
Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches

 ,
,  ,
,  , , , and
, , , and 
Abstract
:1. Introduction
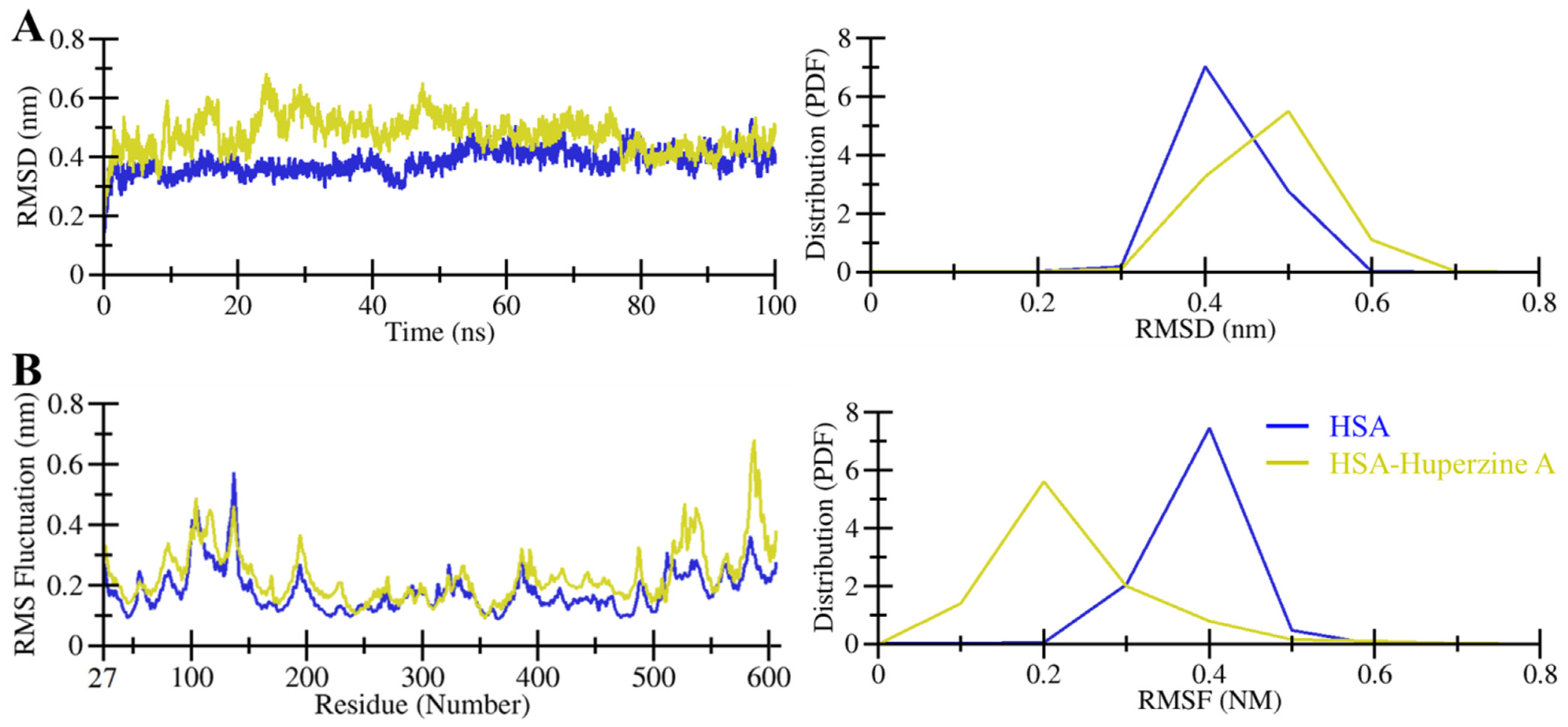
2. Results
2.1. Molecular Docking
2.2. MD Simulations
Dynamics of Intra-/Intermolecular Hydrogen Bonding
2.3. Principal Component and Free Energy Landscape Analyses
2.4. Fluorescence-Based Binding
3. Materials and Methods
3.1. Materials
3.2. Receptor and Ligand Preparation
3.3. Molecular Docking
3.4. Interaction Analysis
3.5. MD Simulations
3.6. Principal Component Analysis and Free Energy Landscape
3.7. Fluorescence-Based Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Rimac, H.; Debeljak, Z.; Bojić, M.; Miller, L. Displacement of Drugs from Human Serum Albumin: From Molecular Interactions to Clinical Significance. Curr. Med. Chem. 2017, 24, 1930–1947. [Google Scholar] [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981, 33, 17–53. [Google Scholar] [PubMed]
- Yadav, D.K. Recent Advances on Small Molecule Medicinal Chemistry to Treat Human Diseases-Part III. Curr. Top. Med. Chem. 2021, 21, 1517–1518. [Google Scholar] [CrossRef]
- Shamsi, A.; Ahmed, A.; Khan, M.S.; Al Shahwan, M.; Husain, F.M.; Bano, B. Understanding the binding between Rosmarinic acid and serum albumin: In vitro and in silico insight. J. Mol. Liq. 2020, 311, 113348. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Teli, M.K.; Kim, M. Ligand-based pharmacophore modeling and docking studies on vitamin D receptor inhibitors. J. Cell. Biochem. 2020, 121, 3570–3583. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Dockal, M.; Carter, D.C.; Rüker, F. Conformational Transitions of the Three Recombinant Domains of Human Serum Albumin Depending on pH. J. Biol. Chem. 2000, 275, 3042–3050. [Google Scholar] [CrossRef] [Green Version]
- Fani, N.; Bordbar, A.; Ghayeb, Y. Spectroscopic, docking and molecular dynamics simulation studies on the interaction of two Schiff base complexes with human serum albumin. J. Lumin. 2013, 141, 166–172. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L., III; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Behl, C.; Moosmann, B. Antioxidant neuroprotection in Alzheimer’s disease as preventive and therapeutic approach. Free Radic. Biol. Med. 2002, 33, 182–191. [Google Scholar] [CrossRef]
- Eckert, A.; Marques, C.A.; Keil, U.; Schüssel, K.; Müller, W.E. Increased apoptotic cell death in sporadic and genetic Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2003, 1010, 604–609. [Google Scholar] [CrossRef]
- Ma, X.; Tan, C.-H.; Zhu, D.; Gang, D.R.; Xiao, P. Huperzine A from Huperzia species—An ethnopharmacolgical review. J. Ethnopharmacol. 2007, 113, 15–34. [Google Scholar] [CrossRef]
- Wang, H.; Tang, X. Anticholinesterase effects of huperzine A, E2020, and tacrine in rats. Zhongguo yao li xue bao = Acta Pharmacol. Sin. 1998, 19, 27–30. [Google Scholar]
- Li, H.; Min, Q. Huperzine A improved the cognition of vascular dementia: A report of 30 patients in therapeutics. Chin. J. Clin. Rehabil. 2001, 5, 59. [Google Scholar]
- Yadav, D.K.; Kumar, S.; Saloni; Misra, S.; Yadav, L.; Teli, M.; Sharma, P.; Chaudhary, S.; Kumar, N.; Choi, E.H.; et al. Molecular insights into the interaction of RONS and Thieno [3, 2-c] pyran analogs with SIRT6/COX-2: A molecular dynamics study. Sci. Rep. 2018, 8, 4777. [Google Scholar] [CrossRef]
- Tiwari, M.K.; Coghi, P.; Agrawal, P.; Shyamlal, B.R.K.; Jun Yang, L.; Yadav, L.; Peng, Y.; Sharma, R.; Yadav, D.K.; Sahal, D.; et al. Design, Synthesis, Structure-Activity Relationship and Docking Studies of Novel Functionalized Arylvinyl-1, 2, 4-Trioxanes as Potent Antiplasmodial as well as Anticancer Agents. ChemMedChem 2020, 15, 1216–1228. [Google Scholar] [CrossRef]
- Shyamlal, B.R.K.; Yadav, L.; Tiwari, M.K.; Mathur, M.; Prikhodko, J.I.; Mashevskaya, I.V.; Yadav, D.K.; Chaudhary, S. Synthesis, Bioevaluation, Structure-Activity Relationship and Docking Studies of Natural Product Inspired (Z)-3-benzylideneisobenzofuran-1(3H)-ones as Highly Potent antioxidants and Antiplatelet agents. Sci. Rep. 2020, 10, 2307. [Google Scholar] [CrossRef] [Green Version]
- Teli, M.K.; Kumar, S.; Yadav, D.K.; Kim, M. In silico identification of prolyl hydroxylase inhibitor by per-residue energy decomposition-based pharmacophore approach. J. Cell. Biochem. 2021, 122, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Siddiqui, S.; Shamsi, A.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M.I. Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies. Molecules 2020, 25, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadhe, C.G.; Kim, M.-H. Insights into the binding modes of CC chemokine receptor 4 (CCR4) inhibitors: A combined approach involving homology modelling, docking, and molecular dynamics simulation studies. Mol. Biosyst. 2015, 11, 618–634. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Gadhe, C.G.; Balupuri, A.; Cho, S.J. In silico characterization of binding mode of CCR8 inhibitor: Homology modeling, docking and membrane based MD simulation study. J. Biomol. Struct. Dyn. 2015, 33, 2491–2510. [Google Scholar] [CrossRef]
- Ito, A.; Mukaiyama, A.; Itoh, Y.; Nagase, H.; Thøgersen, I.B.; Enghild, J.J.; Sasaguri, Y.; Mori, Y. Degradation of Interleukin 1β by Matrix Metalloproteinases. J. Biol. Chem. 1996, 271, 14657–14660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, R.E. Hydrogen Bonds in Proteins: Role and Strength. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal Component Analysis for Protein Folding Dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Mohammad, T.; Jairajpuri, D.S.; Rehman, T.; Hussain, A.; Samim, M.; Ahmad, F.; Alajmi, M.F.; Hassan, I. Identification and evaluation of glutathione conjugate gamma-l-glutamyl-l-cysteine for improved drug delivery to the brain. J. Biomol. Struct. Dyn. 2020, 38, 3610–3620. [Google Scholar] [CrossRef]
- Altis, A.; Otten, M.; Nguyen, P.H.; Hegger, R.; Stock, G. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 2008, 128, 245102. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Shamsi, A.; Shahbaaz, M.; Queen, A.; Khan, P.; Hasan, G.M.; Islam, A.; Alajmi, M.F.; Hussain, A.; Ahmad, F.; et al. Rosmarinic Acid Exhibits Anticancer Effects via MARK4 Inhibition. Sci. Rep. 2020, 10, 10300. [Google Scholar] [CrossRef]
- Soares, S.; Mateus, N.; de Freitas, V. Interaction of Different Polyphenols with Bovine Serum Albumin (BSA) and Human Salivary α-Amylase (HSA) by Fluorescence Quenching. J. Agric. Food Chem. 2007, 55, 6726–6735. [Google Scholar] [CrossRef]
- Klajnert, B.; Stanisławska, L.; Bryszewska, M.; Pałecz, B. Interactions between PAMAM dendrimers and bovine serum albumin. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2003, 1648, 115–126. [Google Scholar] [CrossRef]
- Shamsi, A.; Al Shahwan, M.; Ahamad, S.; Hassan, M.I.; Ahmad, F.; Islam, A. Spectroscopic, calorimetric and molecular docking insight into the interaction of Alzheimer’s drug donepezil with human transferrin: Implications of Alzheimer’s drug. J. Biomol. Struct. Dyn. 2020, 38, 1094–1102. [Google Scholar] [CrossRef]
- Waseem, R.; Anwar, S.; Khan, S.; Shamsi, A.; Hassan, I.; Anjum, F.; Shafie, A.; Islam, A.; Yadav, D.K. MAP/Microtubule Affinity Regulating Kinase 4 Inhibitory Potential of Irisin: A New Therapeutic Strategy to Combat Cancer and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10986. [Google Scholar] [CrossRef]
- Anwar, S.; Mohammad, T.; Shamsi, A.; Queen, A.; Parveen, S.; Luqman, S.; Hasan, G.M.; Alamry, K.A.; Azum, N.; Asiri, A.M.; et al. Discovery of Hordenine as a Potential Inhibitor of Pyruvate Dehydrogenase Kinase 3: Implication in Lung Cancer Therapy. Biomedicines 2020, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Shamsi, A.; Kar, R.K.; Queen, A.; Islam, A.; Ahmad, F.; Hassan, I. Structural and biochemical investigation of MARK4 inhibitory potential of cholic acid: Towards therapeutic implications in neurodegenerative diseases. Int. J. Biol. Macromol. 2020, 161, 596–604. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Nasreen, K.; Hassan, I.; Ahmad, F.; Islam, A. Insight into the binding of PEG-400 with eye protein alpha-crystallin: Multi spectroscopic and computational approach: Possible therapeutics targeting eye diseases. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M. Swiss-Model and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Mohammad, T.; Mathur, Y.; Hassan, I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Briefings Bioinform. 2021, 22, bbaa279. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Van Aalten, D.M.F.; Bywater, R.; Findlay, J.B.C.; Hendlich, M.; Hooft, R.W.W.; Vriend, G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Choi, E.H.; Chaudhary, S.; Kim, M.H. Computational Modeling on Aquaporin-3 as Skin Cancer Target: A Virtual Screening and Molecular Dynamic Simulation Study. Front Chem. 2019, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.T.; Mohammad, T.; Hasan, G.M.; Hassan, I. Advancements in Docking and Molecular Dynamics Simulations Towards Ligand-receptor Interactions and Structure-function Relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef]
- Shamsi, A.; Anwar, S.; Mohammad, T.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Hasan, G.M.; Islam, A.; Hassan, M.I. MARK4 inhibited by AChE inhibitors, donepezil and Rivastigmine tartrate: Insights into Alzheimer’s disease therapy. Biomolecules 2020, 10, 789. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Ksv (104 M−1) | K 105 M−1 | n | ∆G kcal mol−1 | ∆S cal mol−1 K−1 | ∆H kcal mol−1 | T∆S kcal mol−1 |
|---|---|---|---|---|---|---|---|
| 15 | 1.8 | 0.36 | 1.07 | −7.78 | −165.26 | −55.38 | −47.59 |
| 20 | 3.33 | 1.17 | 1.10 | −6.96 | −48.42 | ||
| 25 | 5.19 | 9.35 | 1.25 | −6.13 | −49.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamsi, A.; Shahwan, M.; Khan, M.S.; Alhumaydhi, F.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Abdullaev, B.; Yadav, D.K. Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches. Molecules 2022, 27, 797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030797
Shamsi A, Shahwan M, Khan MS, Alhumaydhi FA, Alsagaby SA, Al Abdulmonem W, Abdullaev B, Yadav DK. Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches. Molecules. 2022; 27(3):797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030797
Chicago/Turabian StyleShamsi, Anas, Moyad Shahwan, Mohd Shahnawaz Khan, Fahad A. Alhumaydhi, Suliman A. Alsagaby, Waleed Al Abdulmonem, Bekhzod Abdullaev, and Dharmendra Kumar Yadav. 2022. "Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches" Molecules 27, no. 3: 797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030797