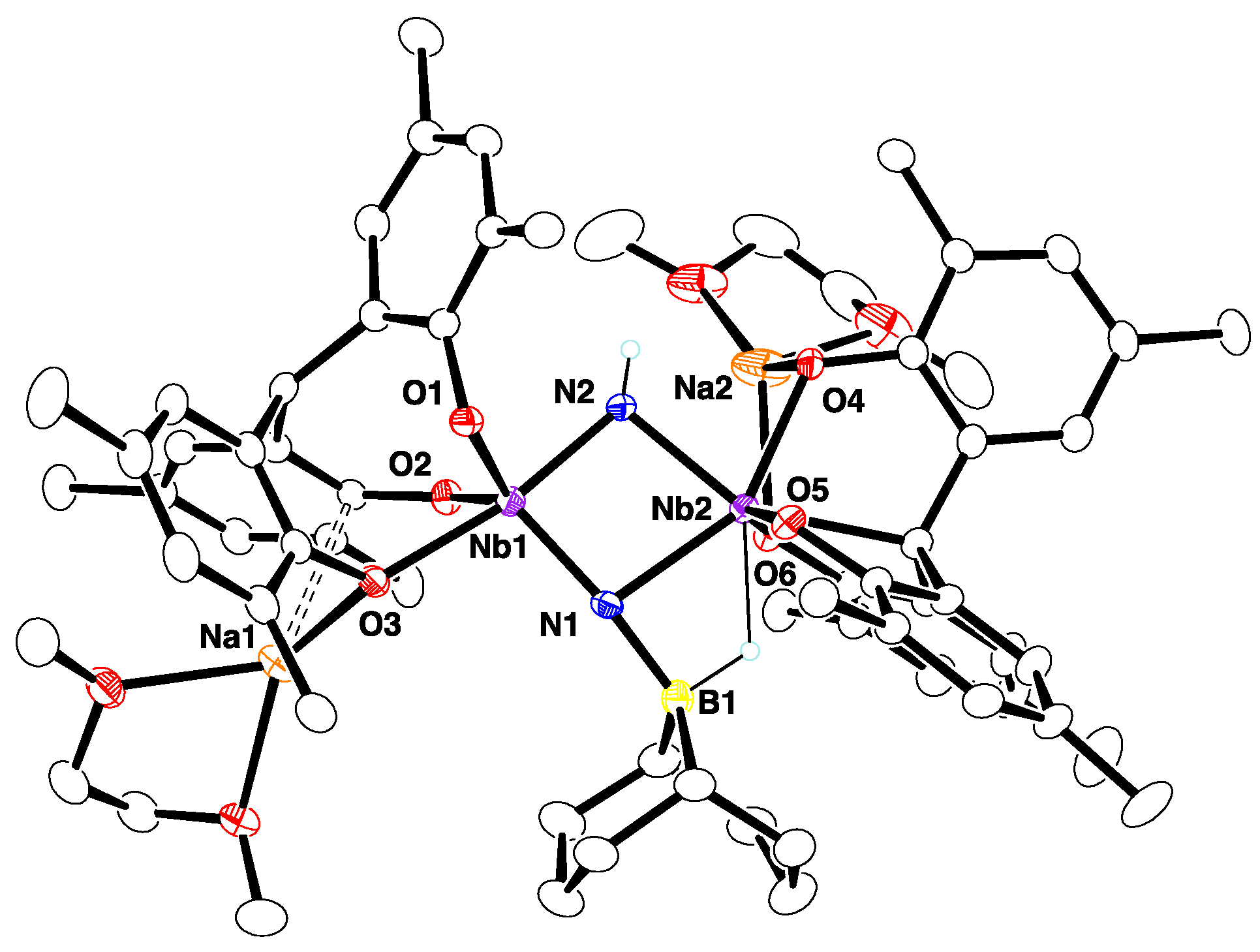
On the NMR tube scale, no reaction was observed at room temperature between a 1:1 mixture of 2-Na and 9-BBN in C6D6. Heating at 80 °C for 3 days led to the consumption of all of the 9-BBN and the formation of [{Na(dme)}2{(O3)Nb}(μ-NH)(μ-NBC8H14){Nb(O3C)}] (4) (Scheme 1). However, a small amount of 2-Na remained unreacted because 9-BBN partially decomposed during the reaction. Therefore, further addition of 9-BBN gave complete conversion to 4. The reaction was scaled up using excess 9-BBN to give 4 in a 62% isolated yield. The X-ray crystal structure of 4 (Figure 2) confirmed its composition and revealed that the product was the result of an N–B and N–H bond formation and a [O3] methine C–H bond scission.
The diniobium complex 4 is C1 symmetric. The original [O3] ligand, which was bound to the Nb2 atom, was transformed into a tetradentate [O3C] ligand via C–H activation of the bridgehead methine, while the other [O3] ligand bound to the Nb1 atom remained intact. The two Nb atoms were bridged by a parent imide and a 9-BBN-bound nitride ligand, with bond lengths being Nb1–N1 = 1.855(2), Nb1–N2 = 1.942(2), Nb2–N1 = 2.202(2), and Nb2–N2 = 2.111(2) Å. A 9-BBN unit was attached to a nitride group and exhibited a B1–H1–Nb1 agnostic interaction. The B1–N1 bond length of 1.541(4) Å was in the range expected for an N→B dative bond [34,35]. These geometrical parameters are consistent with the bonding sequence shown in Scheme 1. The central Nb2N2 core of 4 is structurally related to the motif formed via the hydroboration of coordinated N2 in [{(NPN)Ta}2(μ-η1:η2-N2)(μ-H)2] [31,32]. Attempts to observe and isolate any intermediates in the conversion of 2-Na to 4 were unsuccessful, preventing elucidation of the mechanism of the reaction. The source of the NH proton remains to be determined.
The 1H NMR spectrum of 4 indicates a highly asymmetric compound, with twelve inequivalent tert-butyl singlets and one methine singlet (5.82 ppm). The N–H resonance was observed at 10.6 ppm and split into a doublet (1JNH = 72.2 Hz) when the 15N-labeled complex was used. The B–H resonance could not be clearly assigned due to overlap with resonances from the BC8H14 group. The 11B NMR spectrum contained a resonance at 50.3 ppm. The 15N NMR spectrum of the 15N-labeled complex displayed 374 and 445 ppm, which were shifted upfield in comparison to that of 2-Na-15N at 680 ppm. The resonance at 374 ppm was assigned to the NH ligand, but the large quadruple moment of the 93Nb nucleus prevented observation of the N–H scalar coupling.
2.2. Reactions with ZnMe2 and AlMe3
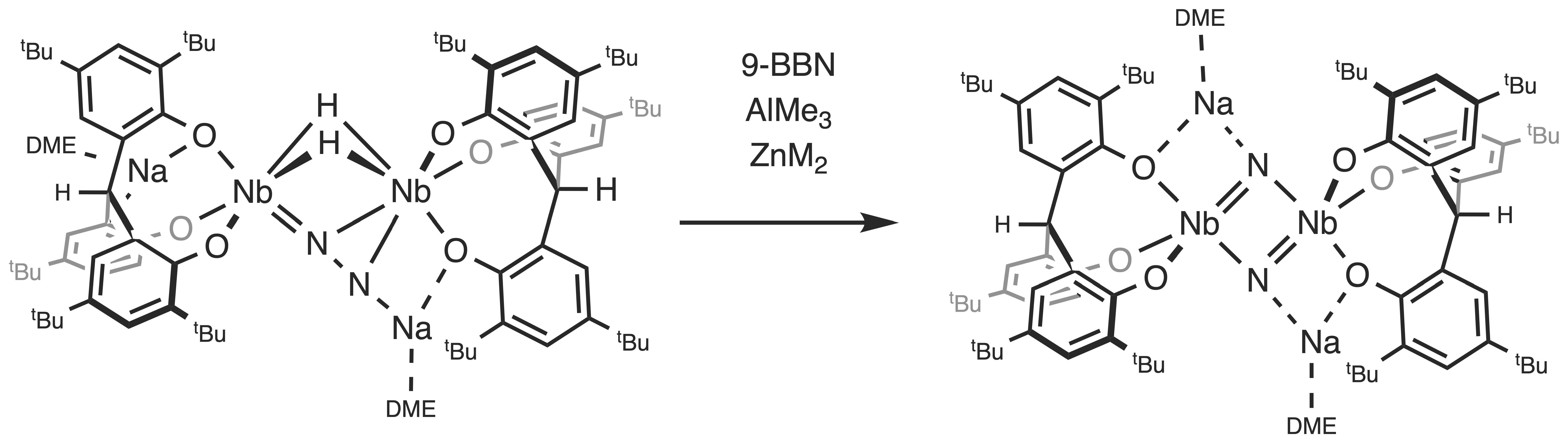
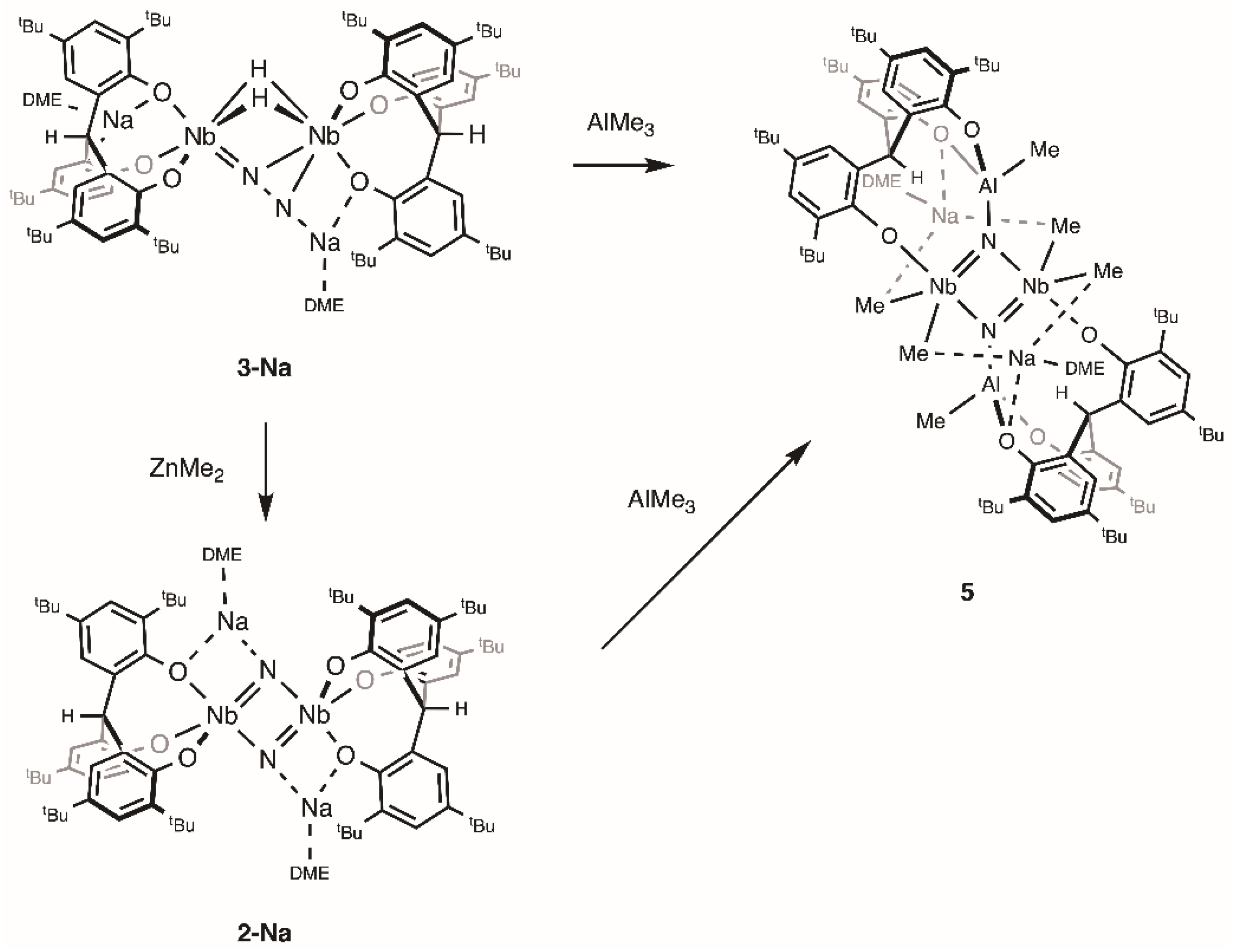
The borane 9-BBN mediates the conversion of 3-Na to 2-Na by behaving as a Lewis acid. Prior to reductive elimination of H2, the reaction may proceed through a putative borane adduct. The exact role of 9-BBN is unclear, because the interaction may occur at the coordinated N2 unit, the phenoxide groups, or the hydride ligands. Aiming to isolate acid–base adducts and confirm where the interaction occurs, we studied the behavior of the side-on end-on N2 complex 3-Na with other Lewis acids, ZnMe2 and AlMe3 (Scheme 2). However, we found that these Lewis acids also promote N–N bond cleavage of 3-Na.
Addition of ZnMe2 to 3-Na in toluene at room temperature resulted in an immediate color change from dark green to brown and the formation of a mixture of products. The products contained 2-Na according to 1H NMR spectroscopy, but they proved to be inseparable upon workup. In contrast to the reaction with 9-BBN, ZnMe2 was consumed during the reaction. The formation of CH4 was observed but attempts to identify any product containing Zn were unsuccessful.
When two equivalents of AlMe3 were added to a solution of 2-Na in toluene, the reaction mixture immediately turned brown with the concomitant liberation of H2. Workup of the reaction mixture led to the isolation of [{Na(dme)}2{(O3)AlMe}2(NbMe2)2(μ-N)2] (5) as a brown powder in 69%. The complex 5 was also prepared by the reaction of 3-Na with two equivalents of AlMe3 in toluene at room temperature. This suggests that the reaction of 3-Na with AlMe3 initially yields the nitride complex 2-Na in a manner analogous to the reaction with 9-BBN. The nitride complex 2-Na would then form the Lewis acid–base adducts with AlMe3, which would undergo further arrangements to produce 5. The potassium salt was also prepared by the analogous reaction of 2-K with AlMe3.
The X-ray crystal structure of 5 (Figure 3) revealed that the molecule lies on a crystallographic inversion center in the middle of the Nb2N2 core. Each niobium atom adopts a trigonal bipyramidal geometry, with two methyl groups, two nitride groups, and one phenoxide group of the [O3] ligand. The complex 5 contains two Al atoms, each of which is tetrahedrally coordinated by two phenoxide groups of the [O3] ligand, one methyl group, and one nitride group. The Nb2N2Al2 core closely approaches planarity, imparting a trigonal plane geometry to each nitrogen atom. Two solvated sodium cations are retained as a part of a cluster structure, each being bridged to the Nb2Al core by two methyl groups and one phenoxide group. The Nb–N bond lengths of 1.8719(13) and 2.0870(13) Å are comparable with those of 2-Na. The Al–N bond length of 1.8980(14) Å is longer than those for Al–N(amide) bonds [36,37] and shorter than those for Al–N dative bonds [38,39].
The 1H NMR spectrum of 5 shows four singlets for the tert-butyl groups in a 1:1:2:2 ratio, which is consistent with the average C2h symmetry in the molecule. The methine protons of the [O3] ligand appear as a singlet at 7.00 ppm. The two singlets for the Nb–Me and Al–Me groups are observed at 1.46 and −0.02 ppm in a 2:1 ratio, respectively. The 15N NMR spectrum of the 15N-labeled complex displays a single resonance upfield shifted to 553 ppm, indicative of coordination to aluminum.
3. Conclusions
We demonstrated that the side-on end-on dinitrogen hydride complex, 3-Na, readily underwent reductive elimination of H2 in the presence of Lewis acids such as 9-BBN, AlMe3, and ZnMe2, followed by N–N bond cleavage to produce 2-Na. The reactions are proposed to proceed via initial formation of Lewis acid adducts with 3-Na at the β-N atom of the coordinated dinitrogen, taking into account the nucleophilicity of the β-N atom of the side-on end-on N2 unit and the silylation of the β-N atom by the reaction of 3-Na with Me3SiCl [24]. However, attempts to observe and isolate any adducts failed, because the Lewis acid adducts of 3-Na are quite prone to reductive elimination of H2. When other Lewis acids such as HBpin and B(C6F5)3 were used, the reactions yielded intractable mixtures. The resulting nitride complex, 2-Na, was found to further react with 9-BBN and AlMe3 to produce 4 and 5, respectively. Further work will focus on the functionalization of the coordinated N2 unit in 3-Na.
4. Materials and Methods
4.1. General Information
All manipulations were carried out using standard Schlenk techniques or in a glovebox under an atmosphere of argon. Anhydrous hexane, pentane, and toluene were dried by passage through two columns of activated alumina and a Q-5 column, while anhydrous THF, Et2O, and DME were dried by passage through two columns of activated alumina. Anhydrous deuterated benzene (benzene-d6) was dried and degassed over a potassium mirror prior to use. NMR spectra were recorded on a JEOL ECX-500 spectrometer (JEOL Ltd., Tokyo, Japan). 1H NMR spectra were reported with reference to solvent resonances of C6D6 residual protons (δ = 7.16 ppm). 13C NMR spectra were referenced to solvent (peaks δ = 128.06 (C6D6) ppm). 11B chemical shifts were referenced to BF3•OEt2 (neat at 0.0 ppm) as an external standard. 15N chemical shifts were referenced to 90% formamide in dimethyl sulfoxide-d6 (112.7 ppm with respect to NH3 at 0.0 ppm) as an external standard. Complexes 2-K and 3-Na were prepared following the literature procedure [23,24]. Elemental analyses (C, H, and N) were carried out on an Elementar vario MICRO Cube (Elementar, Frankfurt, Germany). IR spectra were recorded on a JASCO VIR-200 spectrometer (JASCO, Tokyo, Japan). Spectra and structures are available in Supplementary Materials.
4.2. Synthesis of 2-Na
To a solution of 3-Na (170 mg, 0.100 mmol) in toluene (6 mL) was added 9-BBN (25.0 mg, 0.205 mmol). The solution was stirred at room temperature overnight and changed color from green to brown. The volatile materials were removed under vacuum. The residue was washed with hexane and was then dried under vacuum, leaving a brown powder of 2-Na (67% yield, 114 mg). 1H NMR (C6D6, 400 MHz, 323 K): δ 1.23 (s, 54H, tBu), 1.69 (s, 54H, tBu), 2.83 (s, 12H, DME), 2.86 (s, 8H, DME), 5.66 (s, 2H, CH), 7.14 (d, J = 2.6 Hz, 6H, ArH), and 7.37 (d, J = 2.6 Hz, 6H, ArH). 13C NMR (C6D6, 125.8 MHz): δ 31.5, 31.8, 34.3, 36.0 (tBu), 59.5 (DME), 60.1 (CH), 70.3 (DME), 122.5, 125.7, 138.8, 140.5, and 161.3. One peak for the aromatic carbon was not assigned, probably due to overlapping. Calcd for C94H142N2Na2Nb2O10: C, 66.73; H, 8.46; N, 1.66. Found: C, 66.44; H, 8.38; N 0.75.
4.3. Synthesis of 2-Na-15N
The 15N-labeled analogue was prepared in a manner identical to that used for 2-Na, except for using the 15N-labeled precursor 3-Na-15N. 15N NMR (C6H6, 50.7 MHz): δ 679.1.
4.4. Synthesis of 4
To a solution of 3-Na (42.4 mg, 25.0 μmol) in toluene (6 mL) was added 9-BBN (12.2 mg, 100 μmol). The solution was stirred at room temperature overnight and was then allowed to warm to 80 °C. The color of the solution turned from green to brown to orange. After stirring for 2 days, the volatile materials were removed under vacuum. The residue was washed with hexane, leaving 4 as a yellow powder (62%, 28.0 mg). 1H NMR (C6D6, 400 MHz): δ 1.20, 1.21, 1.24, 1.36, 1.39, 1.50, 1.56, 1.62, 1.66, 1.67, 1.69, 1.71 (s, 9H each, tBu), 0.93–3.84 (complicated overlapping resonances, 14H total, BC8H14), 2.40 (br, 12H, DME), 2.53 (br, 8H, DME), 5.82 (s, 1H, CH), 7.06, 7.10, 7.18, 7.23, 7.30, 7.32, 7.36, 7.43, 7.63, 7.66, 8.45, 8.53 (d, J = 2 Hz, 1H each, ArH), 10.57 (s, 1H, NH). 13C NMR (C6D6, 125.8 MHz): δ 31.2, 31.5, 31.6 (2C), 31.7, 31.95, 31.99, 32.1, 32.3, 32.4, 32.47, 32.53, 33.7 (2C), 34.15, 34.21, 34.5 (2C), 34.8, 34.9, 35.3, 35.6, 36.0, 36.3 (tBu), 25.1, 25.9 (2C), 28.0, 33.5, 36.1, 39.9 (9–BBN), 58.9 (CH), 58.8, 70.2 (DME), 119.7, 120.1, 120.4, 122.0, 122.2, 122.7, 122.8, 124.3, 124.9, 125.8, 126.6, 129.0, 130.7, 132.1, 133.7, 135.1, 135.9, 138.2, 140.1, 140.16, 140.24, 140.9, 141.1, 141.2, 142.0, 142.8, 143.7, 147.2, 148.5, 149.7, 159.1, 160.2, 160.9, 163.6, 164.0, 164.8. One peak for 9-BBN was not assigned, probably due to overlapping. 11B NMR (C6D6, 160.5 MHz) δ 50.3. Anal. Calcd for C102H157BN2Na2Nb2O10: C, 67.54; H, 8.72; N, 1.54. Found: C, 67.10; H, 8.55; N, 1.57. IR (KBr; ν/cm–1) 3447 (N–H).
4.5. Reaction of 2-Na with 9-BBN
To a solution of 2-Na (15.2 mg, 8.98 μmol) in C6D6 (0.6 mL) was added 9-BBN (1.1 mg, 9.0 μmol). The 1H NMR spectrum was identical to that of a sample of isolated 4.
4.6. Reaction of 2-K-15N with 9-BBN
To a solution of 2-K (14.5 mg, 8.40 μmol) in C6D6 (0.6 mL) was added 9-BBN (1.0 mg, 8.2 μmol). The 1H NMR spectrum was similar to that of a sample of isolated 4, except for a resonance attributed to the N–H group. 1H NMR (C6D6, 500.2 MHz): 1.19, 1.22, 1.30, 1.40, 1.43, 1.46, 1.56, 1.60, 1.67, 1.71, 1.74, 1.75 (s, 9H each, tBu), 0.94–3.76 (complicated overlapping resonances, 14H total, BC8H14), 2.66 (s, 15H, DME), 2.72 (s, 10H, DME), 5.83 (s, 1H), 7.06, 7.22, 7.24, 7.39, 7.40, 7.47, 7.57, 7.61, 8.36, 8.57 (br, 1H each, ArH) 10.96 (d, 1JNH = 72.2 Hz, 1H, NH). Two peak for Ar–H were not assigned, probably due to overlapping. 11B NMR (C6D6, 160.5 MHz,); δ 60.8. 15N NMR (C6D6, 50.7 MHz): δ 374.4 (NH), 444.7 (NB).
4.7. Reaction of 3-Na with ZnMe2
To a solution of 3-Na (16.8 mg, 9.92 μmol) in C6D6 (0.6 mL) was added ZnMe2 (1.05 M in hexane, 10.0 μL, 10.5 μmol). The 1H NMR spectrum contained resonances identical to that of a sample of isolated 2-Na.
4.8. Synthesis of 5
To a solution of 3-Na (104 mg, 61.5 μmol) in toluene (4 mL) was added a solution of AlMe3 (1.05 M in hexane, 117 μL, 123 μmol) at room temperature. The solution turned brown instantly and was stirred overnight. The volatile materials were removed under vacuum, and then the residue was washed with hexane. The product 5 was isolated as an orange solid (76.8 mg, 41.8 μmol, 69%). 1H NMR (C6D6, 500.2 MHz): δ −0.02 (s, 6H, AlCH3), 1.32 (s, 36H, tBu), 1.46 (s, 12H, NbCH3), 1.50 (s, 18H, tBu), 1.59 (s, 36H, tBu), 1.67 (s, 18H, tBu), 2.39 (br, 12H, DME), 2.55 (br, 8H, DME), 7.00 (s, 2H, CH), 7.28 (d, J = 2.6 Hz, 4H, ArH), 7.40 (br, 4H, ArH), 7.49 (d, J = 2.5 Hz, 2H, ArH), 8.23 (br, 2H, ArH). Anal. Calcd for C108H180Al2N2Na2Nb2O14: C, 64.33; H, 9.00; N, 1.39. Found: C, 64.61; H, 9.28; N, 1.59. The low solubility of 5 in solvent prevented us from acquiring 13C NMR spectra.
4.9. Reaction of 2-Na with AlMe3
To a solution of 2-Na (16.8 mg, 9.93 μmol) in toluene (2 mL) was added AlMe3 (1.05 M in hexane, 20 μL, 21 μmol). The 1H NMR spectrum was identical to that of a sample of isolated 5.
4.10. Reaction of 2-K-15N with AlMe3
To a solution of 2-K-15N (10.3 mg, 5.97 μmol) in C6D6 (0.6 mL) was added AlMe3 (1.05 M in hexane, 11 μL mg, 11.6 μmol). The 1H NMR spectrum was identical to that of a sample of isolated 5. 15N NMR (C6D6, 50.7 MHz): δ 552.9.
4.11. X-ray Crystallography
Single crystals were immersed in immersion oil on a micromount and transferred to a Rigaku Varimax with a Saturn system or a Rigaku XtaLAB Synergy-DW system equipped with a Rigaku GNNP low temperature device (Tokyo, Japan). Data were collected under a cold nitrogen stream at 123 K or 173 K using graphite-monochromated MoKα (λ = 0.71073 Å) or CuKα (λ = 1.54184 Å) radiation. Equivalent reflections were merged, and the images were processed with the CrysAlisPro software 1.171.42.64a (Rigaku Oxford Diffraction, Japan). Empirical absorption corrections were applied. All structures were solved by direct method using SHELXT [40] and refined by full-matrix least-squares method on F2 for all data using SHELXL [41] with the Olex2 program [42]. All hydrogen atoms were placed at their geometrically calculated positions. For 4, the hydrogen atoms of the NH group and the BH group were located in the Fourier map and refined isotropically. For 2-Na and 4, some residual electron density was difficult to model, the program SQUEEZE [43] was used to remove the contribution of the electron density in the solvent region from the intensity data. For 2-Na, one tert-butyl group was disordered. A void space contains 175 electrons per unit cell, which could be attributed to distorted pentane molecules (one molecule in the asymmetric unit). A large residual peak in the final difference map was located in the Nb2N2 core. This peak is believed to be due to a small amount of twinning in the crystal. For 4, two tert-butyl groups and one DME molecule were disordered. For 4, a void space contains 1013 electrons per unit cell, which could be attributed to distorted pentane molecules (three molecules in the asymmetric unit). For 5, one benzene molecule was disordered.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules27175553/s1, NMR spectra (Figures S1–S11), crystallographic details (Table S1), (Figures S12–S14), structures and IR spectra (Figures S15–S17) for complexes 2-Na, 4, and 5. The CCDC deposition numbers 2195593–2195595 contain the supplementary crystallographic data for this paper, which can obtained free of charge via emailing [email protected], or by contacting The Cambridge Crystallographic Data Centre at 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
N.S. performed the experiments and analyzed the spectroscopic data; Y.I. and H.K. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI (Grant 22H02092).
Data Availability Statement
The original data presented in this study are available from the authors.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- Allen, A.D.; Senoff, C.V. Nitrogenpentammineruthenium(II) complexes. Chem. Commun. 1965, 621–622. [Google Scholar] [CrossRef]
- Hidai, M.; Mizobe, Y. Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev. 1995, 95, 1115–1133. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A. The Continuing Story of Dinitrogen Activation. Coord. Chem. Rev. 2000, 200, 379–409. [Google Scholar]
- Walter, M.D. Recent Advances in Transition Metal-Catalyzed Dinitrogen Activation. Adv. Organomet. Chem. 2016, 65, 261–377. [Google Scholar]
- Burford, R.J.; Fryzuk, M.D. Examining the Relationship between Coordination Mode and Reactivity of Dinitrogen. Nat. Chem. Rev. 2017, 1, 1–13. [Google Scholar]
- Kim, S.; Loose, F.; Chirik, P.J. Beyond Ammonia: Nitrogen–Element Bond Forming Reactions with Coordinated Dinitrogen. Chem Rev. 2020, 120, 5637–5681. [Google Scholar]
- Singh, D.; Buratto, W.R.; Torres, J.F.; Murray, L.J. Activation of Dinitrogen by Polynuclear Metal Complexes. Chem. Rev. 2020, 120, 5517–5581. [Google Scholar]
- Masero, F.; Perrin, M.A.; Dey, S.; Mougel, V. Dinitrogen Fixation: Rationalizing Strategies Utilizing Molecular Complexes. Chem. Eur. J. 2021, 27, 3892–3928. [Google Scholar]
- Tanabe, Y.; Nishibayashi, Y. Comprehensive Insights into Synthetic Nitrogen Fixation Assisted by Molecular Catalysts under Ambient or Mild Conditions. Chem. Soc. Rev. 2021, 50, 5201–5242. [Google Scholar]
- Lv, Z.-J.; Wei, J.; Zhang, W.-X.; Chen, P.; Deng, D.; Shi, Z.-J.; Xi, Z. Direct Transformation of Dinitrogen: Synthesis of N-Containing Organic Compounds via N–C bond Formation. Natl. Sci. Rev. 2020, 7, 1564–1583. [Google Scholar]
- Fryzuk, M.D.; Johnson, S.A.; Retting, S.J. New Mode of Coordination for the Dinitrogen Ligand: A Dinuclear Tantalum Complex with a Bridging N2 Unit That Is Both Side-On and End-On. J. Am. Chem. Soc. 1998, 120, 11024–11025. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A.; Patrick, B.O.; Albinati, A.; Mason, S.A.; Koetzle, T.F. New Mode of Coordination for the Dinitrogen Ligand: Formation, Bonding, and Reactivity of a Tantalum Complex with a Bridging N2 Unit That Is Both Side-On and End-On. J. Am. Chem. Soc. 2001, 123, 3960–3973. [Google Scholar] [CrossRef] [PubMed]
- Studt, F.; MacKay, B.A.; Fryzuk, M.D.; Tuczek, F. Spectroscopic Properties and Quantum Chemistry-Based Normal Coordinate Analysis (QCB-NCA) of a Dinuclear Tantalum Complex Exhibiting the Novel Side-On End-On Bridging Geometry of N2: Correlations to Electronic Structure and Reactivity. J. Am. Chem. Soc. 2004, 126, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Burford, R.J.; Yeo, A.; Fryzuk, M.D. Dinitrogen Activation by Group 4 and Group 5 metal Complexes Supported by Phosphine-Amido Containing Ligand Manifolds. Coord. Chem. Rev. 2017, 334, 84–99. [Google Scholar] [CrossRef]
- Wang, B.; Luo, G.; Nishiura, M.; Hu, S.; Shima, T.; Luo, Y.; Hou, Z. Dinitrogen Activation by Dihydrogen and a PNP-Ligated Titanium Complex. J. Am. Chem. Soc. 2017, 139, 1818–1821. [Google Scholar] [CrossRef]
- Mo, Z.; Shima, T.; Hou, Z. Synthesis and Diverse Transformation of a Dinitrogen Dititanium Hydride Complex Bearing Rigid Acridane-Based PNP-Pincer Ligands. Angew. Chem. Int. Ed. 2020, 59, 8635–8644. [Google Scholar] [CrossRef]
- Pun, D.; Lobkovsky, E.; Chirik, P.J. Indenyl Zirconium Dinitrogen Chemistry: N2 Coordination to an Isolated Zirconium Sandwich and Synthesis of Side-on, End-on Dinitrogen Compounds. J. Am. Chem. Soc. 2008, 130, 6047–6054. [Google Scholar] [CrossRef]
- Geri, J.B.; Shanahan, J.P.; Szymczak, N.K. Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc. 2017, 139, 5952–5956. [Google Scholar] [CrossRef] [Green Version]
- Apps, S.L.; White, A.J.P.; Miller, P.W.; Long, N.J. Synthesis and Reactivity of an N-Triphos Mo(0) Dinitrogen Complex. Dalton Trans. 2018, 47, 11386–11395. [Google Scholar] [CrossRef]
- Takahashi, T.; Kodama, T.; Watakabe, A.; Uchida, Y.; Hidai, M. Preparation and Characterization of Novel MU3-N2 Mixed Metal Complexes. J. Am. Chem. Soc. 1983, 105, 1680–1682. [Google Scholar] [CrossRef]
- Klein, H.F.; Ellrich, K.; Ackermann, K. Hetero-Bimetallic Dinitrogen Activation: X-ray Structure of the (Me3P)3CoN2AlMe2 Dimer. J. Chem. Soc. Chem. Commun. 1983, 16, 888–889. [Google Scholar] [CrossRef]
- Chatt, J.; Crabtree, R.H.; Jeffery, E.A.; Richards, R.L. The Basic Strengths of some Dinitrogen Complexes of Molybdenum(0), Tungsten(0), Rhenium(0), and Osmium(II). J. Chem. Soc. Dalton Trans. 1973, 11, 1167–1172. [Google Scholar] [CrossRef]
- Studt, F.; MacKay, B.A.; Johnson, S.A.; Patrick, B.O.; Fryzuk, M.D.; Tuczek, F. Lewis Adducts of the Side-On End-On Dinitrogen-Bridged Complex [{(NPN)Ta}2(μ-H)2(μ-η1:η2-N2)] with AlMe3, GaMe3, and B(C6F5)3: Synthesis, Structure, and Spectroscopic Properties. Chem. Eur. J. 2005, 11, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ishida, Y.; Kameo, H.; Sakaki, S.; Kawaguchi, H. Couterion Dependence of Dinitrogen Activation and Functionalization by a Diniobium Hydride Anion. Angew. Chem. Int. Ed. 2020, 59, 13444–13450. [Google Scholar] [CrossRef] [PubMed]
- Akagi, F.; Matsuo, T.; Kawaguchi, H. Dinitrogen Cleavage by a Diniobium Tetrahydride Complex: Formation of a Nitride and Its Conversion into Imide Species. Angew. Chem. Int. Ed. 2007, 46, 8778–8781. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Solari, E.; Scopelliti, R.; Floriani, C.; Re, N.; Rizzoli, C.; Chiesi-Villa, A. Dinitrogen Rearranging over a Metal–Oxo Surface and Cleaving to Nitride: From the End-On to the Side-On Bonding Mode, to the Stepwise Cleavage of the N≡N Bonds Assisted by NbIII-calix[4]arene. J. Am. Chem. Soc. 2000, 122, 3652–3670. [Google Scholar] [CrossRef]
- Coffinet, A.; Specklin, D.; Vendier, L.; Etienne, M.; Simonneau, A. Frustrated Lewis Pair Chemistry Enables N2 Borylation by Formal 1,3-Addition of a B−H Bond in the Coordination Sphere of Tungsten. Chem. Eur. J. 2019, 25, 14300–14303. [Google Scholar] [CrossRef]
- Coffinet, A.; Zhang, D.; Vendier, L.; Bontemps, S.; Simonneau, A. Borane-Catalysed Dinitrogen Borylation by 1,3-B–H Bond Addition. Dalton Trans. 2021, 50, 5582–5589. [Google Scholar] [CrossRef]
- Semproni, S.; Chirik, P.J. Dinitrogen Borylation with Group 4 Metallocene Complexes. Eur. J. Inorg. Chem. 2013, 2013, 3907–3915. [Google Scholar] [CrossRef]
- Ishino, H.; Ishii, Y.; Hidai, M. Synthsis of Boryldiazenido Complexes from Tungsten Dinitorgen Complexes. Chem. Lett. 1998, 27, 677–678. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; MacKay, B.A.; Johnson, S.A.; Patrick, B.O. Hydroboration of Coordinated Dinitrogen: A New Reaction for the N2 ligand that Results in Its Functionalization and Cleavage. Angew. Chem. Int. Ed. 2002, 41, 3709–3712. [Google Scholar] [CrossRef]
- MacKay, B.A.; Johnson, S.A.; Patrick, B.O.; Fryzuk, M.D. Functionalization and Cleavage of Coordinated Dinitrogen via Hydroboration Using Primary and Secondary Boranes. Can. J. Chem. 2005, 83, 315–323. [Google Scholar] [CrossRef]
- Espada, M.F.; Bennaamane, S.; Liao, Q.; Saffon-Merceron, N.; Massou, S.; Clot, E.; Nebra, N.; Fustier-Boutignon, M.; Mézailles, N. Room-Temperature Functionalization of N2 to Borylamine at a Molybdenum Complex. Angew. Chem. Int. Ind. 2018, 57, 12865–12868. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.W. The Crystal Structure of Ammonia-Borane, H3NBH3. J. Am. Chem. Soc. 1956, 78, 502–503. [Google Scholar] [CrossRef]
- Li, J.; Gao, D.; Hu, H.; Cui, C. Reaction of a Bulky Amine Borane with Lanthanide Trialkyls. Formation of Alkyl Lanthanide Imide Complexes. New J. Chem. 2015, 39, 7567–7570. [Google Scholar] [CrossRef]
- Gardiner, M.G.; Raston, C.L.; Skelton, B.W.; White, A.H. Anionic and Neutral Aluminum Bis(N,N′-di-tert-butylethylenediamide) Complexes: [Al{[N(t-Bu)CH2]2}2]− and [Al{[N(t-Bu)CH2]2}2]•. Inorg. Chem. 1997, 36, 12795–12803. [Google Scholar] [CrossRef]
- Schulz, S.; Thomas, F.; Priesmann, W.M.; Neiger, M. Syntheses and X-ray Structures of Base-Stabilized Iminoalanes. Organometallics 2006, 25, 1392–1398. [Google Scholar] [CrossRef]
- Dagorne, S.; Lavanant, L.; Welter, R.; Chassenieux, C.; Haquette, P.; Jaouen, G. Synthesis and Structural Characterization of Neutral and Cationic Alkylaluminum Complexes Based on Bidentate Aminophenolate Ligands. Organometallics 2003, 22, 3732–3741. [Google Scholar] [CrossRef]
- Rutherford, D.; Atwood, D.A. Unusual Alkylaluminum Amides, Adducts, and Aluminates Containing Lithium. J. Am. Chem. Soc. 1996, 118, 11535–11540. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Scheme 1.
Reaction of 3-Na with 9-BBN.

Figure 1.
Molecular structure of 2-Na with thermal ellipsoids set at 30% probability level. All hydrogen atoms and methyl groups of tert-butyl groups are omitted for clarity.
Figure 1.
Molecular structure of 2-Na with thermal ellipsoids set at 30% probability level. All hydrogen atoms and methyl groups of tert-butyl groups are omitted for clarity.

Figure 2.
Molecular structure of 4 with thermal ellipsoids set at 30% probability level. All hydrogen atoms on carbon and methyl groups of tert-butyl groups are omitted for clarity.
Figure 2.
Molecular structure of 4 with thermal ellipsoids set at 30% probability level. All hydrogen atoms on carbon and methyl groups of tert-butyl groups are omitted for clarity.

Scheme 2.
Reactions of 3-Na with ZnMe2 and AlMe3.

Figure 3.
Molecular structure of 5 with thermal ellipsoids set at 30% probability level. All hydrogen atoms and methyl groups of para-tert-butyl substitutes of aryloxide are omitted for clarity.
Figure 3.
Molecular structure of 5 with thermal ellipsoids set at 30% probability level. All hydrogen atoms and methyl groups of para-tert-butyl substitutes of aryloxide are omitted for clarity.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suzuki, N.; Ishida, Y.; Kawaguchi, H. Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex. Molecules 2022, 27, 5553. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175553
AMA Style
Suzuki N, Ishida Y, Kawaguchi H. Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex. Molecules. 2022; 27(17):5553. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175553
Chicago/Turabian StyleSuzuki, Naofumi, Yutaka Ishida, and Hiroyuki Kawaguchi. 2022. "Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex" Molecules 27, no. 17: 5553. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27175553