
Nucleophilic Substitution at Heteroatoms—Identity Substitution Reactions at Phosphorus and Sulfur Centers: Do They Proceed in a Concerted (SN2) or Stepwise (A–E) Way?


Abstract
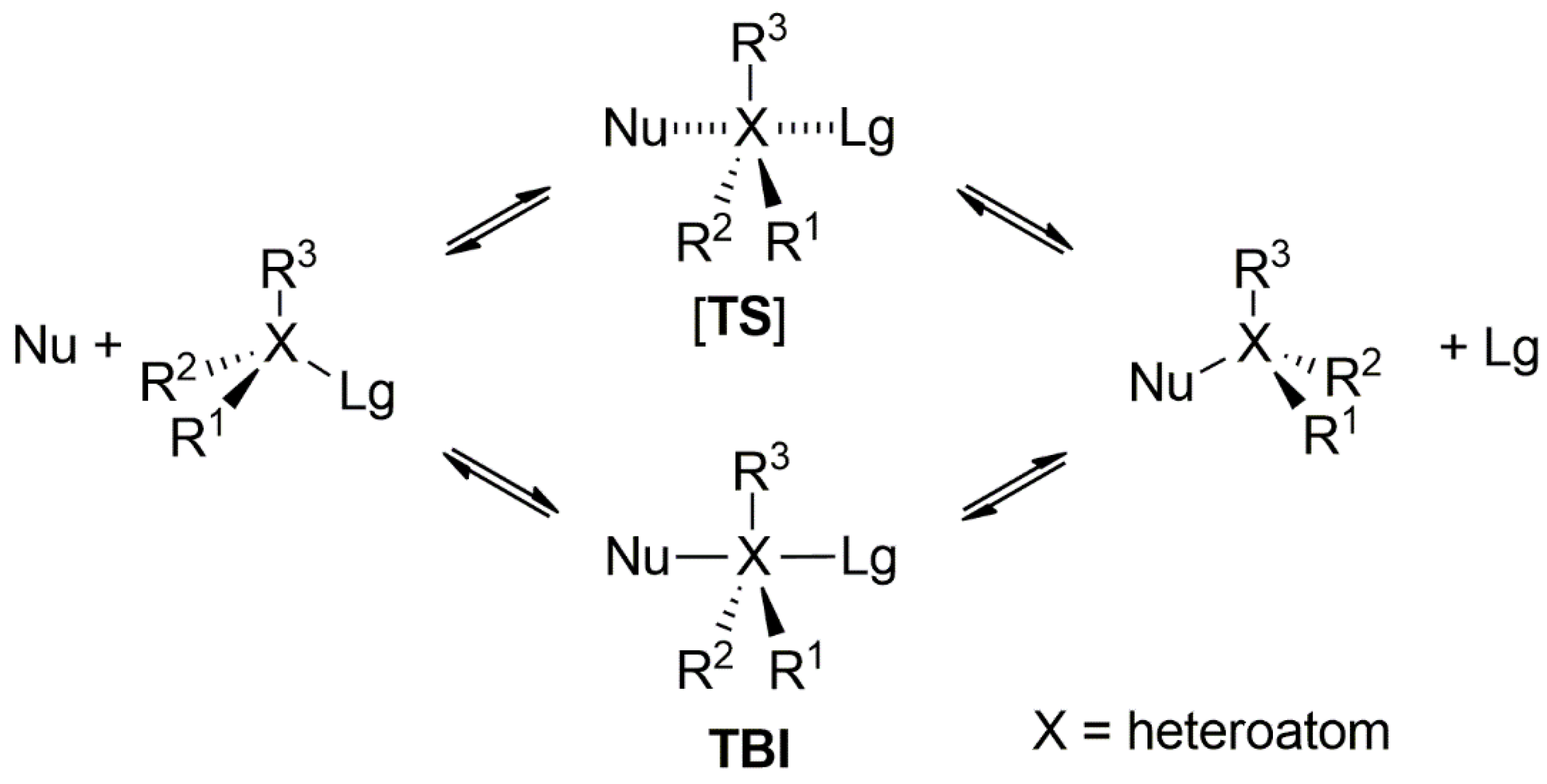
:1. Introduction
2. Results and Discussion
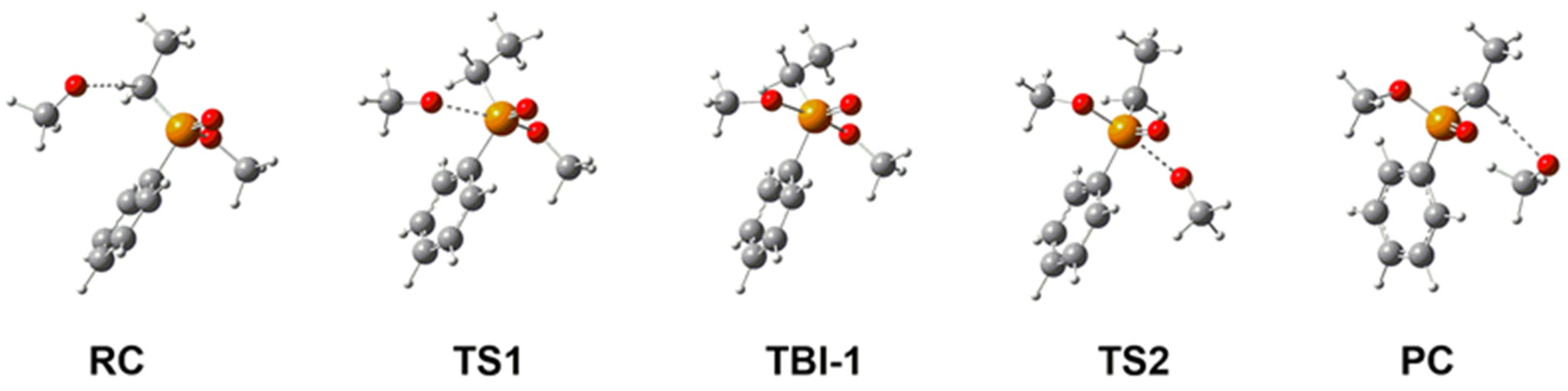
2.1. Stereochemistry and Mechanism of Identity Methoxy Exchange Reaction in Methyl Ethylphenylphosphinate 1
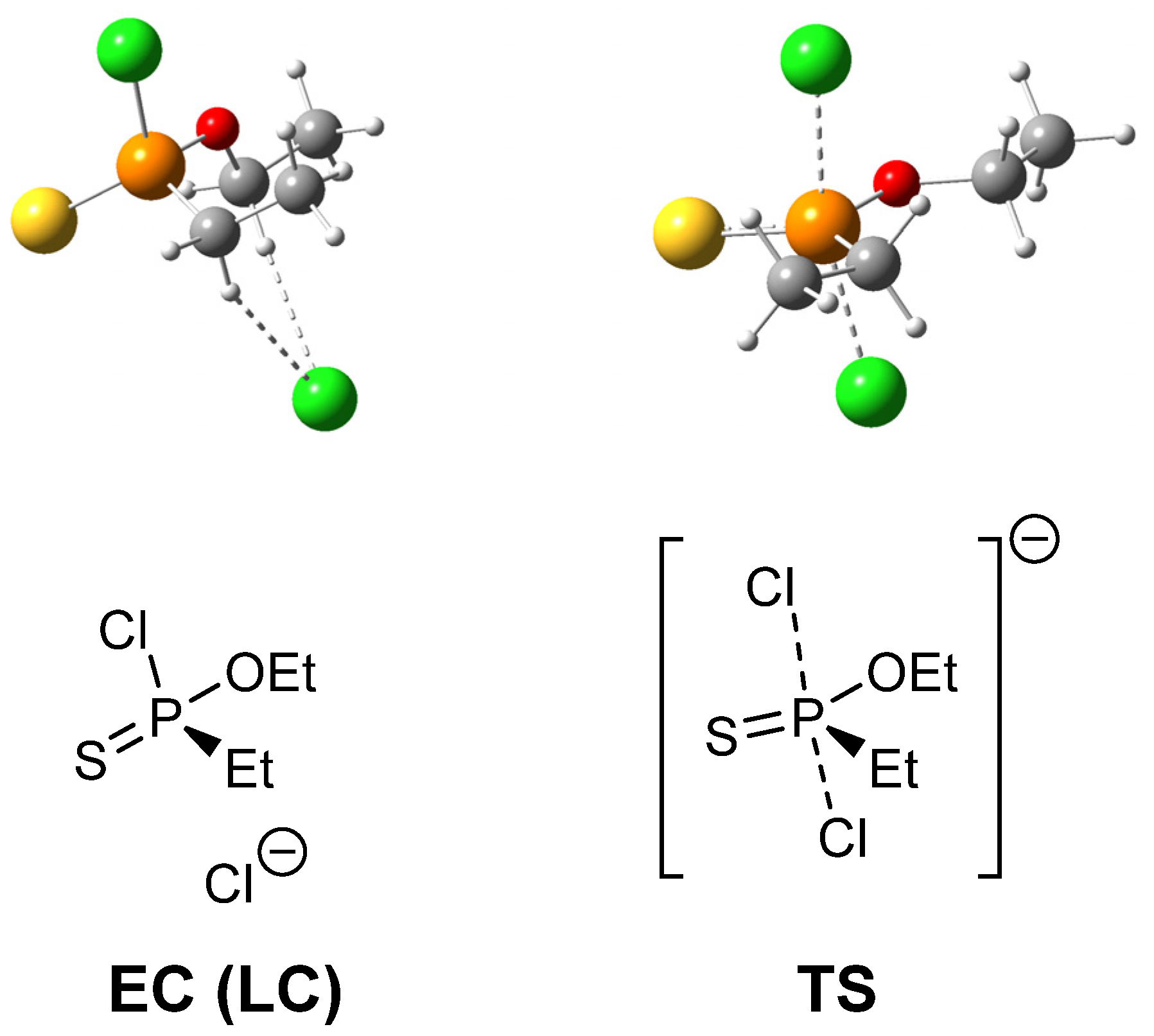
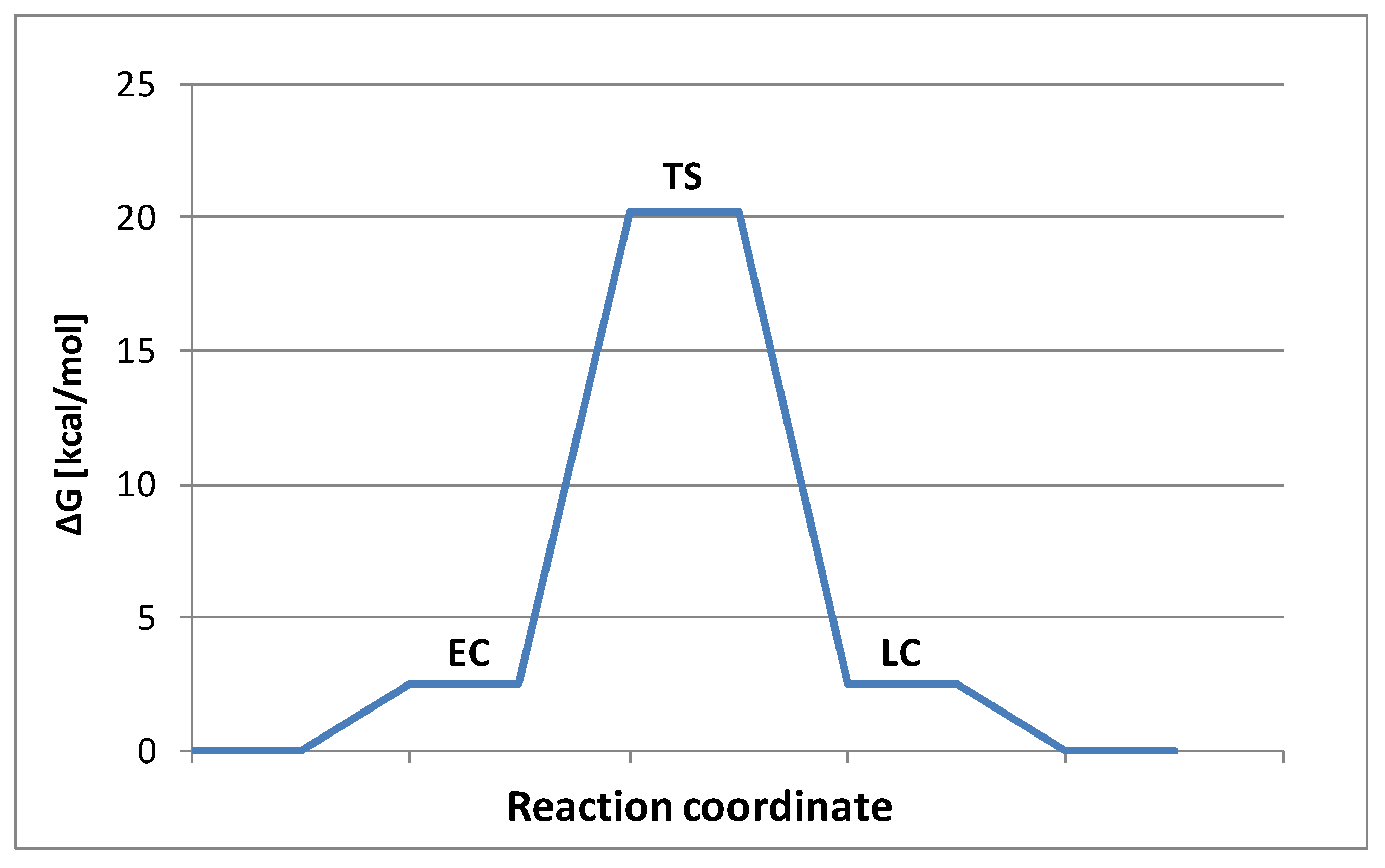
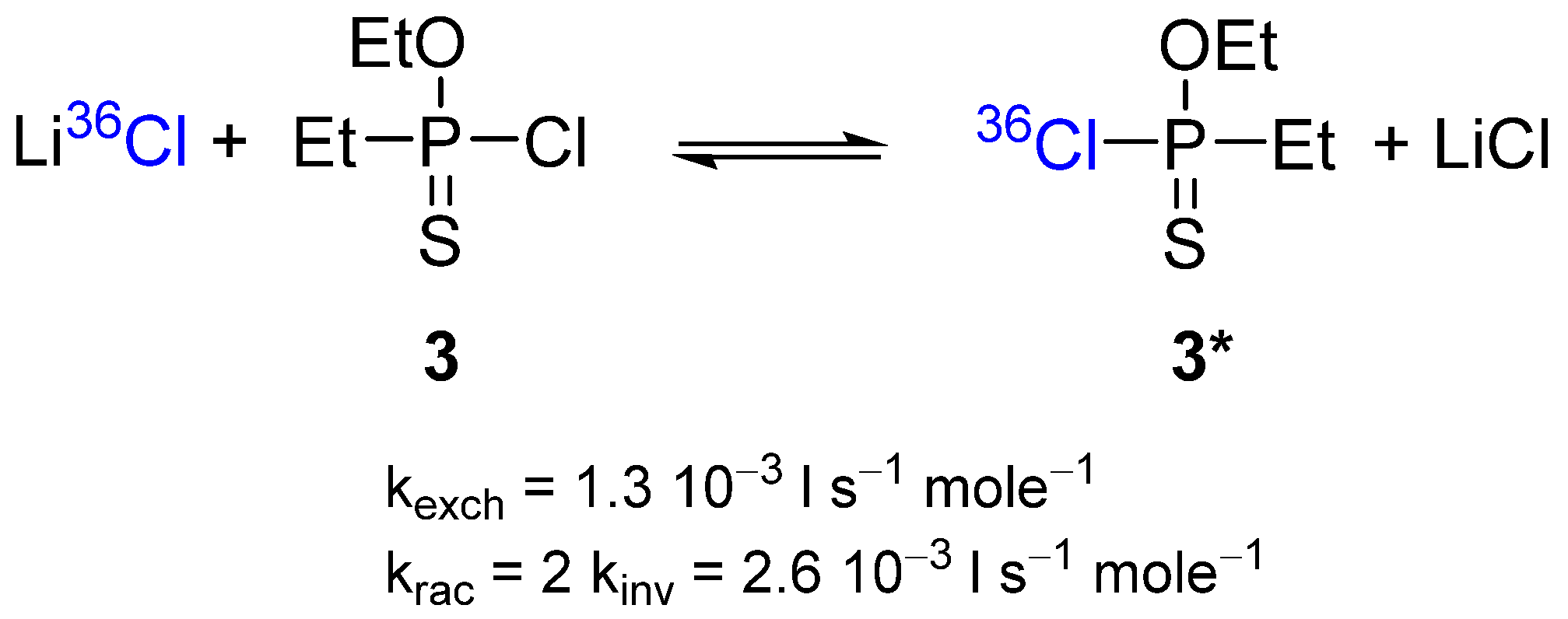
2.2. Stereochemistry and Mechanism of Identity Chloride Exchange Reaction in (Ethoxy)ethylphosphonochloridothionate 3
2.3. Stereochemistry and Mechanism of Identity Methoxy Exchange at Sulfinyl Sulfur in Methyl p-Toluenesulfinate 4
3. Conclusions and Final Remarks
4. Materials and Methods
Theoretical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hughes, E.D.; Juliusburger, F.; Masterman, S.; Topley, B.; Weiss, J. Aliphatic Substitution and the Walden Inversion. Part I. J. Chem. Soc. 1935, 1525–1529. [Google Scholar] [CrossRef]
- Hughes, E.D.; Juliusburger, F.; Scott, A.D.; Topley, B.; Weiss, J. Aliphatic substitution and the Walden inversion. Part II. J. Chem. Soc. B 1936, 1173–1175. [Google Scholar] [CrossRef]
- Cowdrey, W.A.; Hughes, E.D.; Nevell, T.P.; Wilson, C.L. Aliphatic Substitution and the Walden Inversion. Part III. Comparison, using Radioactive Bromine, of the Rates of Inversion and Substitution in the Reaction of Bromide Ions with α-Bromopropionic Acid. J. Chem. Soc. 1938, 209–211. [Google Scholar] [CrossRef]
- Lee, I.; Kim, C.K.; Li, H.G.; Sohn, C.K.; Kim, C.K.; Lee, H.W.; Lee, B.-S. Acyl-Transfer Mechanisms Involving Various Acyl Functional Groups: >X=Y with X = C, S, P and Y = O, S, J. Am. Chem. Soc. 2000, 122, 11162–11172. [Google Scholar] [CrossRef]
- Sung, D.D.; Kim, T.J.; Lee, I. Theoretical Studies of the Nucleophilic Substitution of Halides and Amine at a Sulfonyl Centre. J. Phys. Chem. A 2009, 113, 7073–7079. [Google Scholar] [CrossRef]
- van Bochove, M.A.; Swart, M.; Bickelhaupt, M.F. Nucleophilic Substitution at Phosphorus (SN2@P): Disappearance and Reappearance of Reaction Barriers. J. Am. Chem. Soc. 2006, 128, 10738–10744. [Google Scholar] [CrossRef]
- van Bochove, M.A.; Swart, M.; Bickelhaupt, M.F. Stepwise Walden Inversion in Nucleophilic Substitution at Phosphorus. Phys. Chem. Chem. Phys. 2009, 11, 259–267. [Google Scholar] [CrossRef]
- Mikolajczyk, M.; Gajl, M.; Blaszczyk, J.; Cypryk, M.; Gostynski, B. Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin. Molecules 2020, 25, 1428. [Google Scholar] [CrossRef] [Green Version]
- Green, M.; Hudson, R.F. Optically active phosphorus compounds. Part II. The configurational change accompanying transesterification. J. Chem. Soc. 1963, 540–547. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Smith, S.F.; Jorgensen, W.L. SN2 reaction profiles in the gas phase and aqueous solution. J. Am. Chem. Soc. 1984, 106, 3049–3050. [Google Scholar] [CrossRef]
- Michalski, J.; Mikołajczyk, M.; Omelańczuk, J. Stereochemistry of nucleophilic displacement reaction at thiophosphoryl centre. An example of a Walden cycle involving phosphorus. Tetrahedron Lett. 1965, 6, 1779–1784. [Google Scholar] [CrossRef]
- Michalski, J.; Mikołajczyk, M. Optically Active O-Ethyl Ethylphosphonochloridothiones: A New Route to Optically Active Organophosphorus Compounds. Chem. Ind. 1964, 6, 661–662. [Google Scholar]
- Michalski, J.; Mikołajczyk, M. Stereochemistry of the Reaction of O-Ethyl Ethylphosphonothioic Acid with Phosphorus Pentachloride. Chem. Commun. 1965, 35–36. [Google Scholar] [CrossRef]
- Michalski, J.; Mikołajczyk, M.; Halpern, A.; Prószyńska, K. Stereochemistry of nucleophilic displacement reactions at the thiophosphoryl centre. Chloride-chloride exchange at the asymmetric phosphorus atom in O-ethylethylphosphonochloridothionate. Tetrahedron Lett. 1966, 18, 1919–1924. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J. Chiral Organosulfur Compounds. Top. Stereochem. 1982, 13, 333–468. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Bujnicki, B.; Drabowicz, J. Inversion of the Sulphinamide Configuration. Bull Pol. Acad. Sci. Ser. Chem. 1977, 25, 267–269. [Google Scholar]
- Mikołajczyk, M.; Drabowicz, J. Asymmetric Synthesis of Sulphinic Esters with the Sulphur Atom As a Sole Chirality Centre. J. Chem. Soc. Chem. Commun. 1974, 14, 547–548. [Google Scholar] [CrossRef]
- Mikolajczyk, M.; Drabowicz, J.; Slebocka-Tilk, H. Nucleophilic substitution at sulfur. Kinetic evidence for inversion of configuration at sulfinyl sulfur in acid-catalyzed transesterification of sulfinates. J. Am. Chem. Soc. 1979, 101, 1302–1303. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Jens, A.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
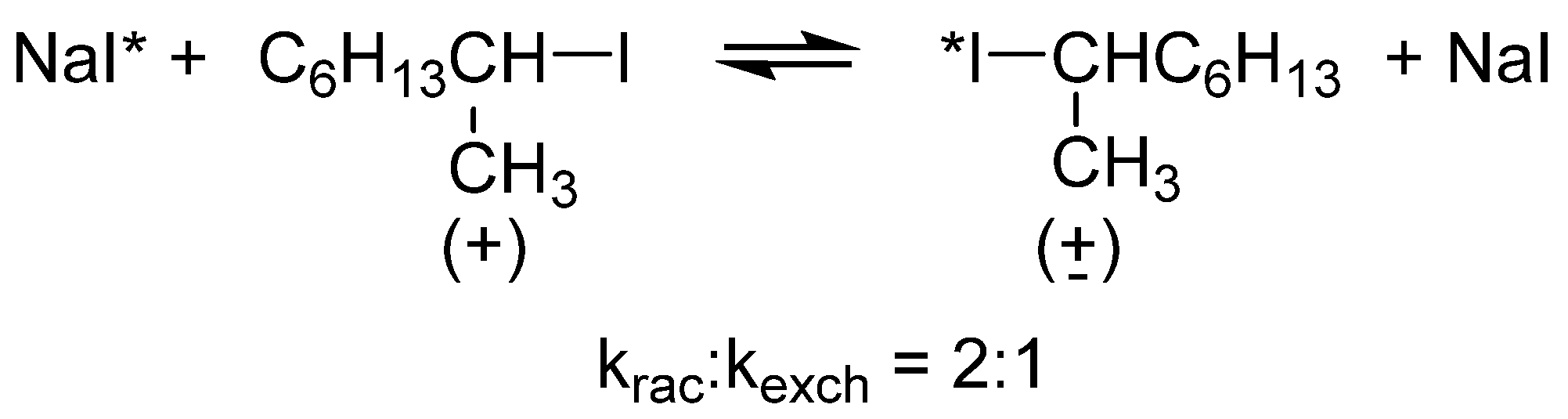{kind=link}
{kind=link}
{kind=link}
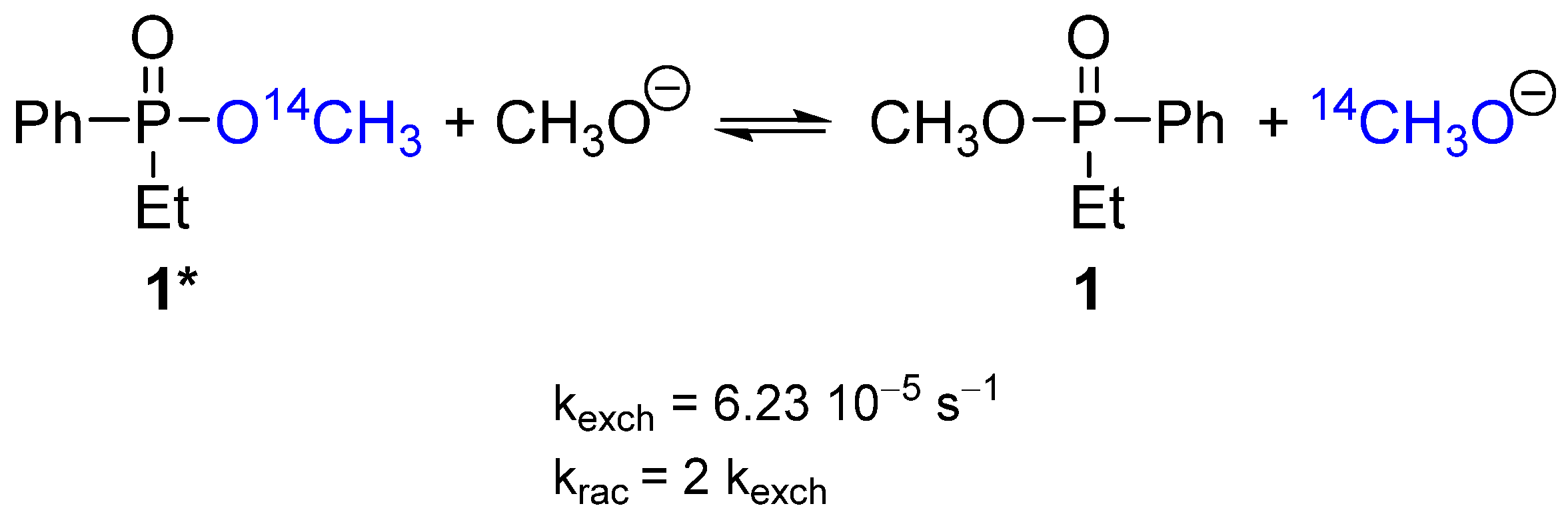{kind=link}
{kind=link}
{kind=link}
{kind=link}
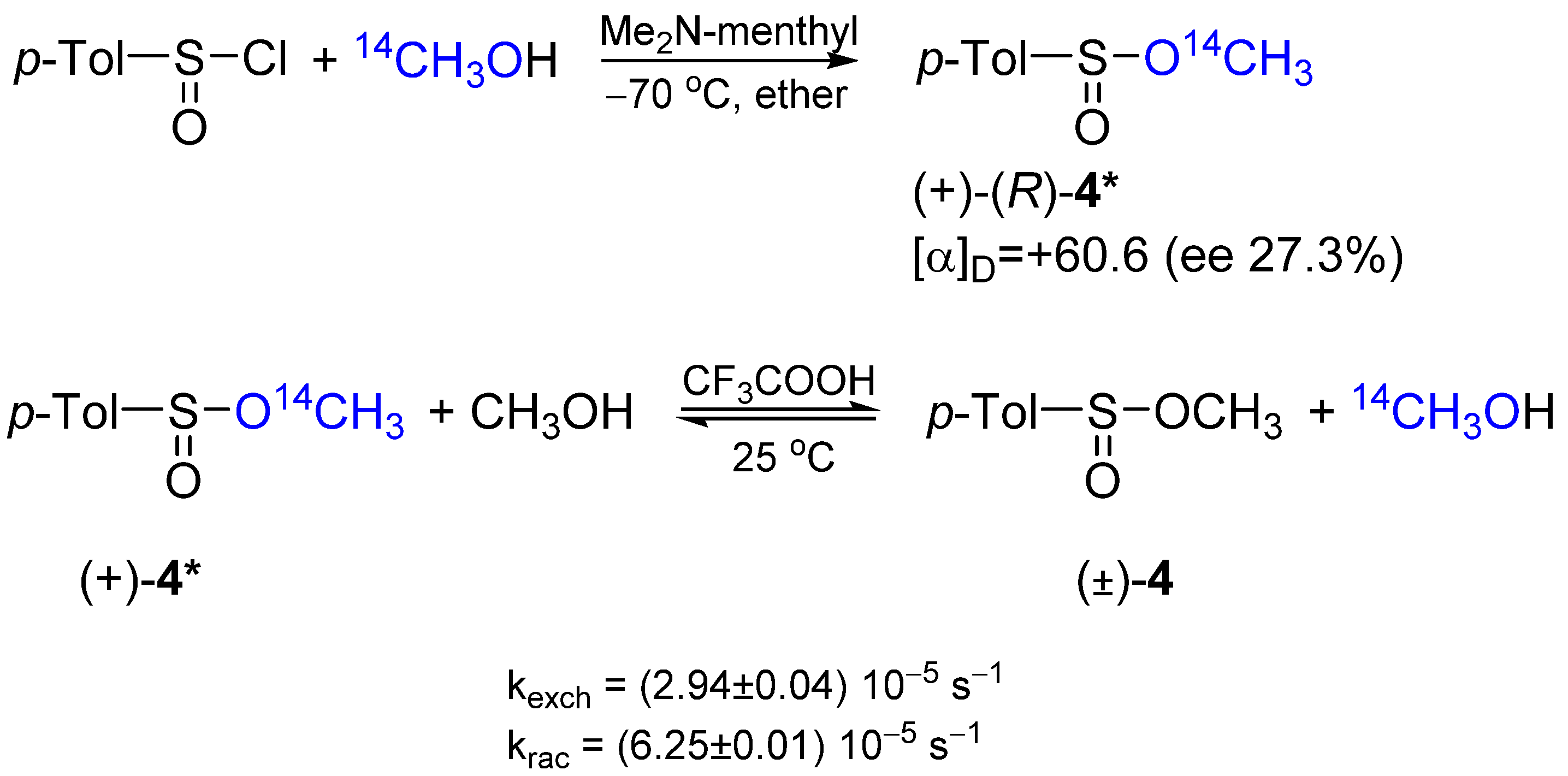{kind=link}
{kind=link}
{kind=link}
| ΔH | ΔG | |
|---|---|---|
| RC | −6.5 | 2.4 |
| TS1 | −2.7 | 9.3 |
| TBI-1 | −4.3 | 7.6 |
| TS2 | −2.7 | 9.3 |
| PC | −6.5 | 2.4 |
| ΔH | ΔG | |
|---|---|---|
| EC | −3.6 | 2.5 |
| TS | 12.5 | 20.2 |
| ΔH | ΔG | ||
|---|---|---|---|
| TolS(O)OMe + CF3COOH + MeOH | free reactants | 0.0 | 0.0 |
| TolS(OH+)OMe + CF3COO(−) + MeOH | 4-H+ | 22.0 | 22.3 |
| TolS(OH)(OMe)2 + CF3COOH | 4-OMe | 26.0 | 39.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikołajczyk, M.; Cypryk, M.; Gostyński, B.; Kowalczewski, J. Nucleophilic Substitution at Heteroatoms—Identity Substitution Reactions at Phosphorus and Sulfur Centers: Do They Proceed in a Concerted (SN2) or Stepwise (A–E) Way? Molecules 2022, 27, 599. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030599
Mikołajczyk M, Cypryk M, Gostyński B, Kowalczewski J. Nucleophilic Substitution at Heteroatoms—Identity Substitution Reactions at Phosphorus and Sulfur Centers: Do They Proceed in a Concerted (SN2) or Stepwise (A–E) Way? Molecules. 2022; 27(3):599. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030599
Chicago/Turabian StyleMikołajczyk, Marian, Marek Cypryk, Bartłomiej Gostyński, and Jakub Kowalczewski. 2022. "Nucleophilic Substitution at Heteroatoms—Identity Substitution Reactions at Phosphorus and Sulfur Centers: Do They Proceed in a Concerted (SN2) or Stepwise (A–E) Way?" Molecules 27, no. 3: 599. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030599