
Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene—Dimethylaniline



Abstract
:
1. Introduction
2. Experimental: Computational Methods
3. Results and Discussion
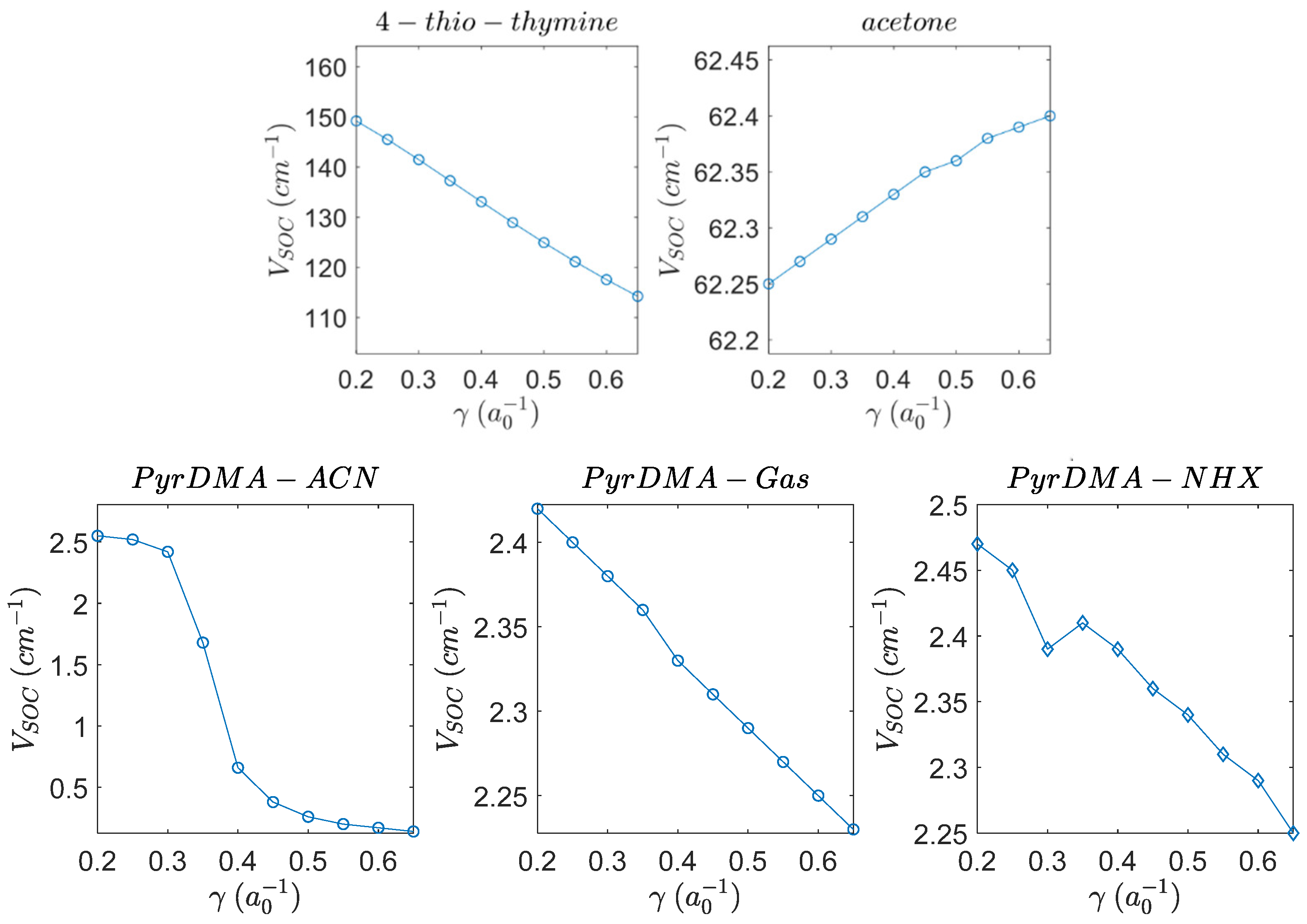
3.1. Benchmarking the SOCME
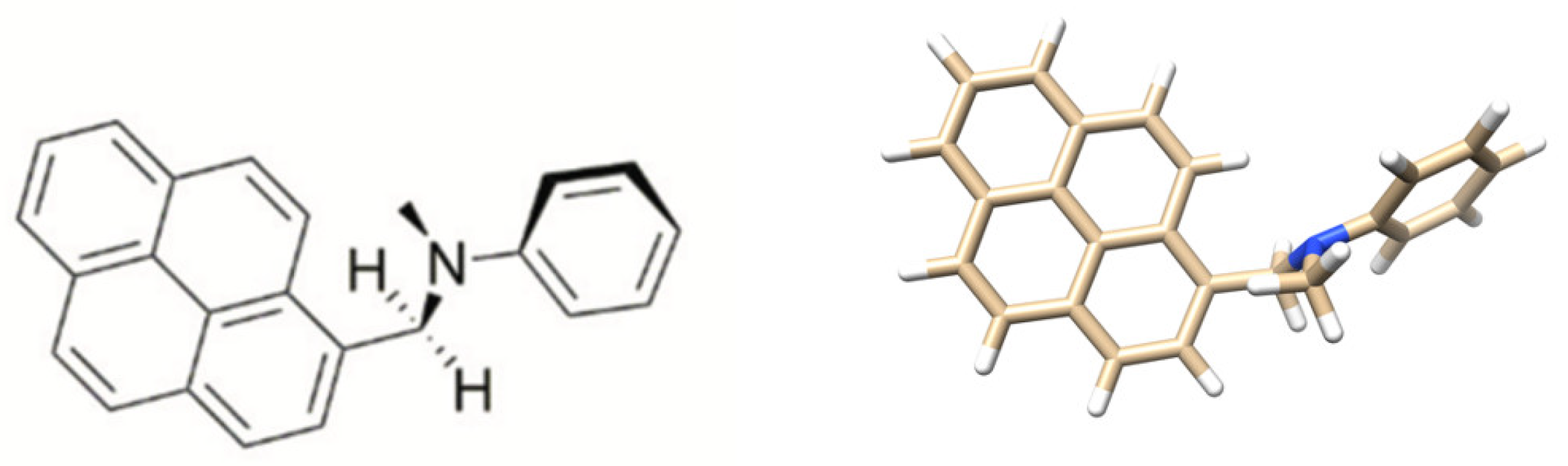
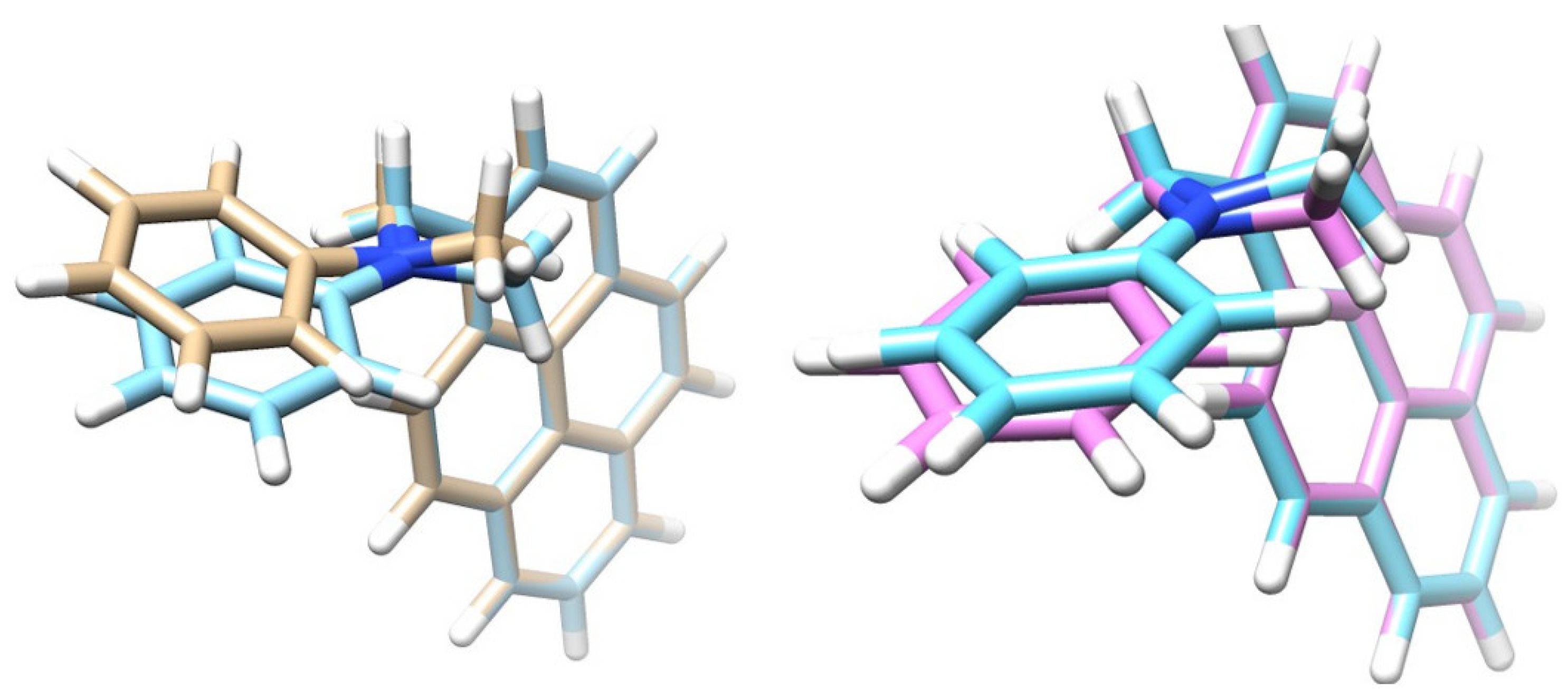
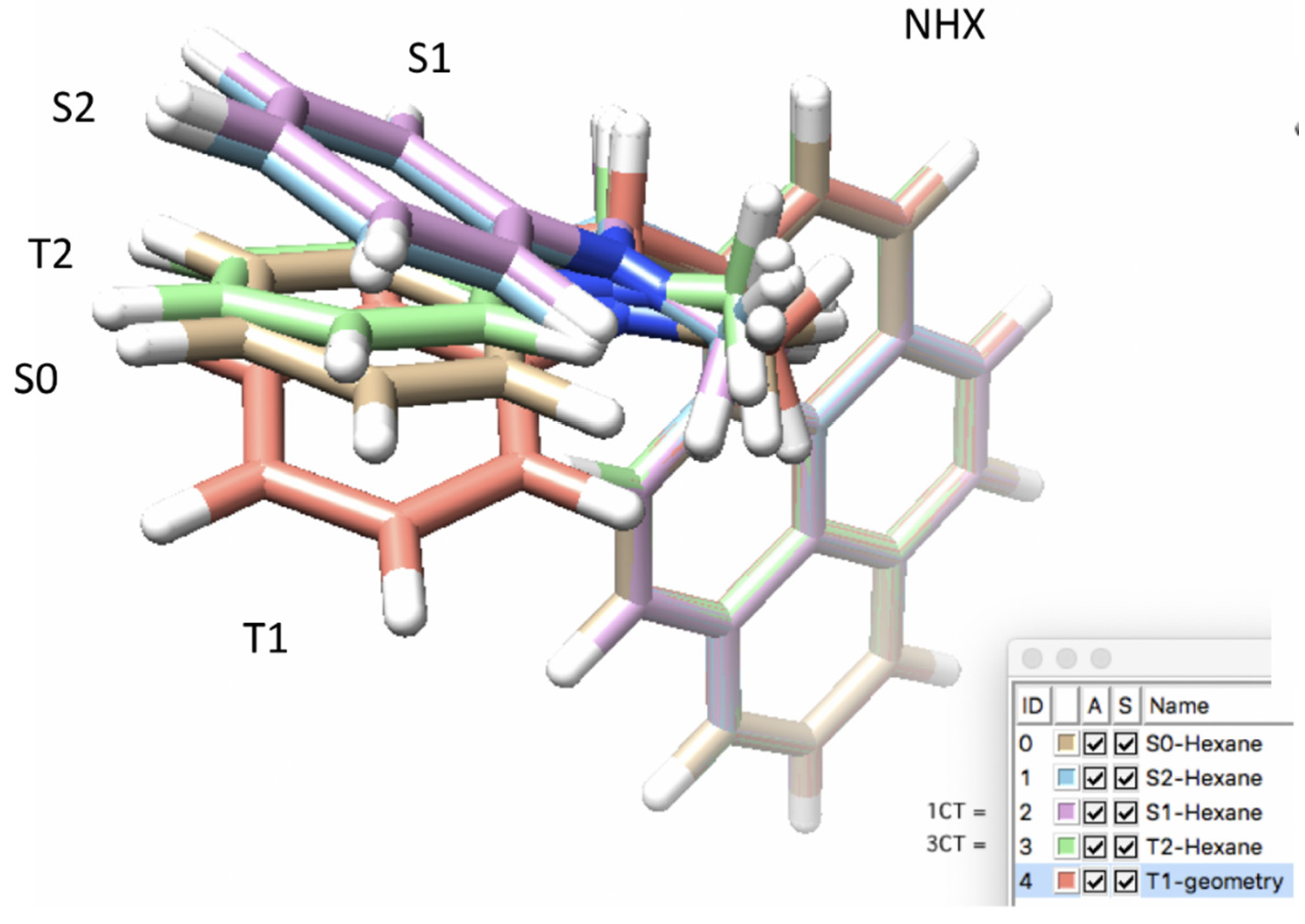
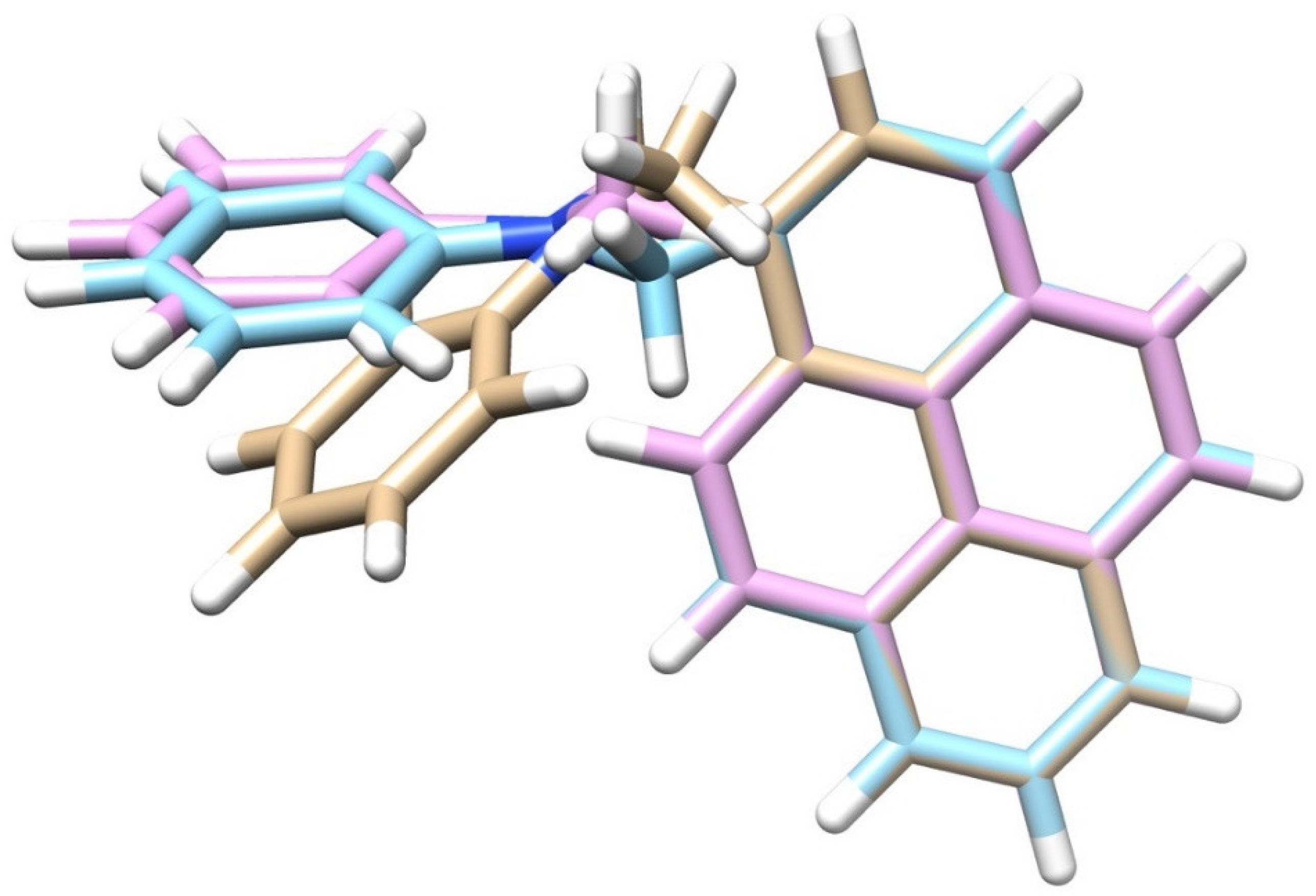
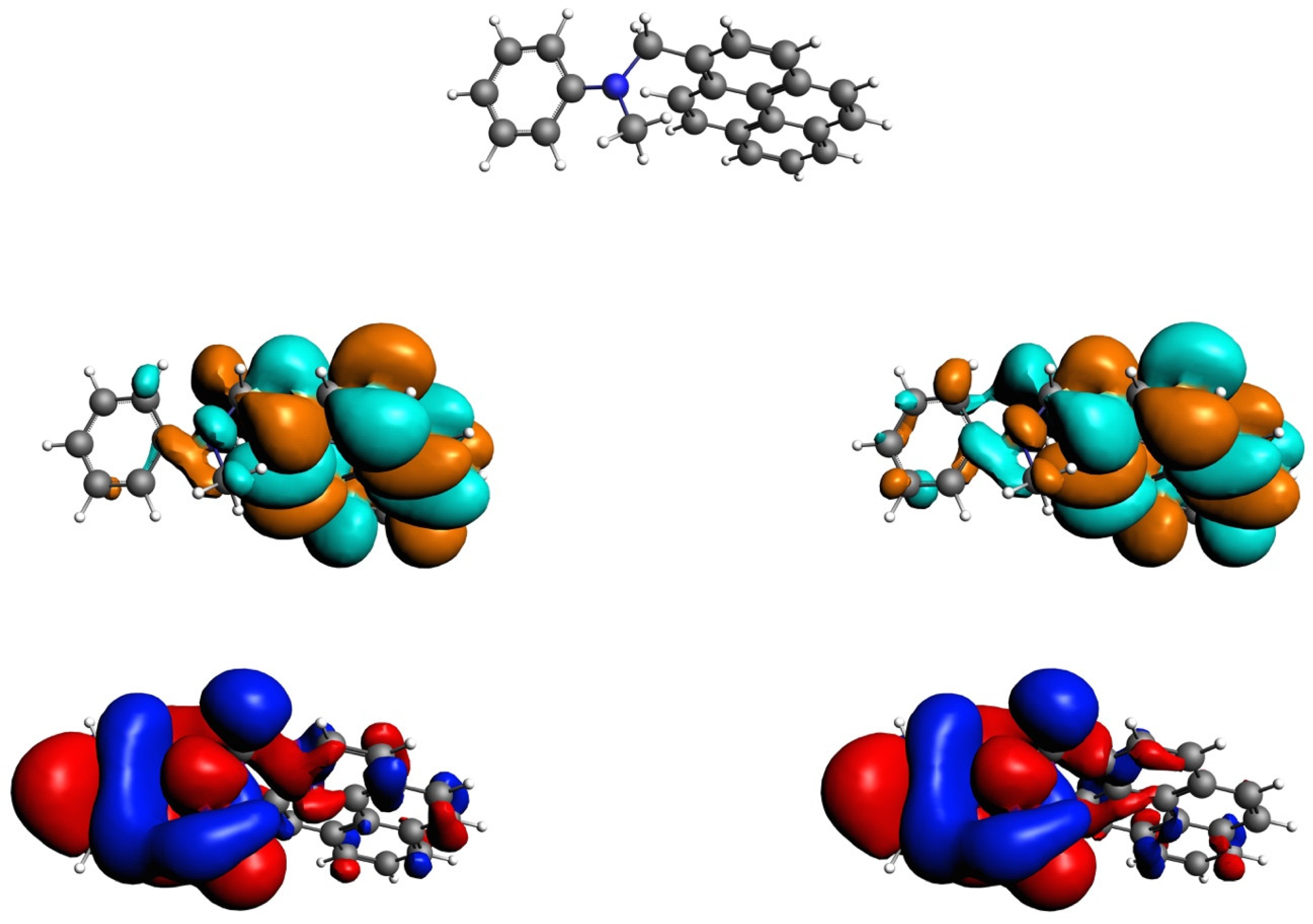
3.2. Excited State Geometries of PyrDMA
3.3. Excitation Energies and Scheme at Optimized Geometries
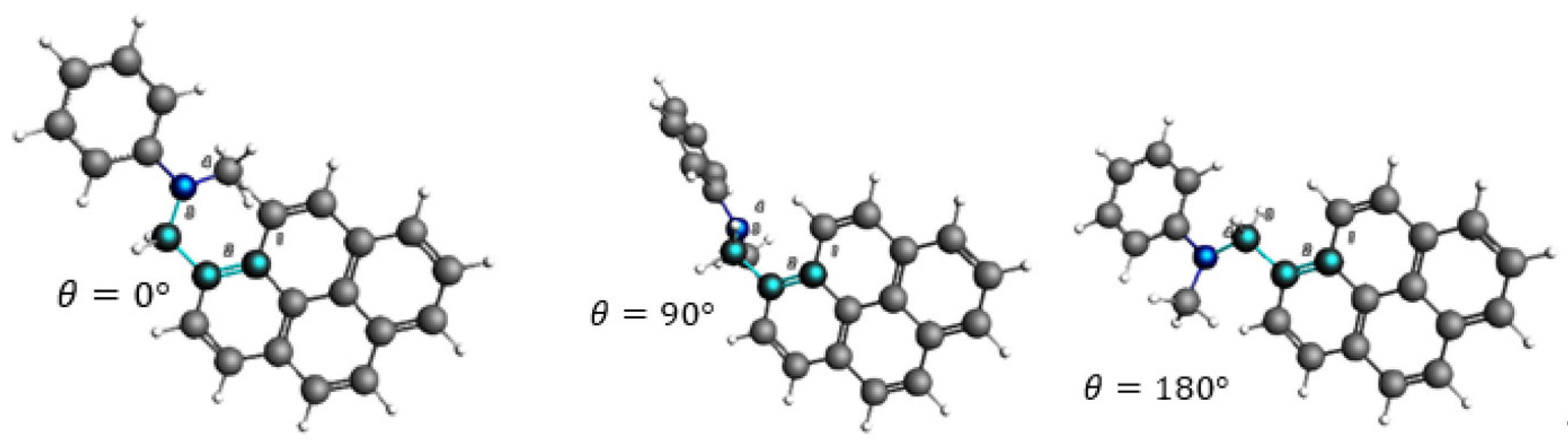
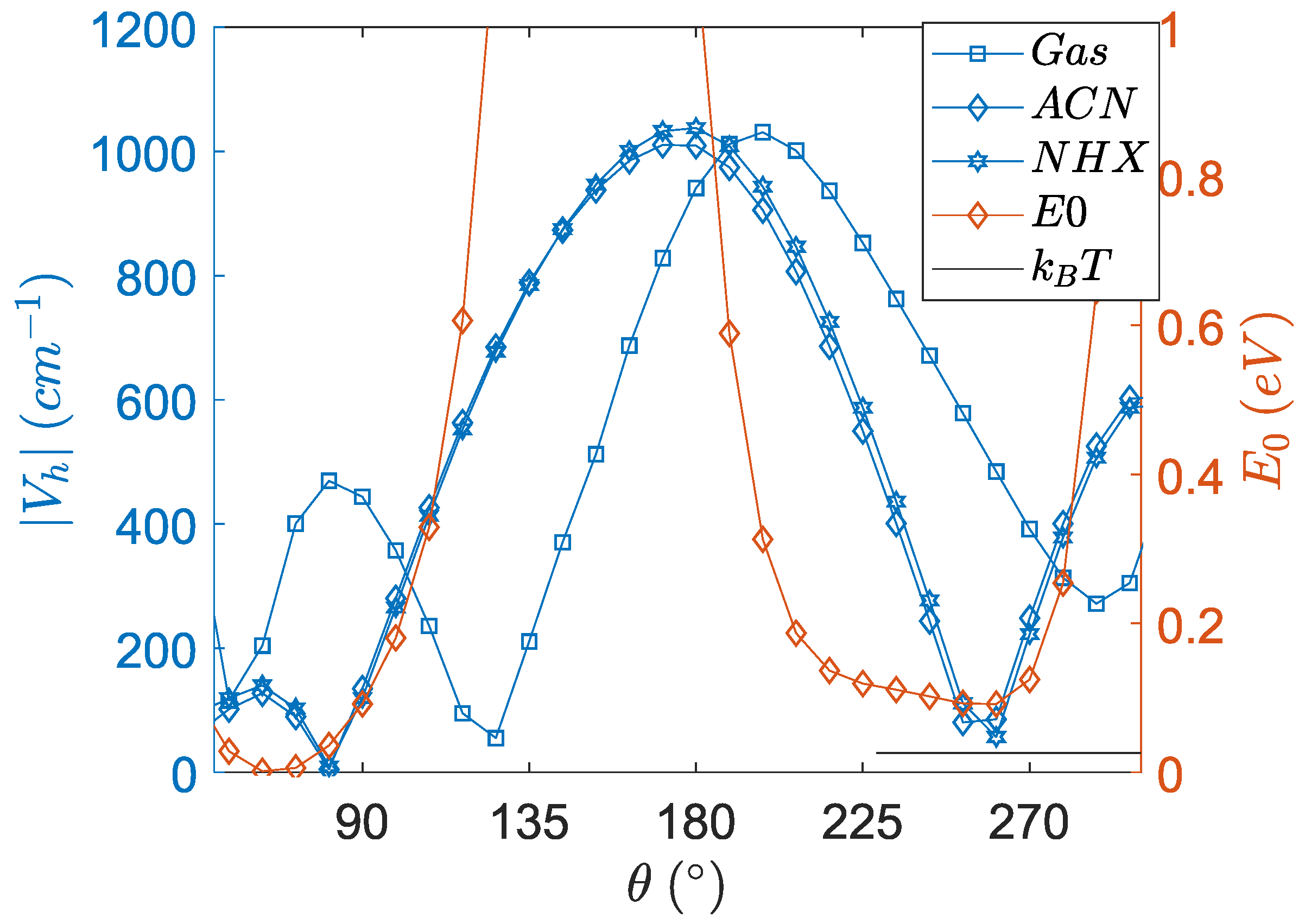
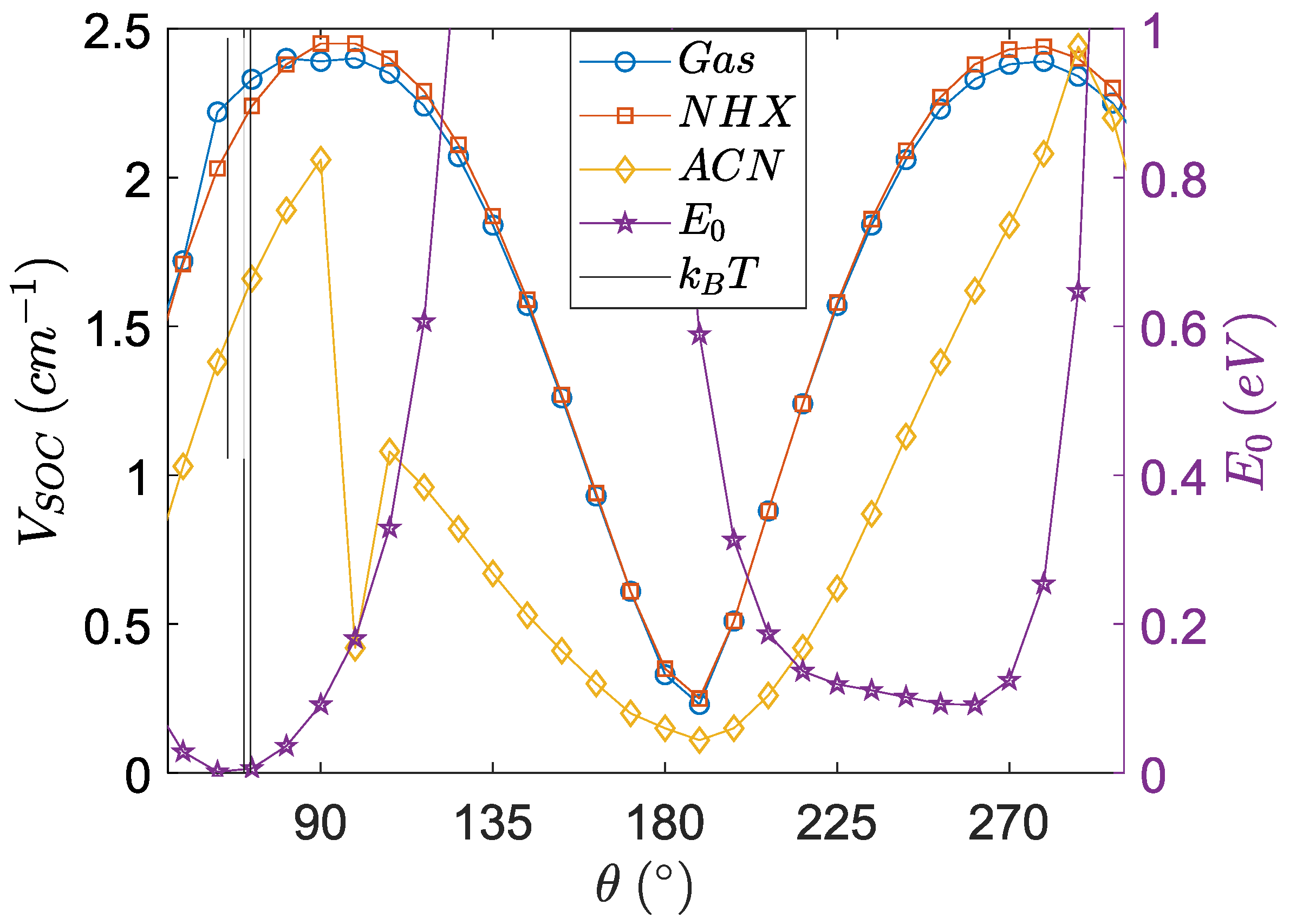
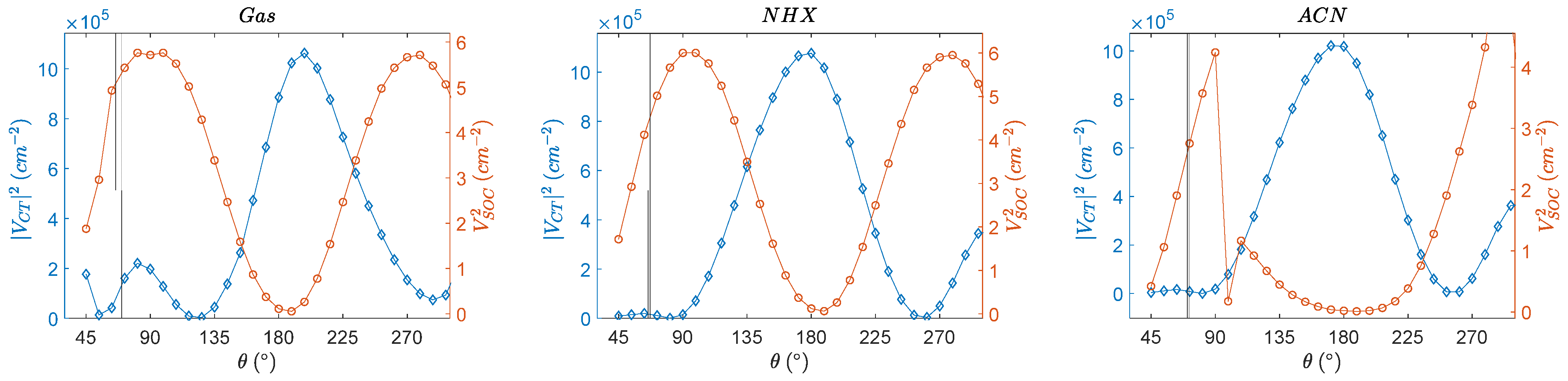
3.4. Angular Dependence of Spin–Orbit Coupling and of the Electronic Coupling
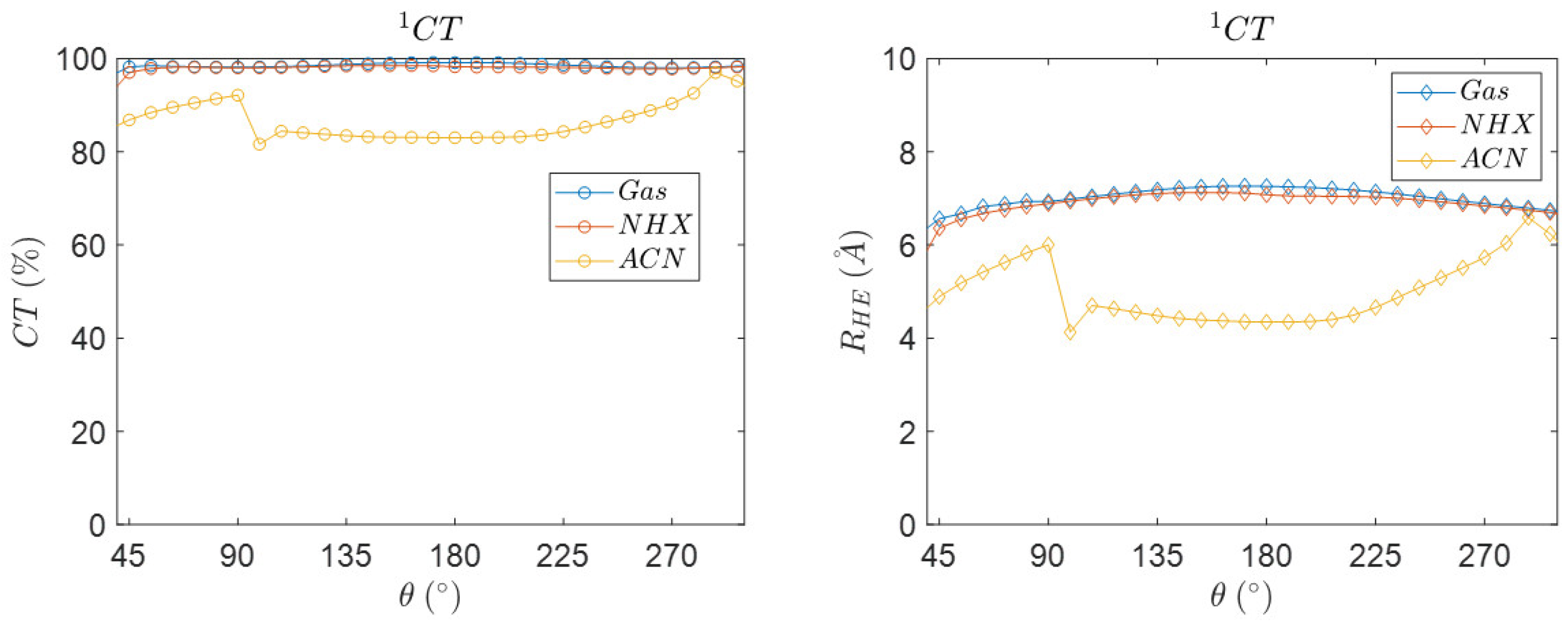
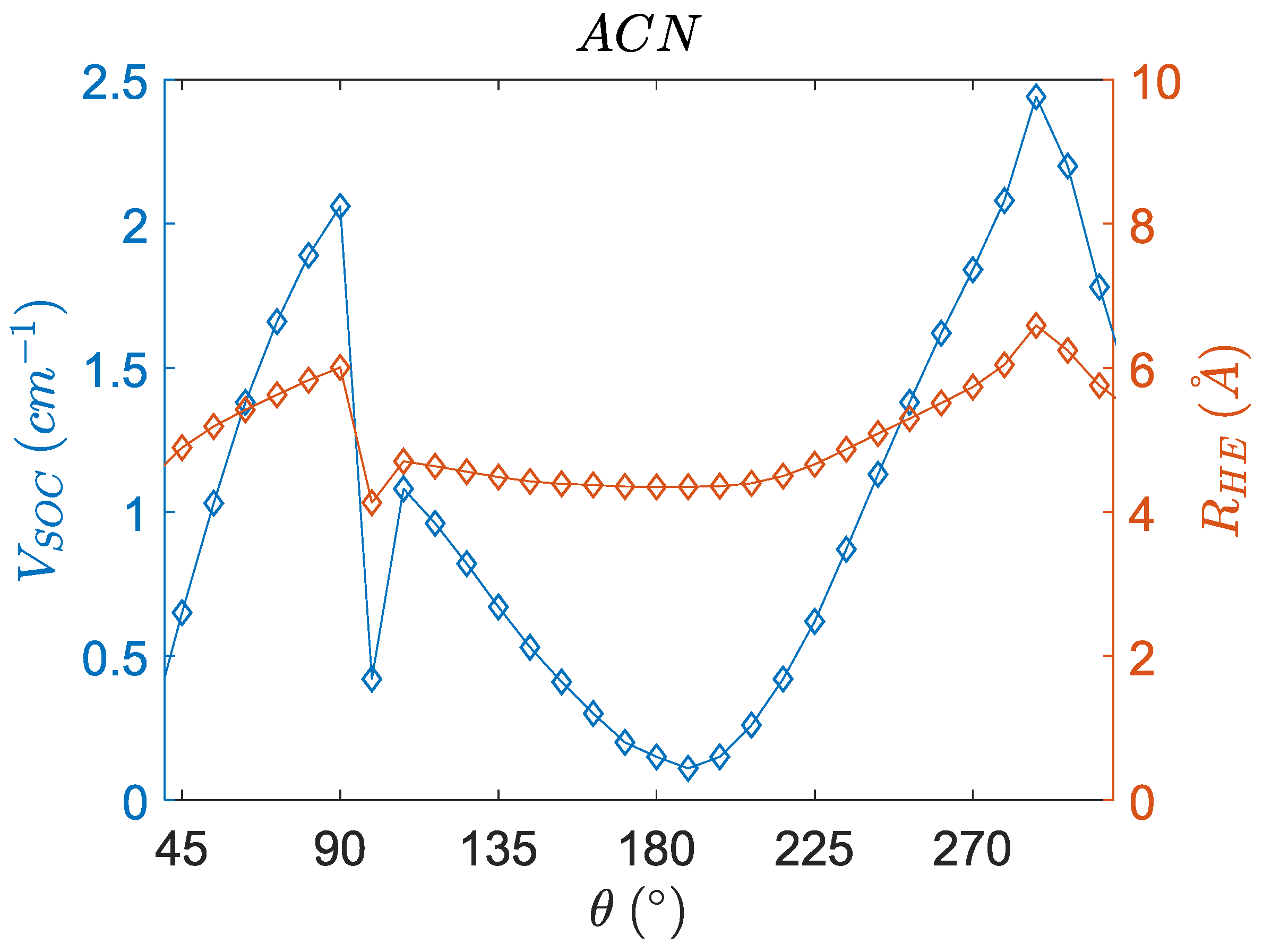
3.5. Angular Dependence of the Charge Transfer Character, Electron Hole Distance and Energetics
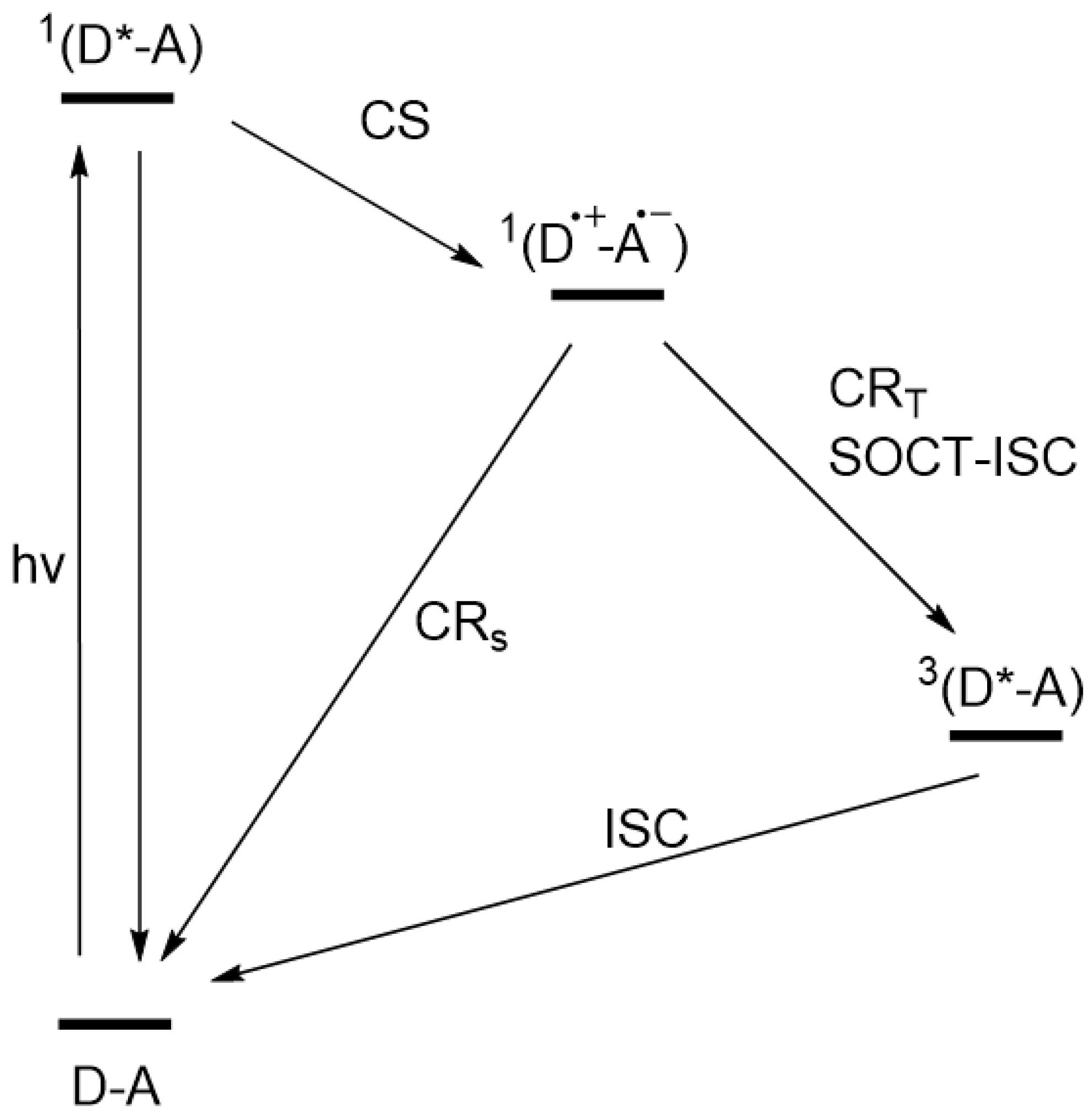
3.6. Energetic and Kinetic Considerations for Charge Separation and Recombination Pathways
4. Conclusions and Future Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TD-DFT | Time-dependent density functional theory |
| ADF | Amsterdam Density Functional |
| DMA | Dimethylaniline |
| Pyr | Pyrene |
| NHX | N-hexane |
| ACN | Acetonitrile |
| DEE | Diethyl ether |
| CS | Charge separation |
| CR | Charge recombination |
| ISC | Intersystem crossing |
| CT | Charge transfer |
| SO-ISC | Spin–orbit coupling intersystem crossing |
| SOCT-ISC | Spin–orbit charge transfer intersystem crossing |
| MO | Molecular orbital |
| SOC | Spin–orbit coupling |
| SOCME | Spin–orbit coupling matrix element |
| GS | Ground state |
| LE | Local excitation |
| Tn | Triplet (state) |
| Sn | Singlet (state) |
| nCS | Charge separated (state) |
| Electronic coupling | |
| Spin–orbit coupling | |
| Temperature | |
| Reduced Planck constant | |
| Boltzmann constant | |
| Speed of light | |
| Reorganization Energy | |
| Lifetime of a state | |
| PES | Potential Energy Surface |
| Gibbs free energy | |
| Huang–Rhys factor | |
| FCWD | Frank Condon Weighted Density |
| Oscillator strength | |
| TDA | Tamm–Dancoff approximation |
| NTO | Natural Transition Orbital |
| COSMO | Conductor-like screening model |
| ZORA | Zero-order regular approximation |
References
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Principles of Molecular Photochemistry: An Introduction; University Science Books: Sausalito, CA, USA, 2009. [Google Scholar]
- Marian, C.M. Spin-Orbit Coupling and Intersystem Crossing in Molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Gao, X.; Bai, S.; Fazzi, D.; Niehaus, T.; Barbatti, M.; Thiel, W. Evaluation of Spin-Orbit Couplings with Linear-Response Time-Dependent Density Functional Methods. J. Chem. Theory Comput. 2017, 13, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Biskup, T.; Rein, S.; Wang, Z.; Bussotti, L.; Russo, N.; Foggi, P.; Zhao, J.; Di Donato, M.; Mazzone, G.; et al. Spin-Orbit Charge Recombination Intersystem Crossing in Phenothiazine-Anthracene Compact Dyads: Effect of Molecular Conformation on Electronic Coupling, Electronic Transitions, and Electron Spin Polarizations of the Triplet States. J. Phys. Chem. C 2018, 122, 27850–27865. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions. J. Chem. Phys. 1965, 43, 679–701. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.A. Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Monti, A.; De Groot, H.J.M.; Buda, F. In-Silico Design of a Donor-Antenna-Acceptor Supramolecular Complex for Photoinduced Charge Separation. J. Phys. Chem. C 2014, 118, 15600–15609. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, X.; Chen, K.; Liu, D.; Wang, Z.; Liu, Q.; Zhao, J.; Barbon, A. Charge Separation, Charge Recombination, Long-Lived Charge Transfer State Formation and Intersystem Crossing in Organic Electron Donor/Acceptor Dyads. J. Mater. Chem. C 2019, 7, 12048–12074. [Google Scholar] [CrossRef]
- Gibbons, D.J.; Farawar, A.; Mazzella, P.; Leroy-Lhez, S.; Williams, R.M. Making Triplets from Photo-Generated Charges: Observations, Mechanisms and Theory. Photochem. Photobiol. Sci. 2020, 19, 136–158. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.M.; Yoon, T.P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Üngördü, A. Electronic, Optical, and Charge Transfer Properties of Porphyrin and Metallated Porphyrins in Different Media. Int. J. Quantum Chem. 2020, 120, e26128. [Google Scholar] [CrossRef]
- Ronca, E.; De Angelis, F.; Fantacci, S. Time-Dependent Density Functional Theory Modeling of Spin-Orbit Coupling in Ruthenium and Osmium Solar Cell Sensitizers. J. Phys. Chem. C 2014, 118, 17067–17078. [Google Scholar] [CrossRef]
- Tsvirko, M.; Solovjev, K.; Gradyushko, A.; Dvornikov, S. Phosphorescence of Porphyrin Free Bases and Their Complexes with Light Metals. Opt. Spectrosc. 1975, 38, 400. [Google Scholar]
- Zhao, J.; Wu, W.; Sun, J.; Guo, S. Triplet Photosensitizers: From Molecular Design to Applications. Chem. Soc. Rev. 2013, 42, 5323–5351. [Google Scholar] [CrossRef]
- Yang, B.; Huang, S.; Luo, S.A. Theoretical Research on Intersystem Crossing, Radiative and Nonradiative Rates of Cyclometalated Platinum(II) Complexes. Theor. Chem. Acc. 2019, 138, 77. [Google Scholar] [CrossRef]
- Minaev, B.; Ågren, H. Theoretical DFT Study of Phosphorescence from Porphyrins. Chem. Phys. 2005, 315, 215–239. [Google Scholar] [CrossRef]
- Filatov, M.A. Heavy-Atom-Free BODIPY Photosensitizers with Intersystem Crossing Mediated by Intramolecular Photoinduced Electron Transfer. Org. Biomol. Chem. 2019, 18, 10–27. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Yu, Y.; Sandoval-Salinas, M.E.; Xu, J.; Lei, Z.; Casanova, D.; Yang, Y.; Chen, J. Engineering the Charge-Transfer State to Facilitate Spin–Orbit Charge Transfer Intersystem Crossing in Spirobis[Anthracene]Diones. Angew. Chemie Int. Ed. 2020, 59, 2–8. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Spin-Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics. J. Chem. Phys. 1963, 38, 2834. [Google Scholar] [CrossRef]
- Okada, T.; Karaki, I.; Matsuzawa, E.; Mataga, N.; Sakata, Y.; Misumi, S. Ultrafast Intersystem Crossing in Some Intramolecular Heteroexcimers. J. Phys. Chem. 1981, 85, 3957–3960. [Google Scholar] [CrossRef]
- Van Willigen, H.; Guilford Jones, I.; Farahat, M.S. Time-Resolved EPR Study of Photoexcited Triplet-State Formation in Electron-Donor-Substituted Acridinium Ions. J. Phys. Chem. 1996, 100, 3312–3316. [Google Scholar] [CrossRef]
- Dance, Z.E.X.; Mickley, S.M.; Wilson, T.M.; Ricks, A.B.; Scott, A.M.; Ratner, M.A.; Wasielewski, M.R. Intersystem Crossing Mediated by Photoinduced Intramolecular Charge Transfer: Julolidine—Anthracene Molecules with Perpendicular π Systems. J. Phys. Chem. A 2008, 112, 4194–4201. [Google Scholar] [CrossRef] [PubMed]
- SCM. Amsterdam Modeling Suite Making Computational Chemistry Work for You Software for Chemistry & Materials. Available online: https://www.scm.com/ (accessed on 9 February 2021).
- Buck, J.T.; Boudreau, A.M.; DeCarmine, A.; Wilson, R.W.; Hampsey, J.; Mani, T. Spin-Allowed Transitions Control the Formation of Triplet Excited States in Orthogonal Donor-Acceptor Dyads. Chem 2019, 5, 138–155. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, D.; John, A.T.; Sunny, J.; Hariharan, M. Access to the Triplet Excited States of Organic Chromophores. Chem. Soc. Rev. 2020, 49, 6122–6140. [Google Scholar] [CrossRef]
- Nguyen, V.-N.; Yan, Y.; Zhao, J.; Yoon, J. Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer. Acc. Chem. Res. 2021, 54, 207–220. [Google Scholar] [CrossRef]
- Sutton, C.; Sears, J.S.; Coropceanu, V.; Brédas, J.L. Understanding the Density Functional Dependence of DFT-Calculated Electronic Couplings in Organic Semiconductors. J. Phys. Chem. Lett. 2013, 4, 919–924. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T. An Implementation of the Conductor-like Screening Model of Solvation within the Amsterdam Density Functional Package. Theor. Chem. Acc. 1999, 101, 396–408. [Google Scholar] [CrossRef]
- Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry; Harper & Row: New York, NY, USA, 1987. [Google Scholar]
- Williams, R.M.; Zwier, J.M.; Verhoeven, J.W. Photoinduced Intramolecular Electron Transfer in a Bridged C60 (Acceptor)-Aniline (Donor) System; Photophysical Properties of the First “Active” Fullerene Diad. J. Am. Chem. Soc. 1995, 117, 4093–4099. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Koeberg, M.; Lawson, J.M.; An, Y.-Z.; Rubin, Y.; Paddon-Row, M.N.; Verhoeven, J.W. Photoinduced Electron Transfer to C60 across Extended 3- and 11-Bond Hydrocarbon Bridges: Creation of a Long-Lived Charge-Separated State. J. Org. Chem. 1996, 61, 5055–5062. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Verhoeven, J.W. Fluorescence of Fullerene-C70 and Its Quenching by Long-Range Intermolecular Electron Transfer. Chem. Phys. Lett. 1992, 194, 6. [Google Scholar] [CrossRef] [Green Version]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- SPARTAN; Wavefunction Inc. 18401 Von Karman Ave, Suite 370, Irvine, CA 92715. Available online: https://www.wavefun.com/ (accessed on 25 January 2022).
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Beljonne, D.; Shuai, Z.; Pourtois, G.; Bredas, J.L. Spin-Orbit Coupling and Intersystem Crossing in Conjugated Polymers: A Configuration Interaction Description. J. Phys. Chem. A 2001, 105, 3899–3907. [Google Scholar] [CrossRef]
- Zhu, Q.; Feng, S.; Guo, X.; Chen, X.; Zhang, J. Strategy for Tuning the Up-Conversion Intersystem Crossing Rates in a Series of Organic Light-Emitting Diodes Emitters Relevant for Thermally Activated Delayed Fluorescence. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 221, 117214. [Google Scholar] [CrossRef]
- Samanta, P.K.; Kim, D.; Coropceanu, V.; Brédas, J.L. Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs. Triplet Excited States. J. Am. Chem. Soc. 2017, 139, 4042–4051. [Google Scholar] [CrossRef]
- Sun, H.; Hu, Z.; Zhong, C.; Chen, X.; Sun, Z.; Brédas, J.L. Impact of Dielectric Constant on the Singlet-Triplet Gap in Thermally Activated Delayed Fluorescence Materials. J. Phys. Chem. Lett. 2017, 8, 2393–2398. [Google Scholar] [CrossRef] [Green Version]
- Cardeynaels, T.; Paredis, S.; Deckers, J.; Brebels, S.; Vanderzande, D.; Maes, W.; Champagne, B. Finding the Optimal Exchange–Correlation Functional to Describe the Excited State Properties of Push–Pull Organic Dyes Designed for Thermally Activated Delayed Fluorescence. Phys. Chem. Chem. Phys. 2020, 22, 16387–16399. [Google Scholar] [CrossRef]
- Hu, Y.; Hou, Y.; Wang, Z.; Li, Y.; Zhao, J. 3,5-Anthryl-Bodipy Dyad/Triad: Preparation, Effect of F-B-F Induced Conformation Restriction on the Photophysical Properties, and Application in Triplet-Triplet-Annihilation Upconversion. J. Chem. Phys. 2020, 153, 224304. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775. [Google Scholar] [CrossRef]
- Gonzalez-Vazquez, J.P.; Burn, P.L.; Powell, B.J. Interplay of Zero-Field Splitting and Excited State Geometry Relaxation in Fac-Ir(Ppy)3. Inorg. Chem. 2015, 54, 10457–10461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhao, J.; Li, X.; Gurzadyan, G.G. Anthracene-Naphthalenediimide Compact Electron Donor/Acceptor Dyads: Electronic Coupling, Electron Transfer, and Intersystem Crossing. J. Phys. Chem. A 2019, 123, 2503–2516. [Google Scholar] [CrossRef]
- Wang, Z.; Sukhanov, A.A.; Toffoletti, A.; Sadiq, F.; Zhao, J.; Barbon, A.; Voronkova, V.K.; Dick, B. Insights into the Efficient Intersystem Crossing of Bodipy-Anthracene Compact Dyads with Steady-State and Time-Resolved Optical/Magnetic Spectroscopies and Observation of the Delayed Fluorescence. J. Phys. Chem. C 2019, 123, 265–274. [Google Scholar] [CrossRef]
- Letrun, R.; Lang, B.; Yushchenko, O.; Wilcken, R.; Svechkarev, D.; Kolodieznyi, D.; Riedle, E.; Vauthey, E. Excited-State Dynamics of a Molecular Dyad with Two Orthogonally-Oriented Fluorophores. Phys. Chem. Chem. Phys. 2018, 20, 30219–30230. [Google Scholar] [CrossRef]
- Liu, T.; Troisi, A. Absolute Rate of Charge Separation and Recombination in a Molecular Model of the P3HT/PCBM Interface. J. Phys. Chem. C 2011, 115, 2406–2415. [Google Scholar] [CrossRef]
- Wen, S.H.; Li, A.; Song, J.; Deng, W.Q.; Han, K.L.; Goddard, W.A. First-Principles Investigation of Anistropic Hole Mobilities in Organic Semiconductors. J. Phys. Chem. B 2009, 113, 8813–8819. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.P.; Trujillo-González, D.E.; Götz, A.W.; Castillo-Alvarado, F.L.; Rodríguez, J.I. Effects of Dispersion Forces on Structure and Photoinduced Charge Separation in Organic Photovoltaics. J. Phys. Chem. C 2017, 121, 20134–20140. [Google Scholar] [CrossRef]
- Kelber, J.B.; Panjwani, N.A.; Wu, D.; Gómez-Bombarelli, R.; Lovett, B.W.; Morton, J.J.L.; Anderson, H.L. Synthesis and Investigation of Donor–Porphyrin–Acceptor Triads with Long-Lived Photo-Induced Charge-Separate States. Chem. Sci. 2015, 6, 6468–6481. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, S.; Hedström, S.; Méndez-Hernández, D.D.; Hendrickson, H.P.; Jung, K.A.; Ho, J.; Batista, V.S. Electron Transfer Assisted by Vibronic Coupling from Multiple Modes. J. Chem. Theory Comput. 2017, 13, 6000–6009. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation Energies in Density Functional Theory: An Evaluation and a Diagnostic Test. J. Chem. Phys. 2008, 128, 044118. [Google Scholar] [CrossRef] [PubMed]
- Plasser, F.; Lischka, H. Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations. J. Chem. Theory Comput. 2012, 8, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Dreuw, A.; Head-Gordon, M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zinc bacteriochlorin-Bacteriochlorin and Bacteriochlorophyll-Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [Google Scholar] [CrossRef]
- D’Souza, F.; Subbaiyan, N.K.; Xie, Y.; Hill, J.P.; Ariga, K.; Ohkubo, K.; Fukuzumi, S. Anion-Complexation-Induced Stabilization of Charge Separation. J. Am. Chem. Soc. 2009, 131, 16138–16146. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of Inner Shell Marcus Terms for Amino Nitrogen Compounds by Molecular Orbital Calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Daswani, U.; Singh, U.; Sharma, P.; Kumar, A. From Molecules to Devices: A DFT/TD-DFT Study of Dipole Moment and Internal Reorganization Energies in Optoelectronically Active Aryl Azo Chromophores. J. Phys. Chem. C 2018, 122, 14390–14401. [Google Scholar] [CrossRef]
- Scherer, P.O.J. Intramolecular Reorganization of the Electron Donor N, N -Dimethylaniline. J. Phys. Chem. A 2003, 107, 8327–8329. [Google Scholar] [CrossRef]
- Scherer, P.O.J.; Tachiya, M. Computer Simulation Studies of Electron Transfer Parameters for Cyanoanthracene/N,N-Dimethylaniline Solutions. J. Chem. Phys. 2003, 118, 4149–4156. [Google Scholar] [CrossRef]
- Schomburg, H.; Staerk, H.; Weller, A. Electron Transfer Reactions and Inhibition of Triplet State Formation in Mixed Fluorescence Quencher Experiments. Chem. Phys. Lett. 1973, 22, 1–4. [Google Scholar] [CrossRef]
- Vân Anh, N.; Schlosser, F.; Groeneveld, M.M.; van Stokkum, I.H.M.; Würthner, F.; Williams, R.M. Photoinduced Interactions in a Pyrene-Calix[4]Arene-Perylene Bisimide Dye System: Probing Ground-State Conformations with Excited-State Dynamics of Charge Separation and Recombination. J. Phys. Chem. C 2009, 113, 18358–18368. [Google Scholar] [CrossRef] [Green Version]
- Leroy-lhez, S.; Belin, C.; D’aleo, A.; Williams, R.M.; De Cola, L.; Fages, F. Extending Excited-State Lifetimes by Interchromophoric Triplet-State Equilibration in a Pyrene-Ru(II)Diimine Dyad System. Supramol. Chem. 2003, 15, 627–637. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Verhoeven, J.W.; Van Ramesdonk, H.J.; Groeneveld, M.M.; Benniston, A.C.; Harriman, A. Long-Lived Charge-Transfer States in Compact Donor-Acceptor Dyads. ChemPhysChem 2005, 6, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Ruhoff, P.T.; Ratner, M.A. Algorithms for Computing Franck-Condon Overlap Integrals. Int. J. Quantum Chem. 2000, 77, 383–392. [Google Scholar] [CrossRef]
- Rujkorakarn, R.; Tanaka, F. Three-Dimensional Representations of Photo-Induced Electron Transfer Rates in Pyrene-(CH2)n-N,N′-Dimethylaniline Systems Obtained by Three Electron Transfer Theories. J. Mol. Graph. Model. 2009, 27, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Staerk, H.; Weller, A. Solvent, Isotope, and Magnetic Field Effects in the Geminate Recombination of Radical Ion Pairs. J. Chem. Phys. 1978, 68, 2419–2426. [Google Scholar] [CrossRef]
- Imran, M.; Sukhanov, A.A.; Wang, Z.; Karatay, A.; Zhao, J.; Mahmood, Z.; Elmali, A.; Voronkova, V.K.; Hayvali, M.; Xing, Y.H.; et al. Electronic Coupling and Spin-Orbit Charge-Transfer Intersystem Crossing in Phenothiazine-Perylene Compact Electron Donor/Acceptor Dyads. J. Phys. Chem. C 2019, 123, 7010–7024. [Google Scholar] [CrossRef]
- Isukapalli, S.V.K.; Lekshmi, R.S.; Samanta, P.K.; Vennapusa, S.R. Formation of Excited Triplet States in Naphthalene Diimide and Perylene Diimide Derivatives: A Detailed Theoretical Study. J. Chem. Phys. 2020, 153, 124301. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | |
|---|---|
| NHX | 2.5 × 1010 |
| DEE | 4.3 × 109 |
| ACN | 7.0 × 108 |
| NHX * | 2.0 × 106 |
| XC | Acetone | 4-thio-thymine | PyrDMA | ||
|---|---|---|---|---|---|
| Gas | Gas | Gas | NHX | ACN | |
| PW91 | 61.85 | 167.50 | 2.68 | 2.75 | 2.85 |
| BLYP | 62.56 | 169.87 | 2.69 | 2.76 | 2.86 |
| CAM-B3LYP | 62.20 | 156.11 | 2.39 | 2.47 | 2.56 |
| CAMY-B3LYP | 62.30 | 138.14 | 2.36 | 2.41 | 1.96 |
| RPBE | 61.00 | 164.18 | 2.63 | 2.69 | 2.79 |
| HTBS | 61.29 | 166.56 | 2.66 | 2.73 | 2.83 |
| S12y | 60.53 | 160.90 | 2.59 | 2.66 | 2.76 |
| LB94 | 67.76 | 181.70 | 3.15 | 3.22 | 3.34 |
| KT1 | 59.55 | 156.83 | 2.59 | 2.65 | 2.75 |
| BhandH | 60.61 | 103.83 | 2.08 | 2.13 | 1.74 |
| Solvent | ETB S0 (eV) | ETB S2 (eV) | ETB S1 (eV) | ETB T2 (eV) | ETB T1 (eV) |
|---|---|---|---|---|---|
| Gas | −389.543 | −385.636 | −386.151 | −386.020 | −387.306 |
| NHX | −389.612 | −385.839 | −385.817 | −386.070 | −387.375 |
| ACN | −389.700 | −386.062 | −386.290 | −386.153 | −387.455 |
| Solvent | ∆ETB S0 (eV) | ∆ETB S2 (eV) | ∆ETB S1 (eV) | ∆ETB T2 (eV) | ∆ETB T1 (eV) |
|---|---|---|---|---|---|
| Gas | 0 | 3.907 | 3.392 | 3.523 | 2.237 |
| NHX | 0 | 3.773 | 3.795 | 3.542 | 2.237 |
| ACN | 0 | 3.638 | 3.410 | 3.547 | 2.245 |
| Solvent | S0 | S1 | S2 | T1 | T2 |
|---|---|---|---|---|---|
| Gas | 67.0 | 69.9 | 65.7 | 65.8 | 64.0 |
| NHX | 66.2 | 65.7 | 67.1 | 65.8 | 63.9 |
| ACN | 66.4 | 71.6 | 70.4 | 66.4 | 64.1 |
| Gas | NHX | ACN | |
|---|---|---|---|
| (cm−1) | 263.11 | 123.24 | 96.40 |
| (cm−1) | 2.36 | 2.41 | 1.95 |
| 197.98 | 179.99 | 171.00 | |
| (cm−1) | 1031.00 | 1037.90 | 1010.70 |
| 81.06 | 90.05 | 89.90 | |
| (cm−1) | 2.40 | 2.45 | 2.06 |
| Solvent | |||
|---|---|---|---|
| Gas | 824.40 | 801.15 | 801.15 |
| NHX | 827.85 | 817.65 | 817.65 |
| ACN | 794.00 | 721.40 | 721.40 |
| Solvent | (eV) | (eV) | ||||
|---|---|---|---|---|---|---|
| Gas | 1.00 | 1.00 | −0.110 | 0.00000 | 0.7817 | 0.7571 |
| NHX | 1.37 | 1.88 | −0.178 | 0.00135 | 0.7618 | 0.7379 |
| ACN | 1.34 | 37.5 | −0.493 | 0.81600 | 0.7187 | 0.6965 |
| Solvent | |||
|---|---|---|---|
| Gas | 0.000 | 0.000 | 0.000 |
| NHX | 0.0018 | 0.0018 | 0.0018 |
| ACN | 1.0294 | 0.9326 | 0.9326 |
| ∆G (eV) | (eV) | (eV) | V (cm−1) | kopt (s−1) | kcalc (s−1) | kexp (s−1) | |
|---|---|---|---|---|---|---|---|
| CRT(L) | −0.73 | 0.7379 | 0.00135 | 2.47 | 1.84 × 109 | 1.84 × 109 | 2.50 × 1010 |
| CRT(H) | −0.73 | 0.7379 | 0.00135 | 3.22 | 3.13 × 109 | 3.12 × 109 | 2.50 × 1010 |
| CRS(C) | −3.08 | 0.7618 | 0.00135 | 159 | 7.84 × 1012 | 2.75 × 10−22 | 7.89 × 106 |
| CRS(E) | −2.87 | 0.7618 | 0.00135 | 159 | 7.50 × 1012 | 1.29 × 10−10 | 7.89 × 106 |
| CS(C) | −0.178 | 0.7000 | 0.00135 | 123.2 | 4.71 × 1012 | 1.01 × 1011 | 5.9 × 1011 |
| CS(E) | −0.49 | 0.7000 | 0.00135 | 123.2 | 4.71 × 1012 | 2.52 × 1012 | 5.9 × 1011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bissesar, S.; van Raamsdonk, D.M.E.; Gibbons, D.J.; Williams, R.M. Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene—Dimethylaniline. Molecules 2022, 27, 891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030891
Bissesar S, van Raamsdonk DME, Gibbons DJ, Williams RM. Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene—Dimethylaniline. Molecules. 2022; 27(3):891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030891
Chicago/Turabian StyleBissesar, Shivan, Davita M. E. van Raamsdonk, Dáire J. Gibbons, and René M. Williams. 2022. "Spin Orbit Coupling in Orthogonal Charge Transfer States: (TD-)DFT of Pyrene—Dimethylaniline" Molecules 27, no. 3: 891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030891