
Halogen-Bond Mediated [2+2] Photodimerizations: À la Carte Access to Unsymmetrical Cyclobutanes in the Solid State


Abstract
:1. Introduction
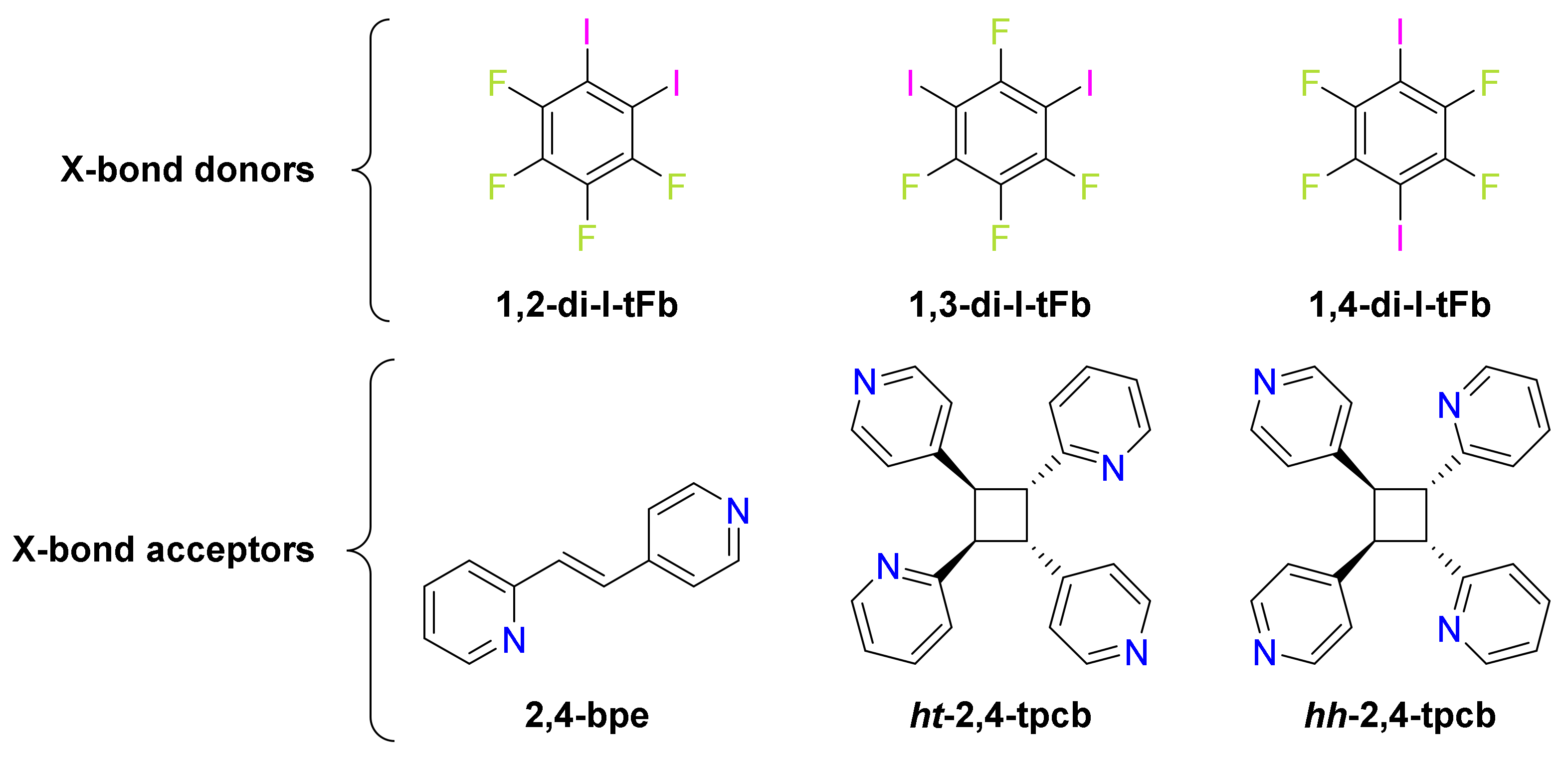
2. Results and Discussion
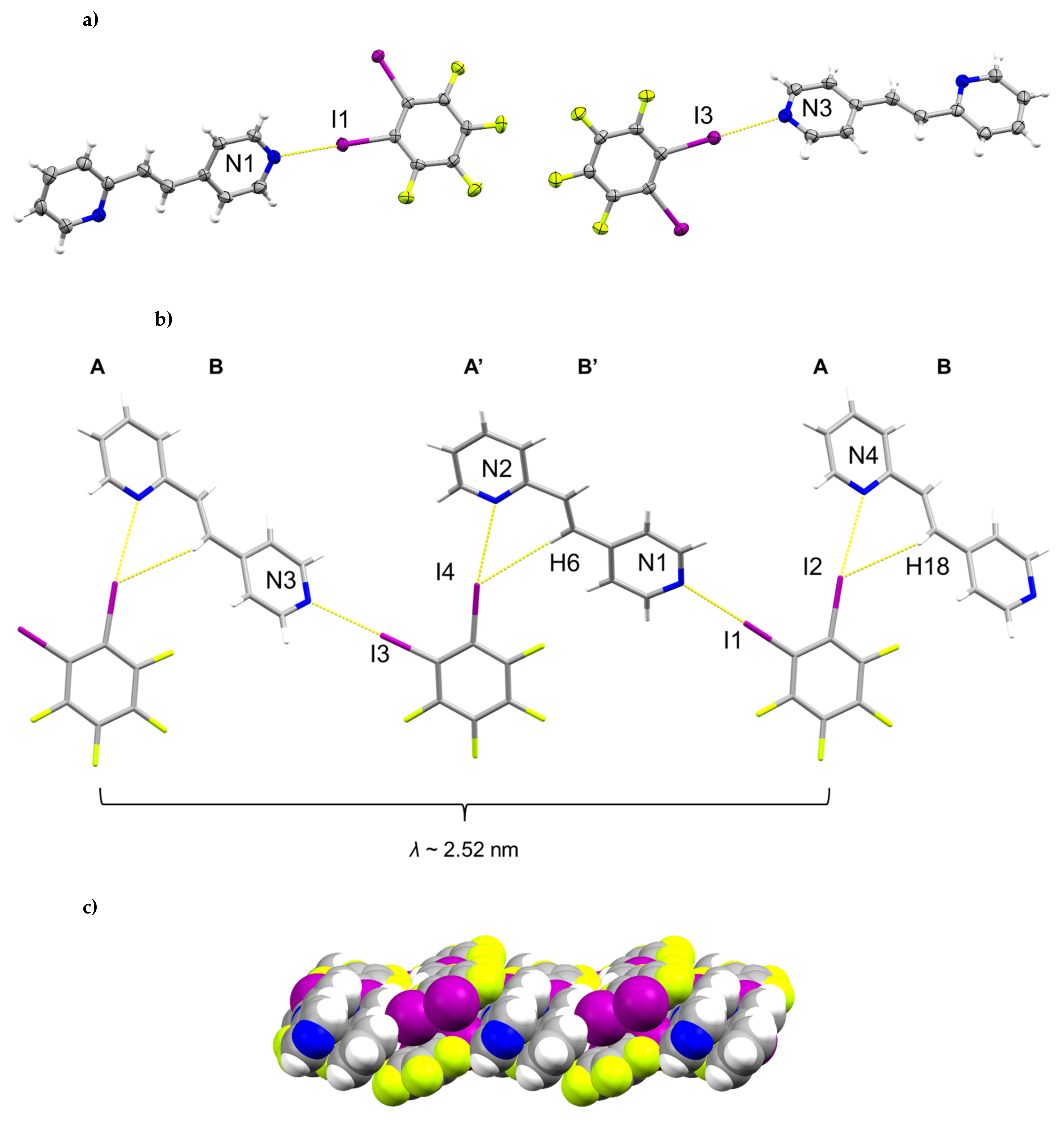
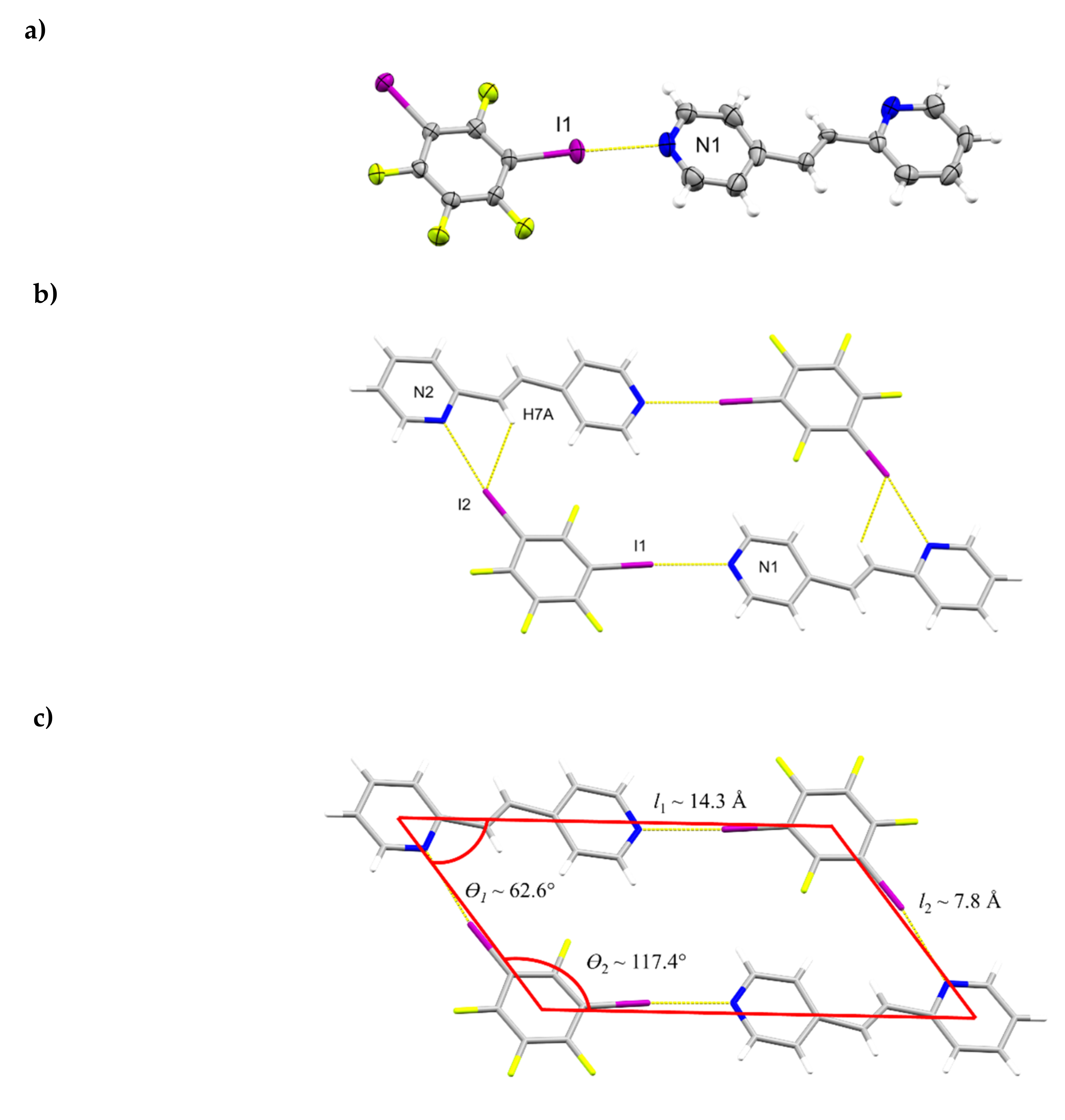
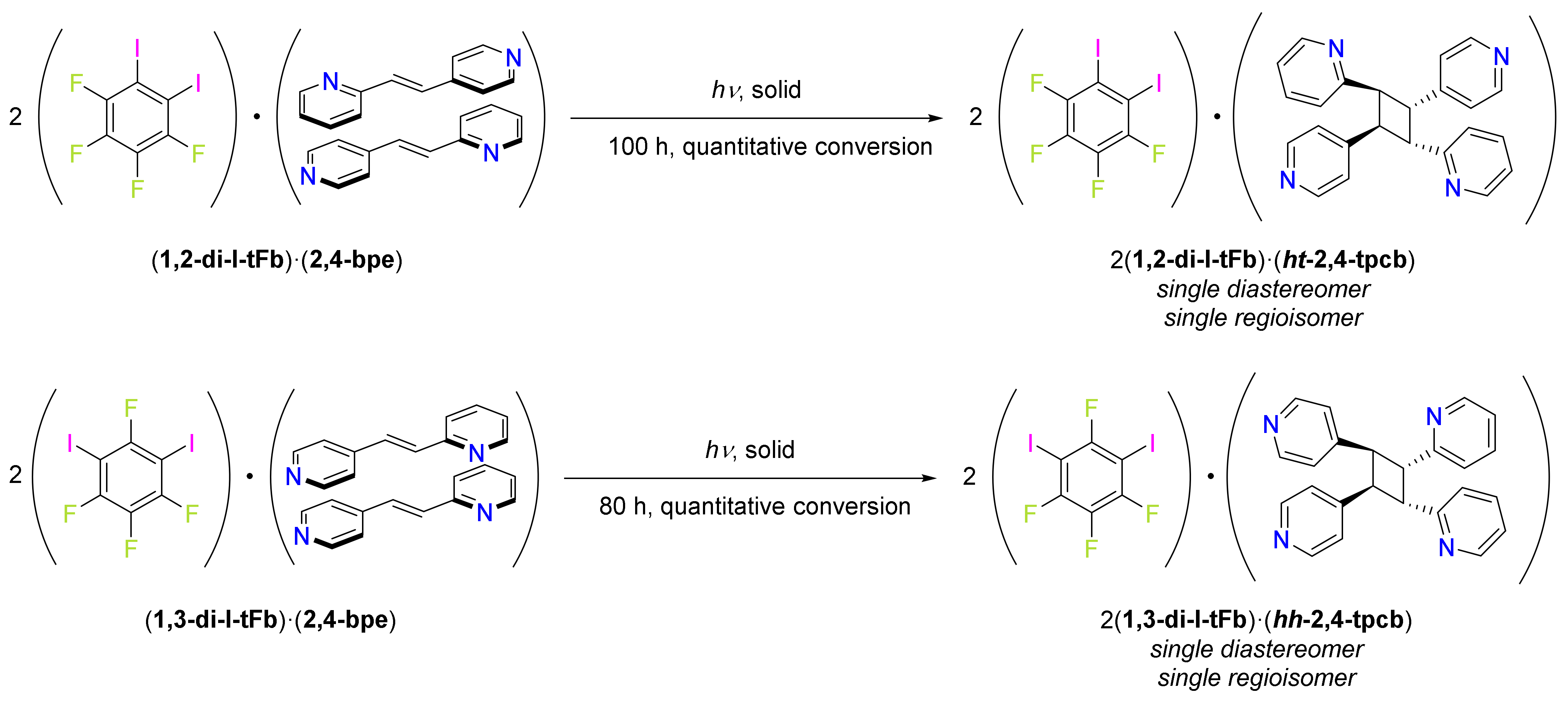
2.1. X-ray Crystal Structure of (1,2-di-I-tFb)∙(2,4-bpe)
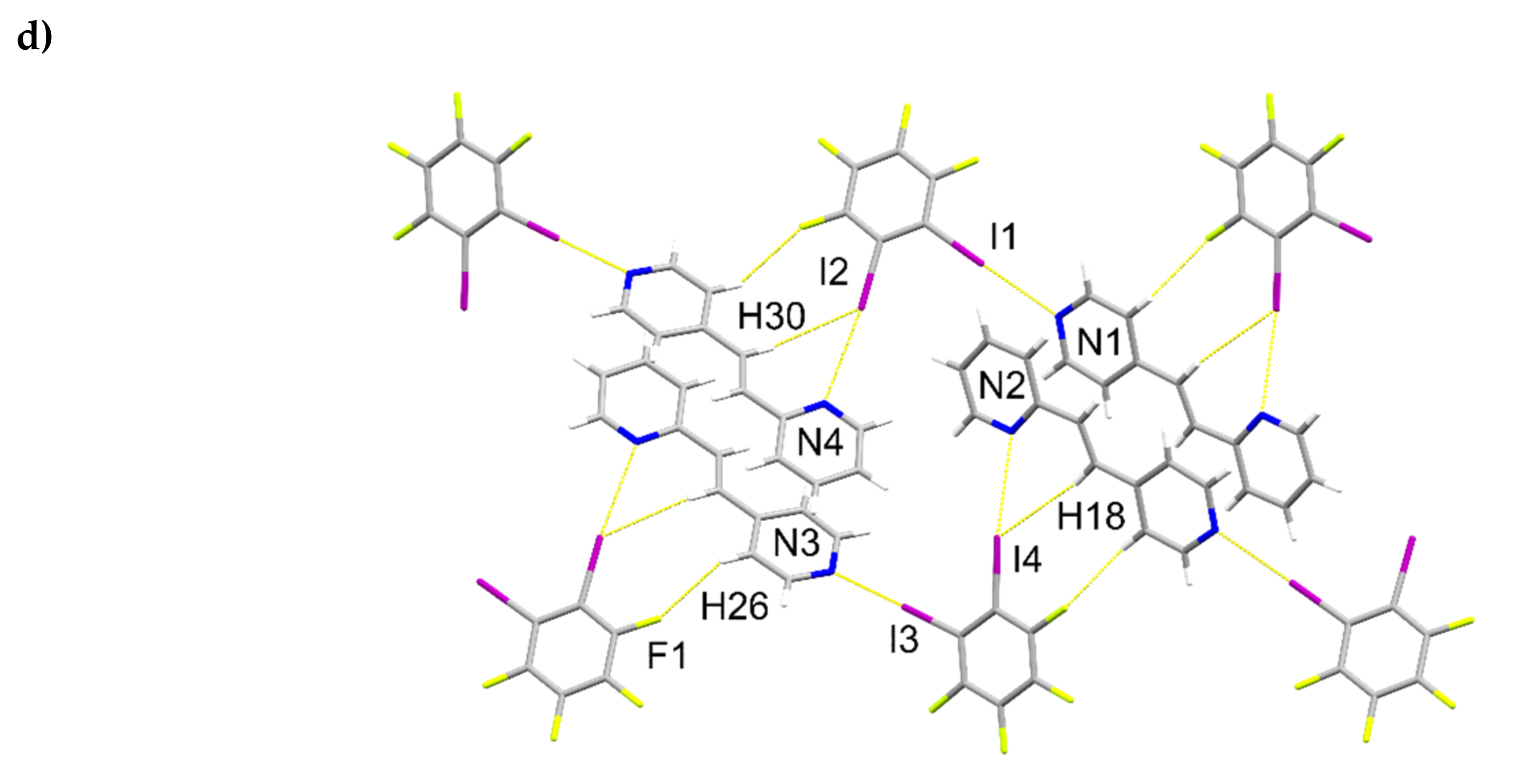
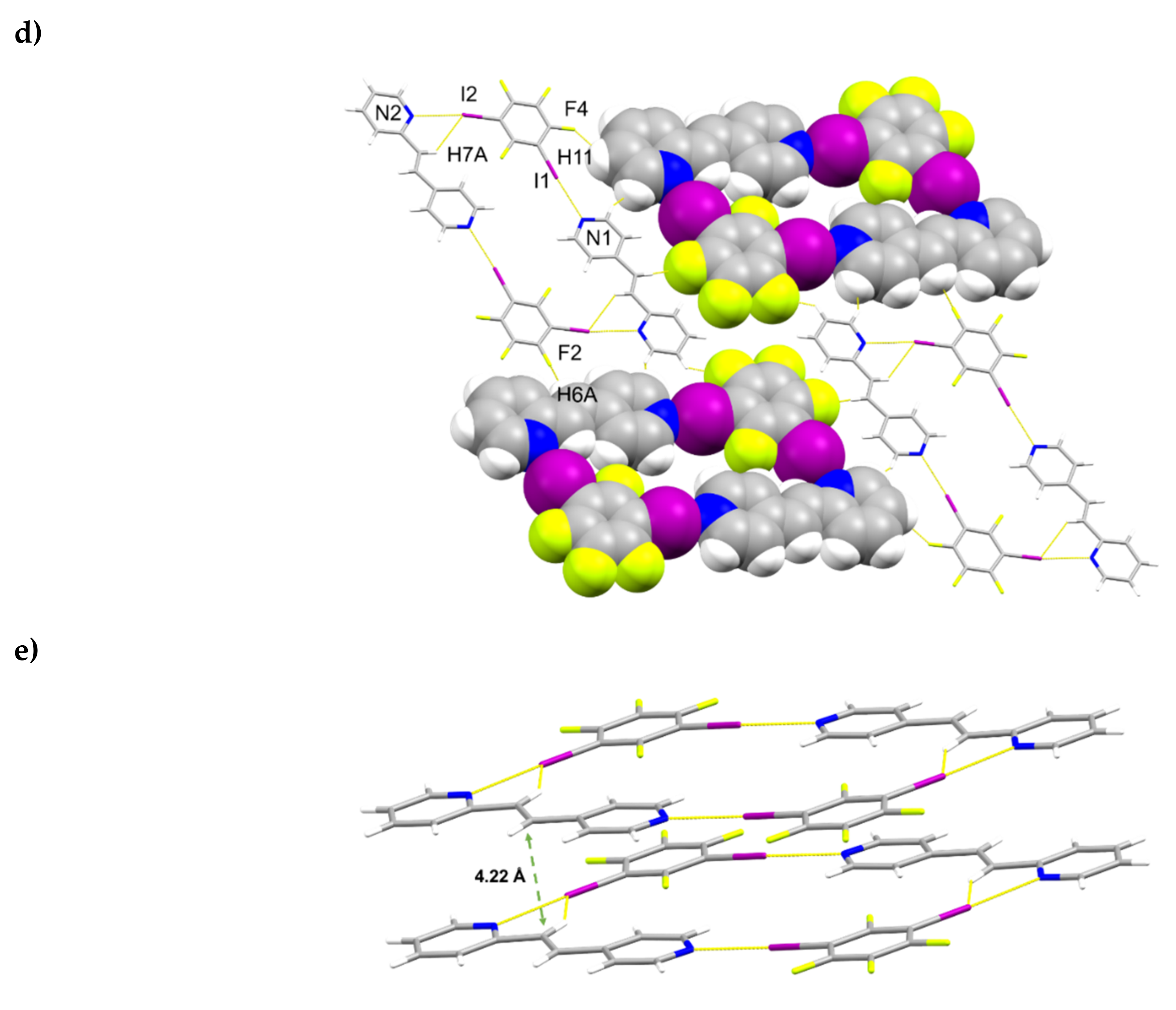
2.2. X-ray Crystal Structure of 2(1,2-di-I-tFb)∙(ht-2,4-tpcb)
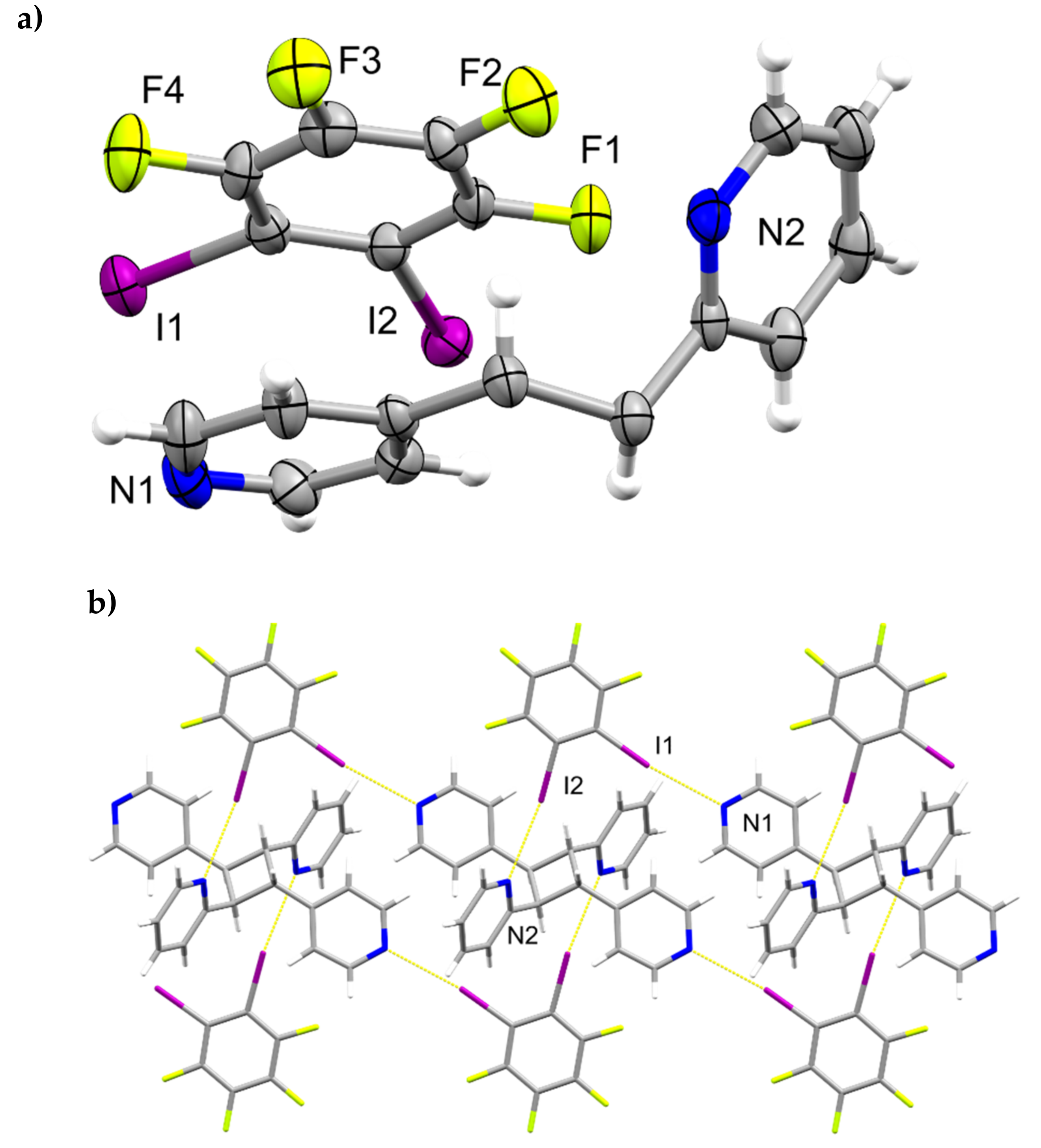
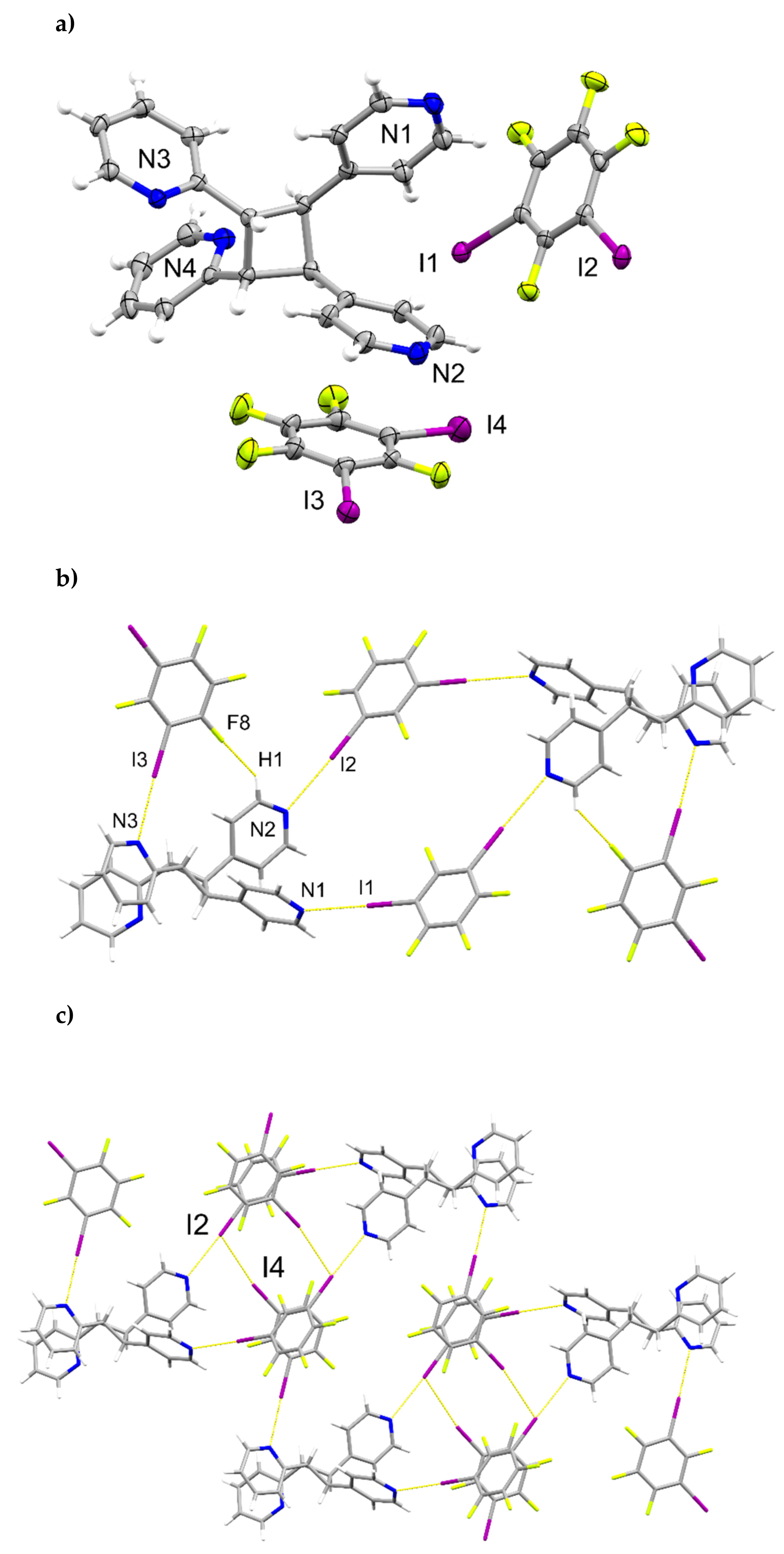
2.3. X-ray Crystal Structure of (1,3-di-I-tFb)∙(2,4-bpe)
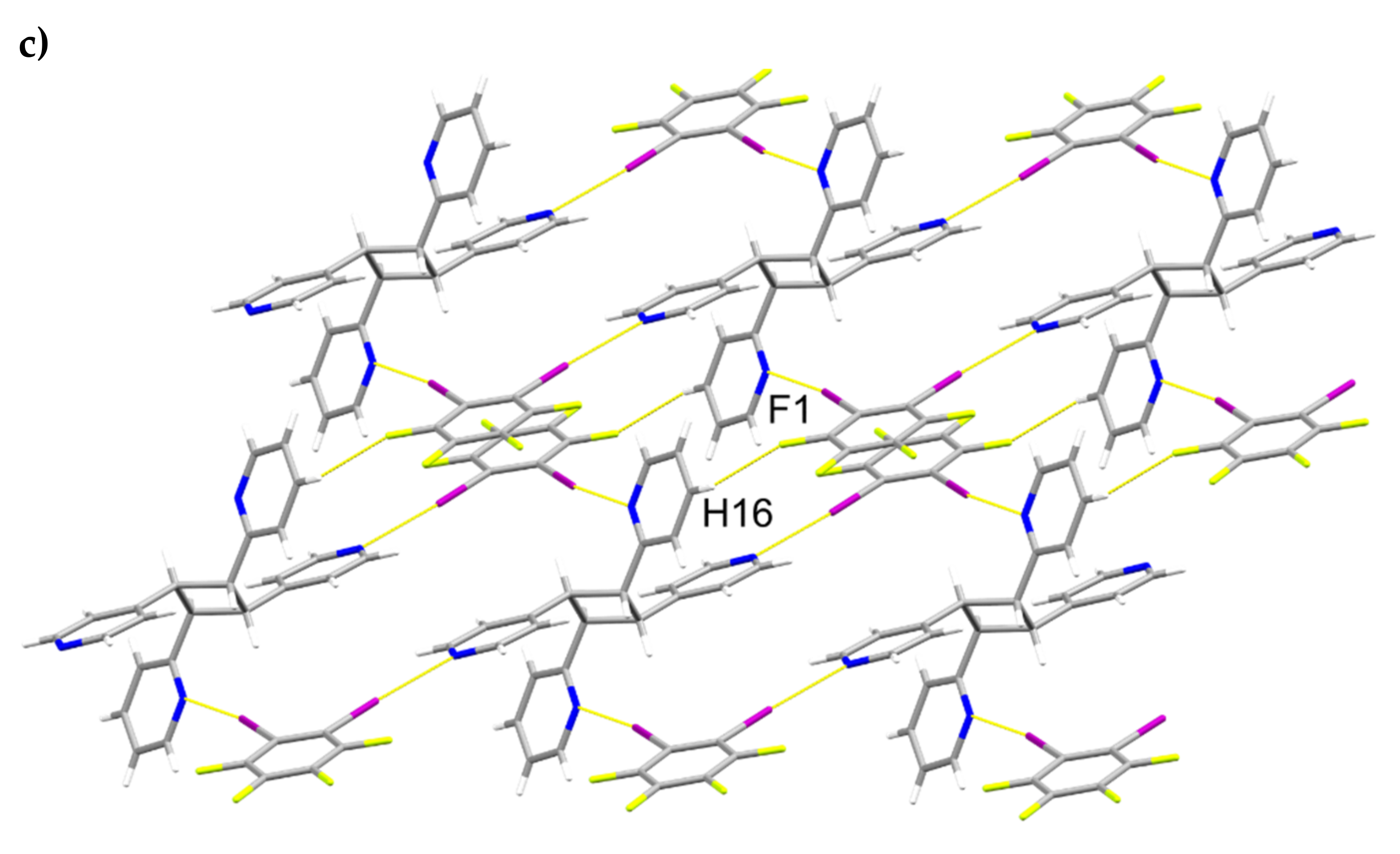
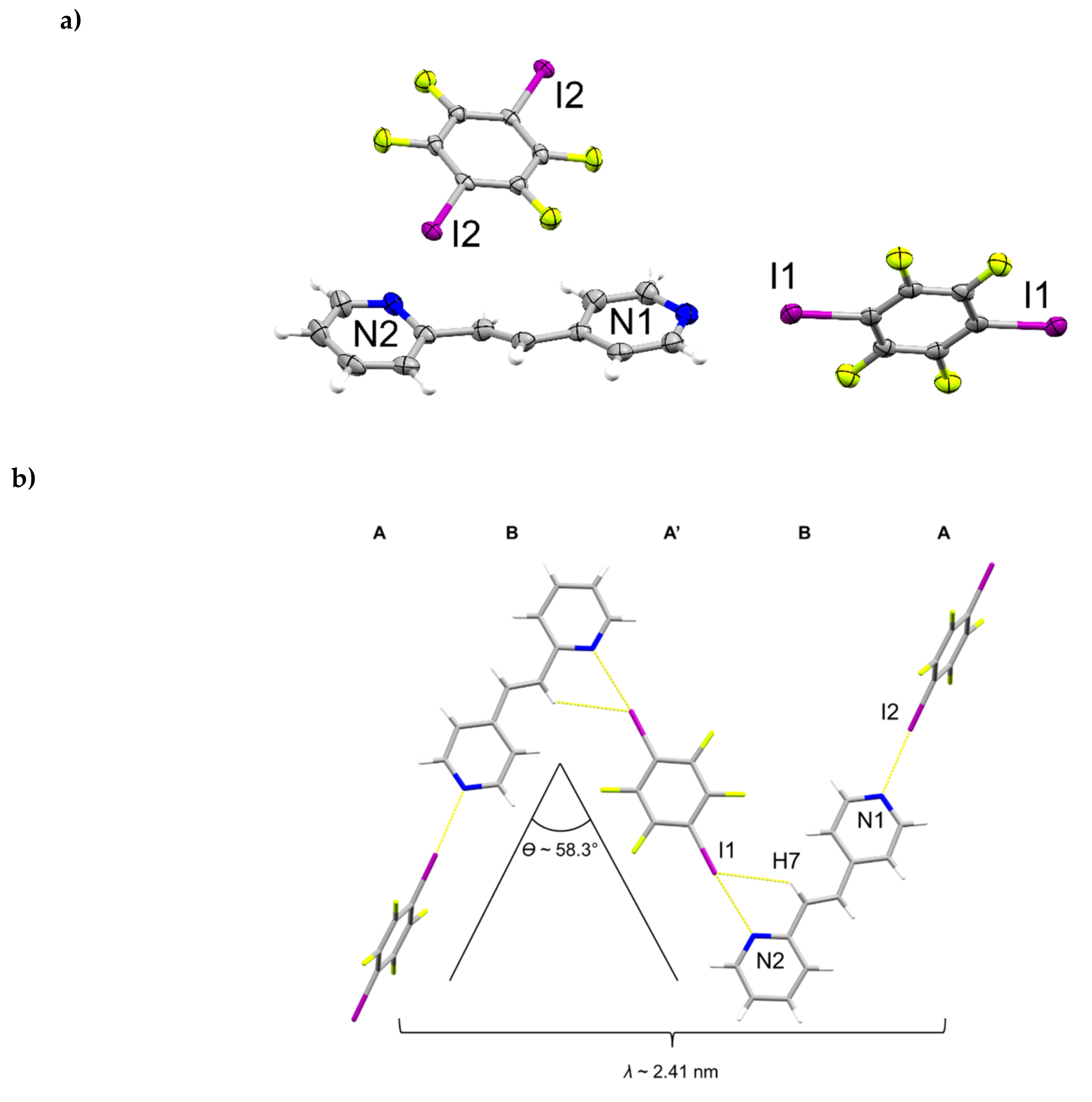
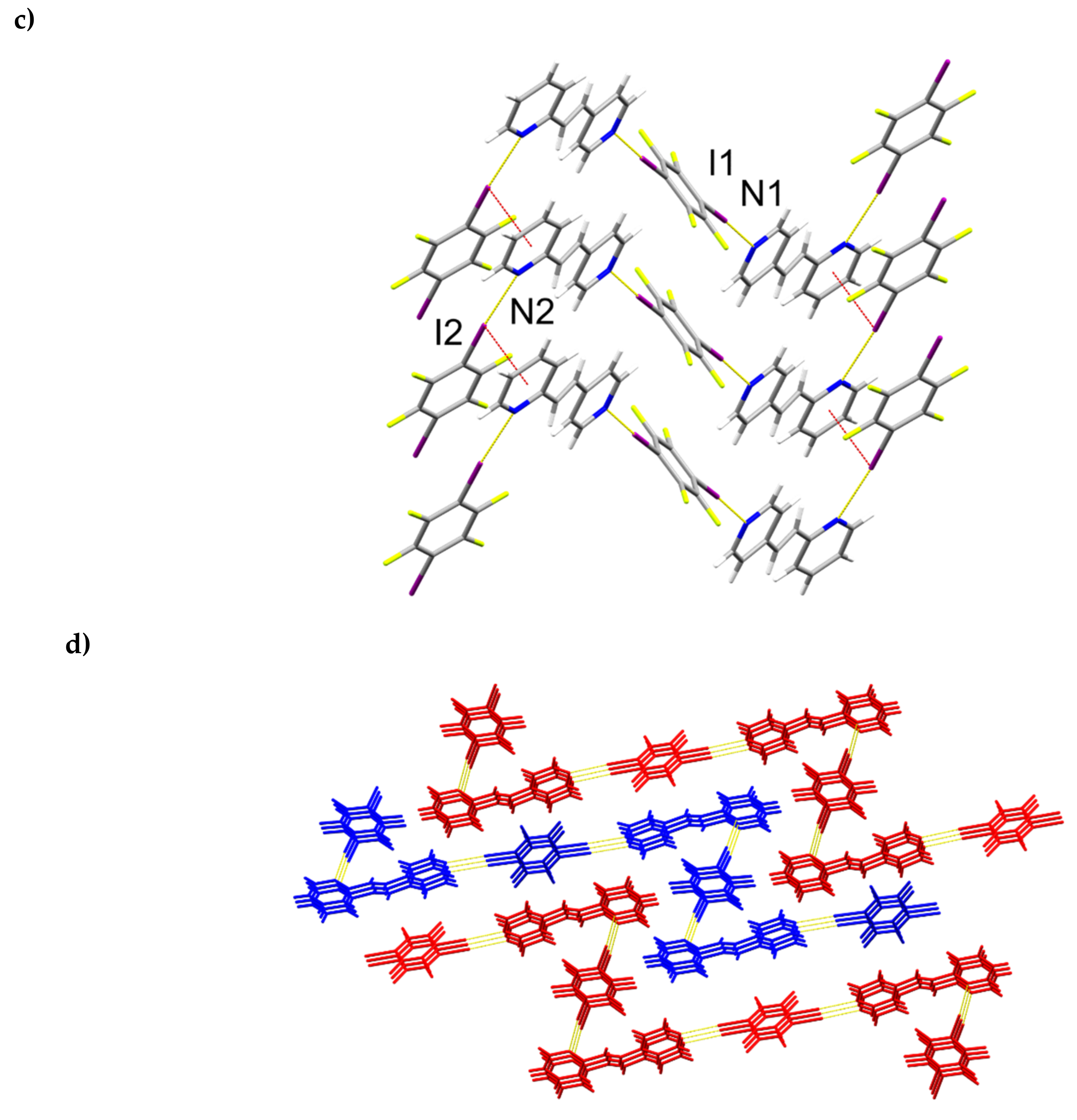
2.4. X-ray Crystal Structure of 2(1,3-di-I-tFb)∙(hh-2,4-tpcb)
2.5. X-ray Crystal Structure of (1,4-di-I-tFb)∙(2,4-bpe)
2.6. Structural Considerations
3. Materials and Methods
3.1. General Experimental
3.2. Synthetic Procedures





3.3. H NMR Spectroscopy
3.4. Powder X-ray Diffraction (pXRD)
3.5. Single-Crystal X-ray Diffraction (scXRD)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dunning, T.J.; Unruh, D.K.; Bosch, E.; Groeneman, R.H. Controlling Topology within Halogen-Bonded Networks by Varying the Regiochemistry of the Cyclobutane-Based Nodes. Molecules 2021, 26, 3152. [Google Scholar] [CrossRef] [PubMed]
- Sinnwell, M.A.; Santana, C.L.; Bosch, E.; MacGillivray, L.R.; Groeneman, R.H. Application of a tetrapyrimidyl cyclobutane synthesized in the organic solid state: A halogen-bonded supramolecular ladder. CrystEngComm 2020, 22, 6780–6782. [Google Scholar] [CrossRef]
- Oburn, S.M.; Sinnwell, M.A.; Ericson, D.P.; Reinheimer, E.W.; Proserpio, D.M.; Groeneman, R.H.; MacGillivray, L. Diversifying molecular and topological space via a supramolecular solid-state synthesis: A purely organic mok net sustained by hydrogen bonds. IUCrJ 2019, 6, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Oburn, S.M.; Santana, C.L.; Elacqua, E.; Groeneman, R.H. A diamondoid net sustained by halogen bonds: Employing a cyclobutane to generate a tetrahedral architecture. CrystEngComm 2020, 22, 4349–4352. [Google Scholar] [CrossRef]
- Cohen, M.D.; Schmidt, G.M.J. Topochemistry. Part I. A Survey. J. Chem. Soc. 1964, 1996–2000. [Google Scholar] [CrossRef]
- Quentin, J.; Swenson, D.C.; MacGillivray, L.R. Supramolecular Sandwiches: Halogen-Bonded Coformers Direct [2+2] Photoreactivity in Two-Component Cocrystals. Molecules 2020, 25, 907. [Google Scholar] [CrossRef] [Green Version]
- Sinnwell, M.A.; MacGillivray, L.R. Halogen-Bond-Templated [2+2] Photodimerization in the Solid State: Directed Synthesis and Rare Self-Inclusion of a Halogenated Product. Angew. Chem. Int. Ed. 2016, 55, 3477–3480. [Google Scholar] [CrossRef]
- Sinnwell, M.A.; Blad, J.N.; Thomas, L.R.; MacGillivray, L.R. Structural flexibility of halogen bonds showed in a single-crystal-to-single-crystal [2+2] photodimerization. IUCrJ 2018, 5, 491–496. [Google Scholar] [CrossRef]
- Bosch, E.; Battle, J.D.; Groeneman, R.H. Crystal structure and photoreactivity of a halogen-bonded cocrystal based upon 1,2-diiodoperchlorobenzene and 1,2-bis(pyridin-4-yl)ethylene. Acta Crystallogr. Sect. C Struct. Chem. 2020, 76, 557–561. [Google Scholar] [CrossRef]
- Bosch, E.; Kruse, S.J.; Krueger, H.R., Jr.; Groeneman, R.H. Role of π-π Stacking and Halogen Bonding by 1,4-Diiodoperchlorobenzene to Organize the Solid State to Achieve a [2 + 2] Cycloaddition Reaction. Cryst. Growth Des. 2019, 19, 3092–3096. [Google Scholar] [CrossRef]
- Carrona, T.; Liantonio, R.; Logothetis, T.A.; Metrangolo, P.; Pilati, T.; Resnati, G. Halogen Bonding and π···π Stacking Control Reactivity in the Solid State. J. Am. Chem. Soc. 2004, 126, 4500–4501. [Google Scholar] [CrossRef] [PubMed]
- Bosch, E.; Kruse, S.J.; Reinheimer, E.W.; Rath, N.P.; Groeneman, R.H. Regioselective [2 + 2] cycloaddition reaction within a pair of polymorphic co-crystals based upon halogen bonding interactions. CrystEngComm 2019, 21, 6671–6675. [Google Scholar] [CrossRef]
- Papaefstathiou, G.S.; Friščić, T.; MacGillivray, L.R. A Regiocontrolled ‘Head-to-Tail’ [2+2] Photodimerization of a Stilbene involving a Ternary Solid based on Catechol. J. Supramol. Chem. 2002, 2, 227–231. [Google Scholar] [CrossRef]
- Hamilton, T.D.; Papaefstathiou, G.S.; MacGillivray, L.R. A Polyhedral Host Constructed Using a Linear Template. J. Am. Chem. Soc. 2002, 124, 11606–11607. [Google Scholar] [CrossRef]
- Grobelny, A.L.; Verdu, F.A.; Groeneman, R.H. Solvent-free synthesis and purification of a photoproduct via sublimation of a tetrahalogenated template. CrystEngComm 2017, 19, 3562–3565. [Google Scholar] [CrossRef]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef] [Green Version]
- Hill, Y.; Briceño, A. Exploiting the use of hydrogen bonding and metal-coordination in the self-assembly of photoreactive multicomponent networks. Chem. Commun. 2007, 38, 3930–3932. [Google Scholar] [CrossRef]
- Quentin, J.; MacGillivray, L.R. Halogen versus Hydrogen Bonding in Binary Cocrystals: Novel Conformation a Coformer with [2+2] Photoreactivity of Criss-Crossed C=C Bonds. ChemPhysChem 2020, 21, 154–163. [Google Scholar] [CrossRef]
- Peedikakkal, A.M.P.; Vittal, J.J. Solid-State Photochemical [2+2] Cycloaddition in a Hydrogen-Bonded Metal Complex Containing Several Parallel and Crisscross C–C bonds. Chemistry 2008, 14, 5329–5334. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Nguyen, T.M.; Retailleau, P. [2+2] Photodimerization of Stilbazoles Promoted by Oxalic Acid in Suspension. Chemistry 2020, 26, 4682–4689. [Google Scholar] [CrossRef]
- Elacqua, E.; Kaushik, P.; Groeneman, R.H.; Sumrak, J.C.; Bučar, D.-K.; MacGillivray, L.R. A Supramolecular Protecting Group Strategy Introduced to the Organic Solid State: Enhanced Reactivity through Molecular Pedal Motion. Angew. Chem. Int. Ed. 2012, 51, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Harada, J.; Ogawa, K. Topics in Stereochemistry; Denmark, S.E., Siegel, J.S., Eds.; Wiley: Chichester, UK, 2006; Volume 25, pp. 31–47. [Google Scholar]
- Harada, J.; Ogawa, K. Invisible but Common Motion in Organic Crystals: A Pedal Motion in Stilbenes and Azobenzenes. J. Am. Chem. Soc. 2001, 123, 10884–10888. [Google Scholar] [CrossRef] [PubMed]
- Harada, J.; Ogawa, K.; Tomoda, S. The Central Bond Length in 1,2-Diphenylethanes. J. Am. Chem. Soc. 1995, 117, 4476–4478. [Google Scholar] [CrossRef]
- Ogawa, K.; Sano, T.; Yoshimura, S.; Takeuchi, Y.; Toriumi, K. Molecular structure and intramolecular motion of (E)-stilbenes in crystals. An interpretation of the unusually short ethylene bond. J. Am. Chem. Soc. 1992, 114, 1041–1051. [Google Scholar] [CrossRef]
- Harada, J.; Ogawa, K.; Tomoda, S. Molecular Motion and Conformational Interconversion of Azobenzenes in Crystals as Studied by X-ray Diffraction. Acta Crystallogr. Sect. B 1997, 53, 662–672. [Google Scholar] [CrossRef]
- Harada, J.; Ogawa, K. Ethane Bond Length in 1,2-Diphenylethanes. Struct. Chem. 2001, 12, 243–250. [Google Scholar] [CrossRef]
- Harada, J.; Ogawa, K. X-ray Diffraction Analysis of Nonequilibrium States in Crystals: Observation of an Unstable Conformer in Flash-Cooled Crystals. J. Am. Chem. Soc. 2004, 126, 3539–3544. [Google Scholar] [CrossRef]
- Harada, J.; Harakawa, M.; Ogawa, K. Conformational Change of N-Benzylideneamines in Crystals. Acta Cryst. 2004, B60, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Ohba, S.; Hosomi, H.; Ito, Y. In Situ X-ray Observation of Pedal-like Conformational Change and Dimerization of trans-Cinnamamide in Cocrystals with Phthalic Acid. J. Am. Chem. Soc. 2001, 123, 6349–6352. [Google Scholar] [CrossRef]
- Gero, A.; Markham, J.J. Studies on Pyridines: I. The Basicity of Pyridine Bases. J. Org. Chem. 1951, 16, 1835–1838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystal | (1,2-di-I-tFb)·(2,4-bpe) | 2(1,2-di-I-tFb)·(ht-2,4-tpcb) |
|---|---|---|
| CCDC deposition number | 2104746 | 2114529 |
| Empirical formula | C18H10F4I2N2 | C36H20F8I4N4 |
| Formula weight/g·mol−1 | 584.08 | 1168.16 |
| Temperature/K | 150(2) | 150(2) |
| Crystal system | Triclinic | Triclinic |
| Space group | P-1 | P-1 |
| a/Å | 7.5879(8) | 9.1499(10) |
| b/Å | 13.1862(13) | 10.6011(11) |
| c/Å | 19.2918(19) | 10.9421(12) |
| α/° | 80.092(5) | 62.054(5) |
| β/° | 89.530(5) | 77.014(5) |
| γ/° | 74.125(5) | 76.828(5) |
| Volume/Å3 | 1827.4(3) | 904.32(17) |
| Z | 4 | 1 |
| ρcalc/g·cm−3 | 2.12 | 2.15 |
| μ/mm−1 | 3.484 | 3.521 |
| F(000) | 1096 | 548 |
| Crystal size/mm3 | 0.11 × 0.06 × 0.03 | 0.10 × 0.09 × 0.06 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 4.21 to 50.25 | 4.25 –to 50.25 |
| Index ranges | −9 ≤ h ≤ 9, −15 ≤ k ≤ 15, −23 ≤ l ≤ 21 | −10 ≤ h ≤ 10, −12 ≤ k ≤ 12, −13 ≤ l ≤ 13 |
| Reflections collected | 43105 | 40307 |
| Independent reflections | 6502 [Rint = 0.0349, Rsigma = 0.0229] | 3220 [Rint = 0.0830, Rsigma = 0.0312] |
| Data/restraints/parameters | 6502/0/549 | 3220/0/235 |
| Goodness-of-fit on F2 | 1.066 | 1.060 |
| Final R indices [I ≥ 2σ (I)] | R1 = 0.0170 | R1 = 0.0358 |
| wR2 = 0.0363 | wR2 = 0.0781 | |
| R indices (all data) | R1 = 0.0211 | R1 = 0.0377 |
| wR2 = 0.0379 | wR2 = 0.0790 | |
| Largest diff. peak/hole/e·Å−3 | 0.66/−0.36 | 2.44/−0.94 |
| Crystal | 2,4-bpe Molecule | Twist Angle ϕ/° |
|---|---|---|
| (1,2-di-I-tFb)·(2,4-bpe) | N1/N2 | 3.4 |
| N3/N4 | 8.0 | |
| (1,3-di-I-tFb)·(2,4-bpe) | N1/N2 | 3.8 |
| (1,4-di-I-tFb)·(2,4-bpe) | N1/N2 | 9.9 |
| Cocrystal | X-Bond | Pyridyl Substitution | d(N···I)/Å | Θ(C-I···N)/° | X-Bond Type | prs * |
|---|---|---|---|---|---|---|
| (1,2-di-I-tFb)·(2,4-bpe) | I1···N1 | 4 | 2.808(2) | 176.6 | I | 20 |
| I2···N4 | 2 | 3.054(2) | 174.0 | I | 13 | |
| I3···N3 | 4 | 2.814(2) | 176.8 | I | 20 | |
| I4···N2 | 2 | 3.147(2) | 171.9 | I | 11 | |
| 2(1,2-di-I-tFb)·(ht-2,4-tpcb) | I1···N1 | 4 | 2.946(4) | 172.6 | I | 17 |
| I2···N2 | 2 | 3.022(4) | 172.1 | I | 14 | |
| (1,3-di-I-tFb)·(2,4-bpe) | I1···N1 | 4 | 2.795(4) | 177.2 | I | 21 |
| I2···N2 | 2 | 2.926(4) | 171.8 | I | 17 | |
| 2(1,3-di-I-tFb)·(hh-2,4-tpcb) | I1···N1 | 4 | 2.826(2) | 174.4 | I | 20 |
| I2···N2 | 4 | 2.892(2) | 177.9 | I | 18 | |
| I3···N3 | 2 | 2.826(2) | 169.0 | I | 20 | |
| (1,4-di-I-tFb)·(2,4-bpe) | I2···N1 | 4 | 2.802(4) | 177.5 | I | 21 |
| I1···N2 | 2 | 2.884(3) | 175.6 | I | 18 |
| Cocrystal | Primary Assembly | Secondary Assembly | Photoreactivity |
|---|---|---|---|
| (1,2-di-I-tFb)·(2,4-bpe) (2104746) | infinite 1D tape based on N∙∙∙I | infinite 2D corrugated layers | active |
| 2(1,2-di-I-tFb)·(rctt-ht-2,4-tpcb) (2114529) | infinite 1D chain based on N∙∙∙I | infinite sheets based on C-H∙∙∙F | - |
| (1,3-di-I-tFb)·(2,4-bpe) (2104747) | discrete, four-component assembly based on N∙∙∙I | infinite sheets based on C-H∙∙∙F | active |
| 2(1,3-di-I-tFb)·(rctt-hh-2,4-tpcb) (2114285) | discrete, six-component assembly based on N∙∙∙I and C-H∙∙∙F | 3D network based on Type II I∙∙∙I | - |
| (1,4-di-I-tFb)·(2,4-bpe) (2104748) | infinite zig-zag chains based on N∙∙∙I | infinite columns based on Type II I∙∙∙I | inert |
| Cocrystal | (1,3-di-I-tFb)·(2,4-bpe) | 2(1,3-di-I-tFb)·(hh-2,4-tpcb) | (1,4-di-I-tFb)·(2,4-bpe) |
|---|---|---|---|
| CCDC deposition number | 2104747 | 2114285 | 2104748 |
| Empirical formula | C18H10F4I2N2 | C36H20F8I4N4 | C18H10F4I2N2 |
| Formula weight/g·mol−1 | 584.08 | 1168.16 | 584.08 |
| Temperature/K | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n | P-1 | P21/c |
| a/Å | 4.2177(4) | 8.1884(6) | 17.7168(18) |
| b/Å | 27.254(3) | 14.2749(14) | 5.2208(5) |
| c/Å | 16.1797(16) | 16.3737(16) | 20.159(2) |
| α/° | 90 | 82.111(5) | 90 |
| β/° | 90.940(5) | 87.336(5) | 103.519(5) |
| γ/° | 90 | 73.759(5) | 90 |
| Volume/Å3 | 1859.6(3) | 1820.1(3) | 1813.0(3) |
| Z | 4 | 2 | 4 |
| ρcalc/g·cm−3 | 2.09 | 2.13 | 2.14 |
| μ/mm−1 | 3.424 | 3.498 | 3.512 |
| F(000) | 1096 | 1096 | 1096 |
| Crystal size/mm3 | 0.15 × 0.15 × 0.05 | 0.07 × 0.06 × 0.05 | 0.27 × 0.09 × 0.02 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 5.036 to 50.244 | 4.152 to 50.246 | 4.156 to 50.246 |
| Index ranges | −5 ≤ h ≤ 5, −32 ≤ k ≤ 32, −18 ≤ l ≤ 19 | −9 ≤ h ≤ 9, −17 ≤ k ≤ 17, −18 ≤ l ≤ 19 | −21 ≤ h ≤ 21, −24 ≤ k ≤ 24, −23 ≤ l ≤ 23 |
| Reflections collected | 38879 | 34954 | 46454 |
| Independent reflections | 3283 [Rint = 0.0379, Rsigma = 0.0213] | 6475 [Rint = 0.0264, Rsigma = 0.0184] | 3233 [Rint = 0.0362, Rsigma = 0.0153] |
| Data/restraints/parameters | 3234/18/253 | 6475/0/469 | 3233/0/235 |
| Goodness-of-fit on F2 | 1.082 | 1.033 | 1.049 |
| Final R indices [I ≥ 2σ (I)] | R1 = 0.0299 | R1 = 0.0174 | R1 = 0.0248 |
| wR2 = 0.0802 | wR2 = 0.0365 | wR2 = 0.0538 | |
| R indices (all data) | R1 = 0.0306 | R1 = 0.0222 | R1 = 0.0271 |
| wR2 = 0.0807 | wR2 = 0.0387 | wR2 = 0.0551 | |
| Largest diff. peak/hole/e·Å−3 | 0.79/−0.63 | 1.20/−1.00 | 0.71/−0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quentin, J.; Reinheimer, E.W.; MacGillivray, L.R. Halogen-Bond Mediated [2+2] Photodimerizations: À la Carte Access to Unsymmetrical Cyclobutanes in the Solid State. Molecules 2022, 27, 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031048
Quentin J, Reinheimer EW, MacGillivray LR. Halogen-Bond Mediated [2+2] Photodimerizations: À la Carte Access to Unsymmetrical Cyclobutanes in the Solid State. Molecules. 2022; 27(3):1048. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031048
Chicago/Turabian StyleQuentin, Jay, Eric W. Reinheimer, and Leonard R. MacGillivray. 2022. "Halogen-Bond Mediated [2+2] Photodimerizations: À la Carte Access to Unsymmetrical Cyclobutanes in the Solid State" Molecules 27, no. 3: 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031048