
Computational Identification of Druggable Bioactive Compounds from Catharanthus roseus and Avicennia marina against Colorectal Cancer by Targeting Thymidylate Synthase

 ,
,  , , , , ,
, , , , , 
 , , and
, , and
Abstract
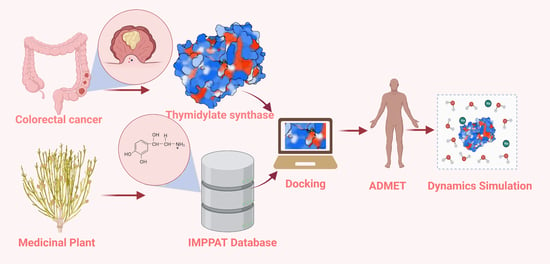
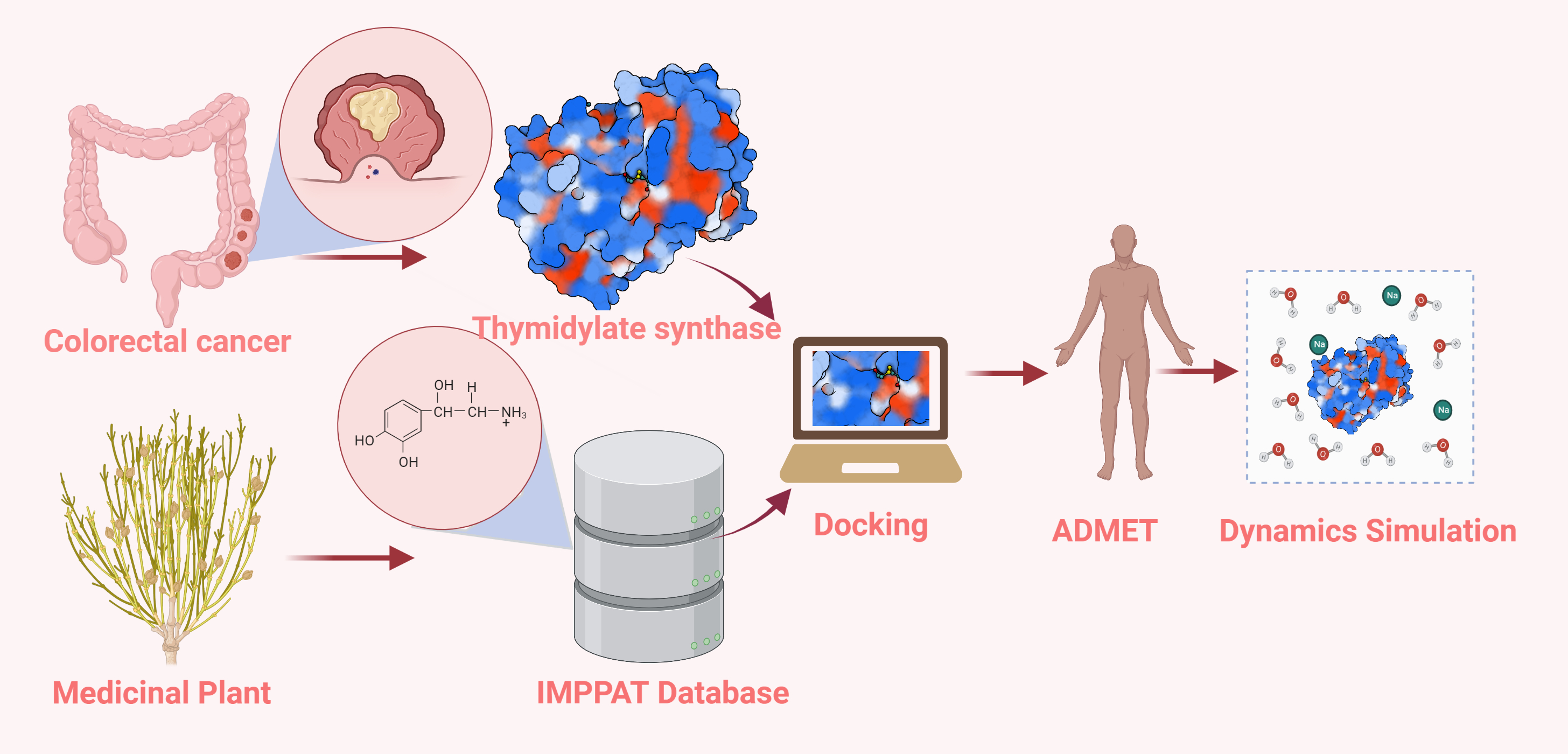
:
1. Introduction
2. Results
2.1. Phytochemical Retrieval and Preparation
2.2. Active Site Identification and Receptor Grid Generation
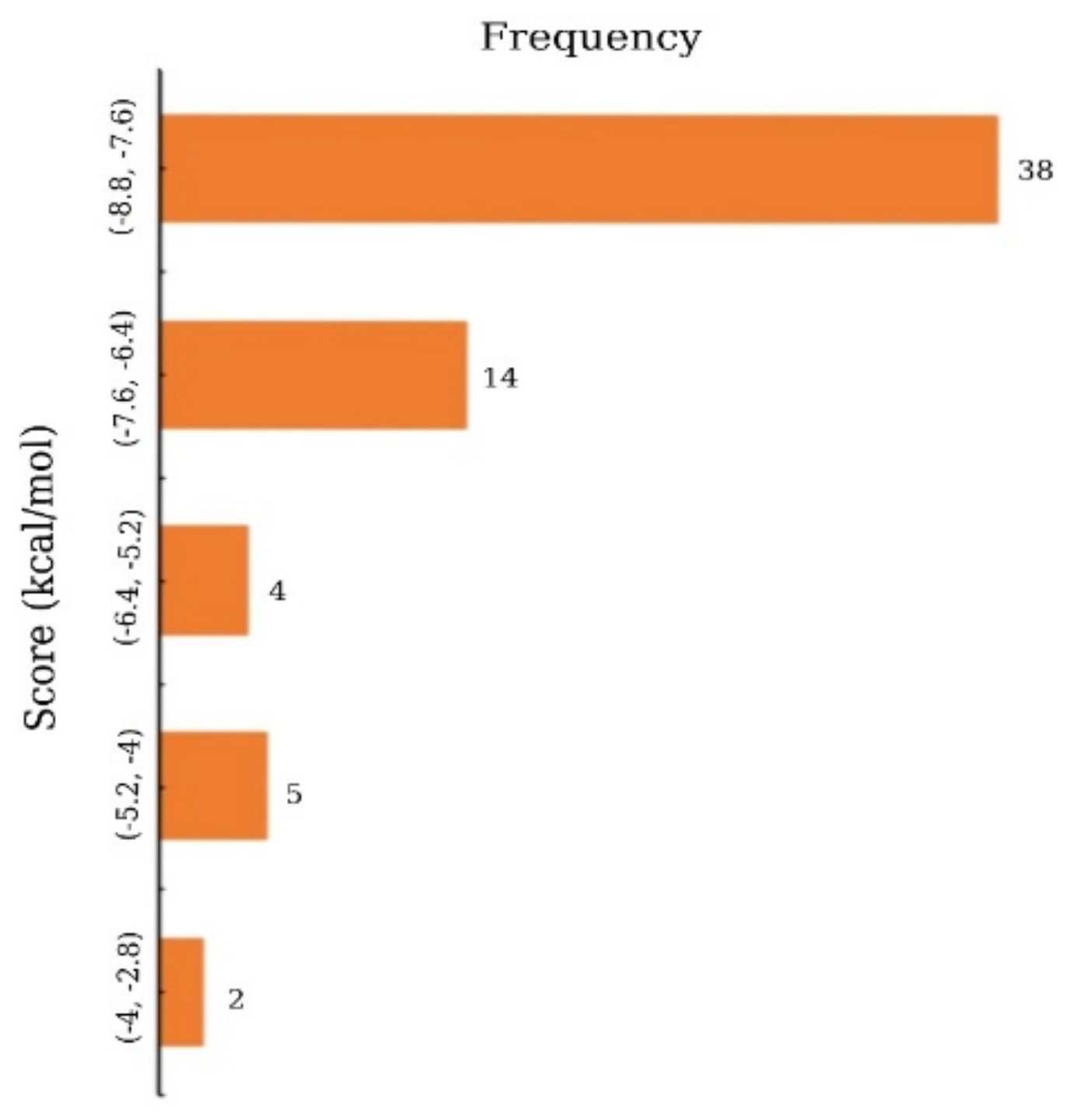
2.3. Molecular Docking Analysis
2.4. PK Properties
2.5. Toxicity Prediction
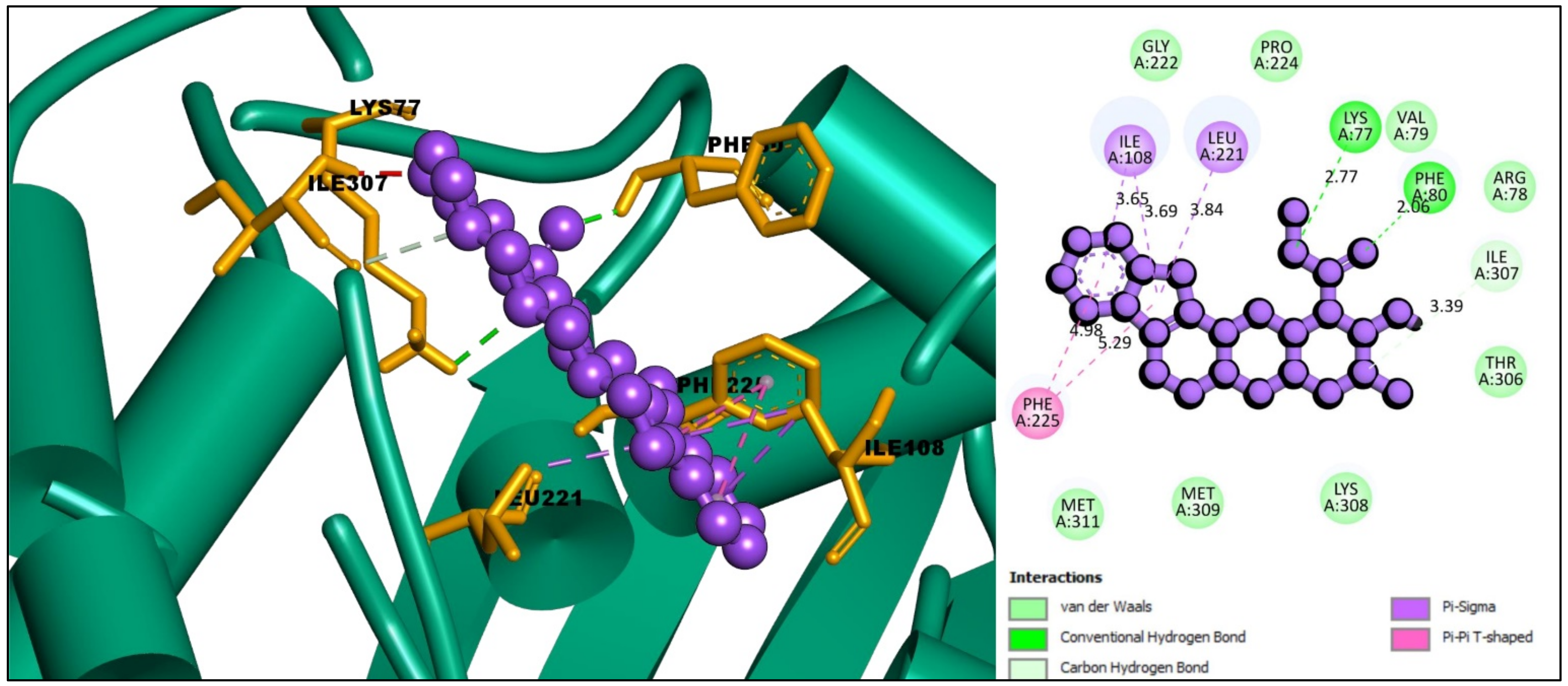
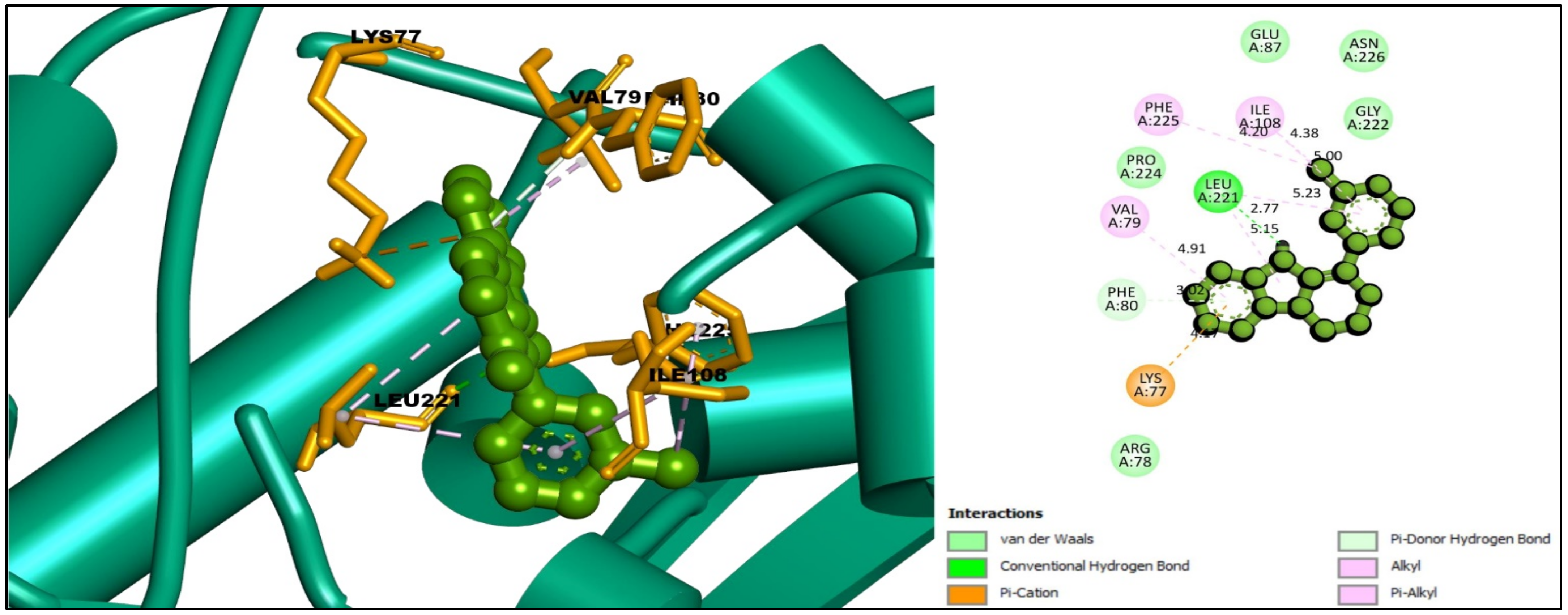
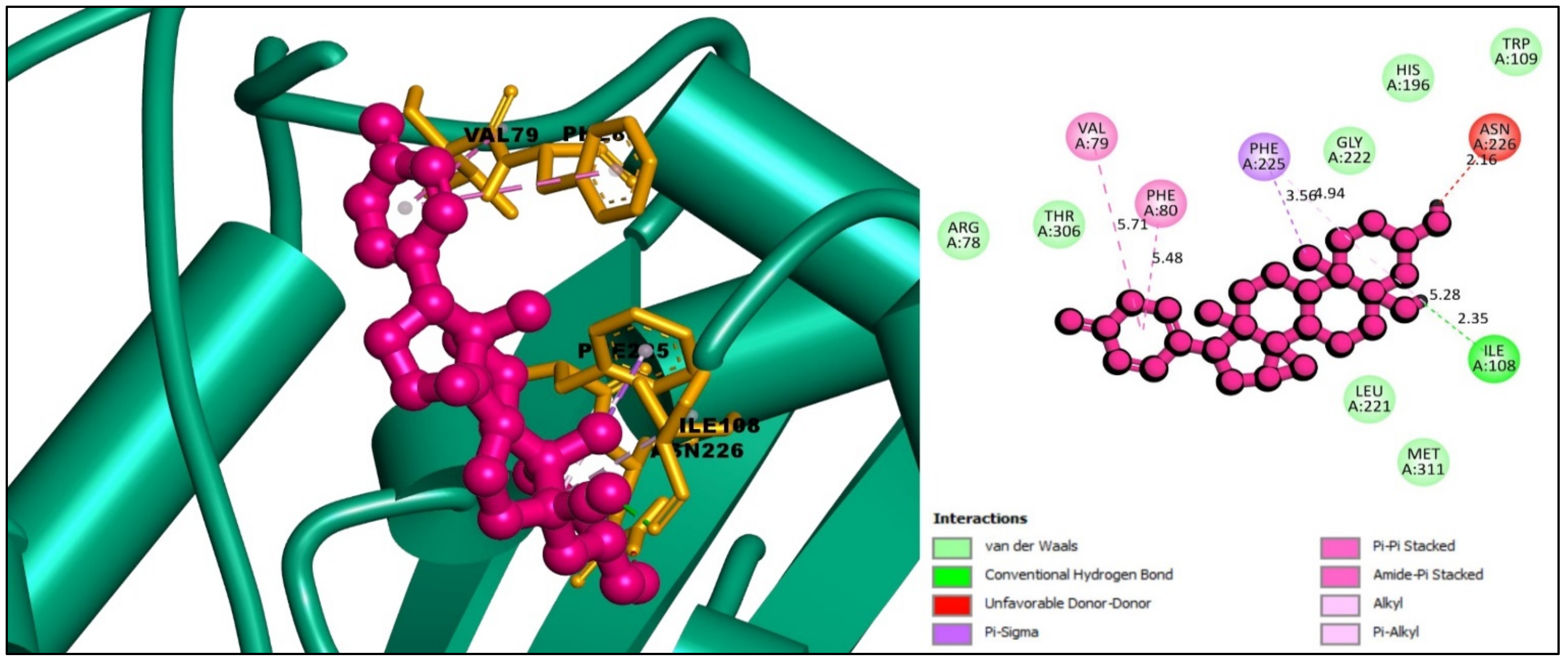
2.6. Interpretations of Protein–Ligands Interaction
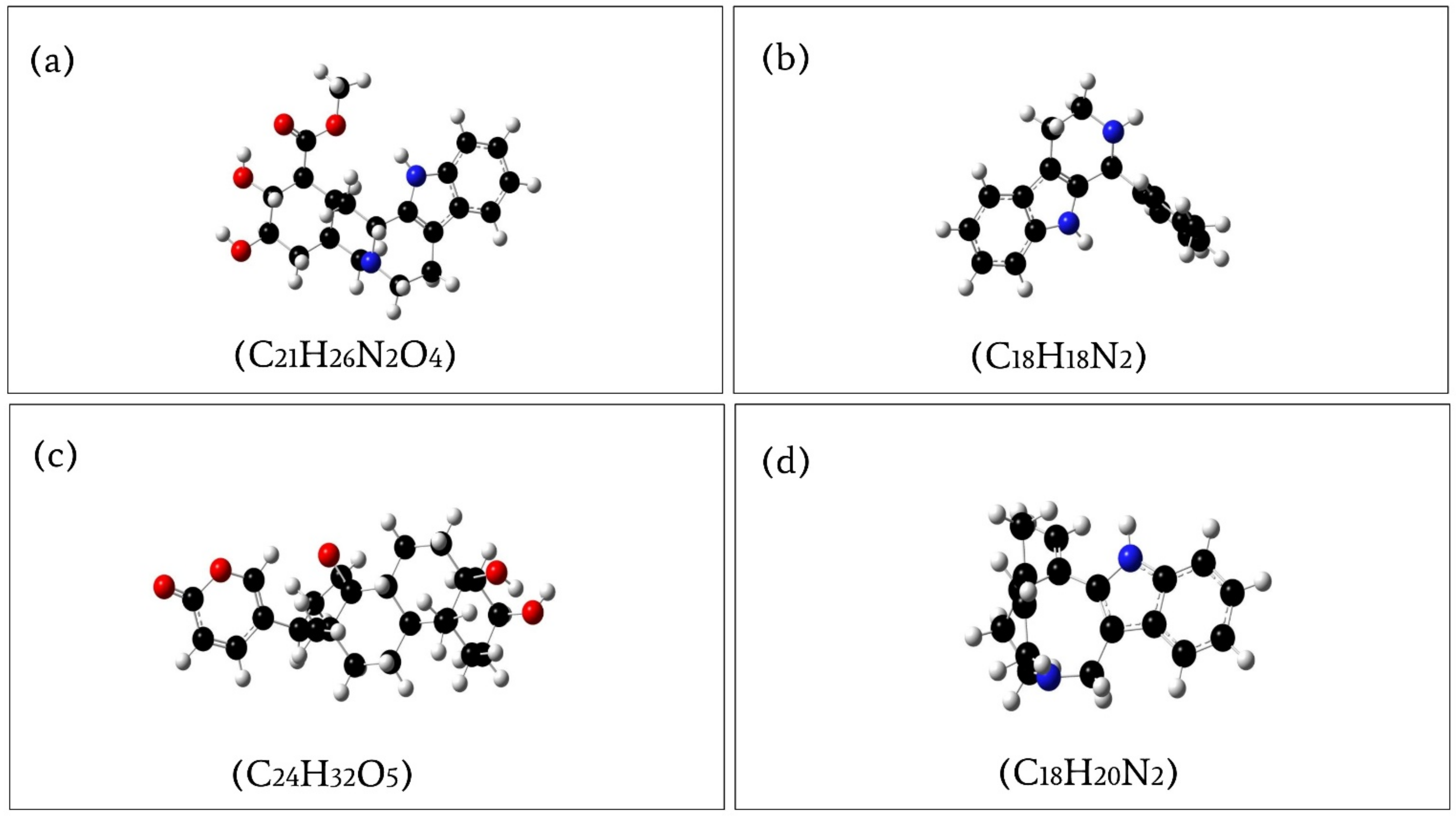
2.7. Geometry Optimization and Theoretical Quantum Chemical Calculation
2.8. Frontier Molecular Orbital HOMO/LUMO Calculation
2.9. MD Simulations Analysis
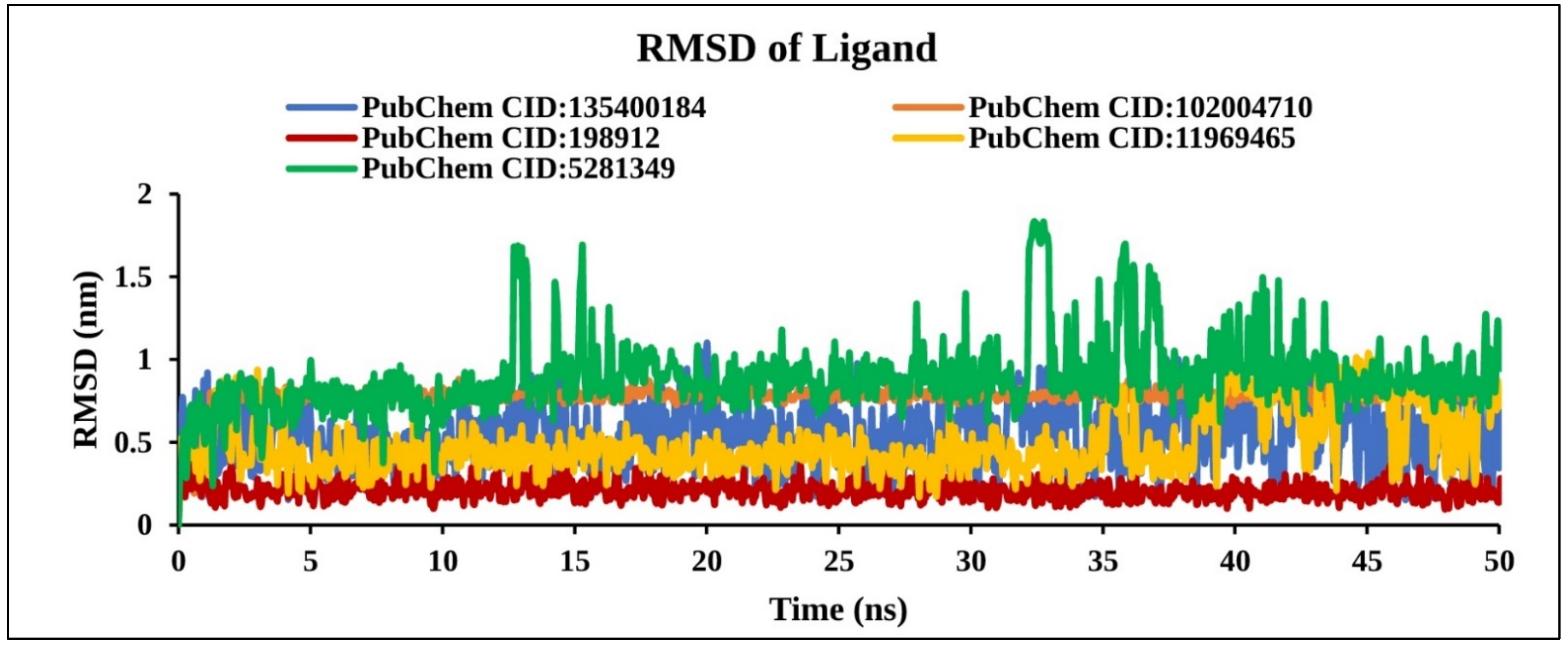
2.9.1. RMSD Analysis
2.9.2. RMSD of Protein
2.9.3. RMSD of Ligand
2.9.4. RMSF Analysis
2.9.5. Solvent Accessible Surface Area Analysis
2.9.6. Protein-Ligand Contact Analysis
3. Discussion
4. Materials and Methods
4.1. Target Preparation
4.2. Compound’s Retrieval and Preparation
4.3. Identification of Binding Site and Receptor Grid Generation
4.4. Docking Analysis
4.5. PK Properties Prediction
4.6. Toxicity Prediction
4.7. DFT Method Based Geometry Optimization
4.8. Frontier Molecular Orbital HOMO/LUMO Calculation
4.9. MD Simulations and Trajectory Analysis
4.9.1. RSMD Analysis
4.9.2. RMSF Analysis
4.9.3. Solvent Accessible Surface Area Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624. [Google Scholar] [CrossRef] [PubMed]
- Van Der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-M.; Yang, Z.-J.; Xie, Q.; Zhang, Z.-K.; Zhang, H.; Ma, J.-Y. Natural products for treating colorectal cancer: A mechanistic review. Biomed. Pharmacother. 2019, 117, 109142. [Google Scholar] [CrossRef] [PubMed]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Mo, J.-L.; Liu, J.-H.; Li, X.; Tan, L.-M.; Zhang, W.; Zhou, H.-H.; Liu, Z.-Q. Pharmacogenomics of 5-fluorouracil in colorectal cancer: Review and update. Cell. Oncol. 2020, 43, 989–1001. [Google Scholar] [CrossRef]
- Koehn, E.M.; Kohen, A. Flavin-dependent thymidylate synthase: A novel pathway towards thymine. Arch. Biochem. Biophys. 2010, 493, 96–102. [Google Scholar] [CrossRef]
- Panczyk, M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J. Gastroenterol. 2014, 20, 9775–9827. [Google Scholar] [CrossRef]
- Liu, J.; Schmitz, J.C.; Lin, X.; Tai, N.; Yan, W.; Farrell, M.; Bailly, M.; Chen, T.-M.; Chu, E. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2002, 1587, 174–182. [Google Scholar] [CrossRef]
- Papamichael, D. The Use of Thymidylate Synthase Inhibitors in the Treatment of Advanced Colorectal Cancer: Current Status. Stem Cells 2000, 18, 166–175. [Google Scholar] [CrossRef]
- Bendardaf, R.; Lamlum, H.; Elzagheid, A.; Ristamäki, R.; Pyrhönen, S. Thymidylate synthase expression levels: A prognostic and predictive role in advanced colorectal cancer. Oncol. Rep. 2005, 14, 657–662. [Google Scholar] [CrossRef]
- Lietava, J. Medicinal plants in a Middle Paleolithic grave Shanidar IV? J. Ethnopharmacol. 1992, 35, 263–266. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Pham, H.N.T.; Vuong, Q.V.; Bowyer, M.C.; Scarlett, C.J. Phytochemicals Derived from Catharanthus roseus and Their Health Benefits. Technologies 2020, 8, 80. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Behbahani, M.; Sadeghi-Aliabadi, H. Antiproliferative Activity and Apoptosis Induction of Crude Extract and Fractions of Avicennia Marina. Iran. J. Basic Med. Sci. 2013, 16, 1203–1208. [Google Scholar] [CrossRef]
- Huang, C.; Lu, C.-K.; Tu, M.-C.; Chang, J.-H.; Chen, Y.-J.; Tu, Y.-H.; Huang, H.-C. Polyphenol-rich Avicennia marina leaf extracts induce apoptosis in human breast and liver cancer cells and in a nude mouse xenograft model. Oncotarget 2016, 7, 35874–35893. [Google Scholar] [CrossRef]
- Sathya Prabhu, D.; Devi Rajeswari, V. Catharanthus roseus: The Cancer-Fighting Medicine. In Catharanthus roseus: Current Research and Future Prospects; Springer: Cham, Switzerland, 2017; pp. 121–151. [Google Scholar] [CrossRef]
- Ahmad, N.H.; Rahim, R.A.; Mat, I. Catharanthus roseus Aqueous Extract is Cytotoxic to Jurkat Leukaemic T-cells but Induces the Proliferation of Normal Peripheral Blood Mononuclear Cells. Trop. Life Sci. Res. 2010, 21, 101–113. [Google Scholar]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Promi, M.M.; Bin Emran, T.; Saleh, A. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Briefings Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Fang, J.; Liu, C.; Wang, Q.; Lin, P.; Cheng, F. In silico polypharmacology of natural products. Briefings Bioinform. 2017, 19, 1153–1171. [Google Scholar] [CrossRef]
- Ahammad, F.; Rashid, T.R.T.A.; Mohamed, M.; Tanbin, S.; Fuad, F.A.A.; Rashid, T.T.A.; Fuad, F.A. Contemporary Strategies and Current Trends in Designing Antiviral Drugs against Dengue Fever via Targeting Host-Based Approaches. Microorganisms 2019, 7, 296. [Google Scholar] [CrossRef]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Bharadvaja, N. Treasuring the computational approach in medicinal plant research. Prog. Biophys. Mol. Biol. 2021, 164, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Jarmula, A. Antifolate inhibitors of thymidylate synthase as anticancer drugs. Mini Rev. Med. Chem. 2010, 10, 1211–1222. [Google Scholar] [CrossRef]
- Panchagnula, R.; Thomas, N.S. Biopharmaceutics and pharmacokinetics in drug research. Int. J. Pharm. 2000, 201, 131–150. [Google Scholar] [CrossRef]
- Raies, A.; Bajic, V.B. In silicotoxicology: Computational methods for the prediction of chemical toxicity. WIREs Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Briefings Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef]
- Hickey, A.L.; Rowley, C.N. Benchmarking Quantum Chemical Methods for the Calculation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A 2014, 118, 3678–3687. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, L.; Muthusamy, K.; Jayaraj, J.M.; Kajamaideen, A.; Balthasar, J.J. In silico insights on tankyrase protein: A potential target for colorectal cancer. J. Biomol. Struct. Dyn. 2019, 37, 3637–3648. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Pârvu, L. QSAR—A piece of drug design. J. Cell. Mol. Med. 2003, 7, 333–335. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, Y.; Lu, A.; Zhang, G. Systems Pharmacology in Small Molecular Drug Discovery. Int. J. Mol. Sci. 2016, 17, 246. [Google Scholar] [CrossRef]
- Costa, M.M.; Hilliou, F.; Duarte, P.; Pereira, L.G.; Almeida, I.; Leech, M.; Memelink, J.; Barceló, A.R.; Sottomayor, M. Molecular Cloning and Characterization of a Vacuolar Class III Peroxidase Involved in the Metabolism of Anticancer Alkaloids in Catharanthus roseus. Plant Physiol. 2007, 146, 403–417. [Google Scholar] [CrossRef]
- Phan, J.; Koli, S.; Minor, W.; Dunlap, R.B.; Berger, S.H.; Lebioda, L. Human Thymidylate Synthase Is in the Closed Conformation When Complexed with dUMP and Raltitrexed, an Antifolate Drug. Biochemistry 2001, 40, 1897–1902. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Albinhassan, T.H.; Saleh, K.A.; Barhoumi, Z.; Alshehri, M.A.; Al-Ghazzawi1, A.M. Anticancer, anti-proliferative activity of Avicennia marina plant extracts. J. Cancer Res. Ther. 2021, 17, 879. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Chu, E.; Callender, M.A.; Farrell, M.P.; Schmitz, J.C. Thymidylate synthase inhibitors as anticancer agents: From bench to bedside. Cancer Chemother. Pharmacol. 2003, 52, 80–89. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with pyrx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Mohammad, T.; Mathur, Y.; Hassan, I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Briefings Bioinform. 2020, 22. [Google Scholar] [CrossRef]
- Umar, A.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. Design of potential anti-melanoma agents against SK-MEL-5 cell line using QSAR modeling and molecular docking methods. SN Appl. Sci. 2020, 2, 1–18. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Guallar, V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu. Rev. Phys. Chem. 2005, 56, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Stability, Reactivity, and Aromaticity of Compounds of a Multivalent Superatom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Flippin, L.A.; Gallagher, D.W.; Jalali-Araghi, K. A convenient method for the reduction of ozonides to alcohols with borane-dimethyl sulfide complex. J. Org. Chem. 1989, 54, 1430–1432. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Electrophilicity-Based Charge Transfer Descriptor. J. Phys. Chem. A 2007, 111, 1358–1361. [Google Scholar] [CrossRef]
- Li, Y.; Evans, J.N.S. The Fukui Function: A Key Concept Linking Frontier Molecular Orbital Theory and the Hard-Soft-Acid-Base Principle. J. Am. Chem. Soc. 1995, 117, 7756–7759. [Google Scholar] [CrossRef]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 40, 14–30. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
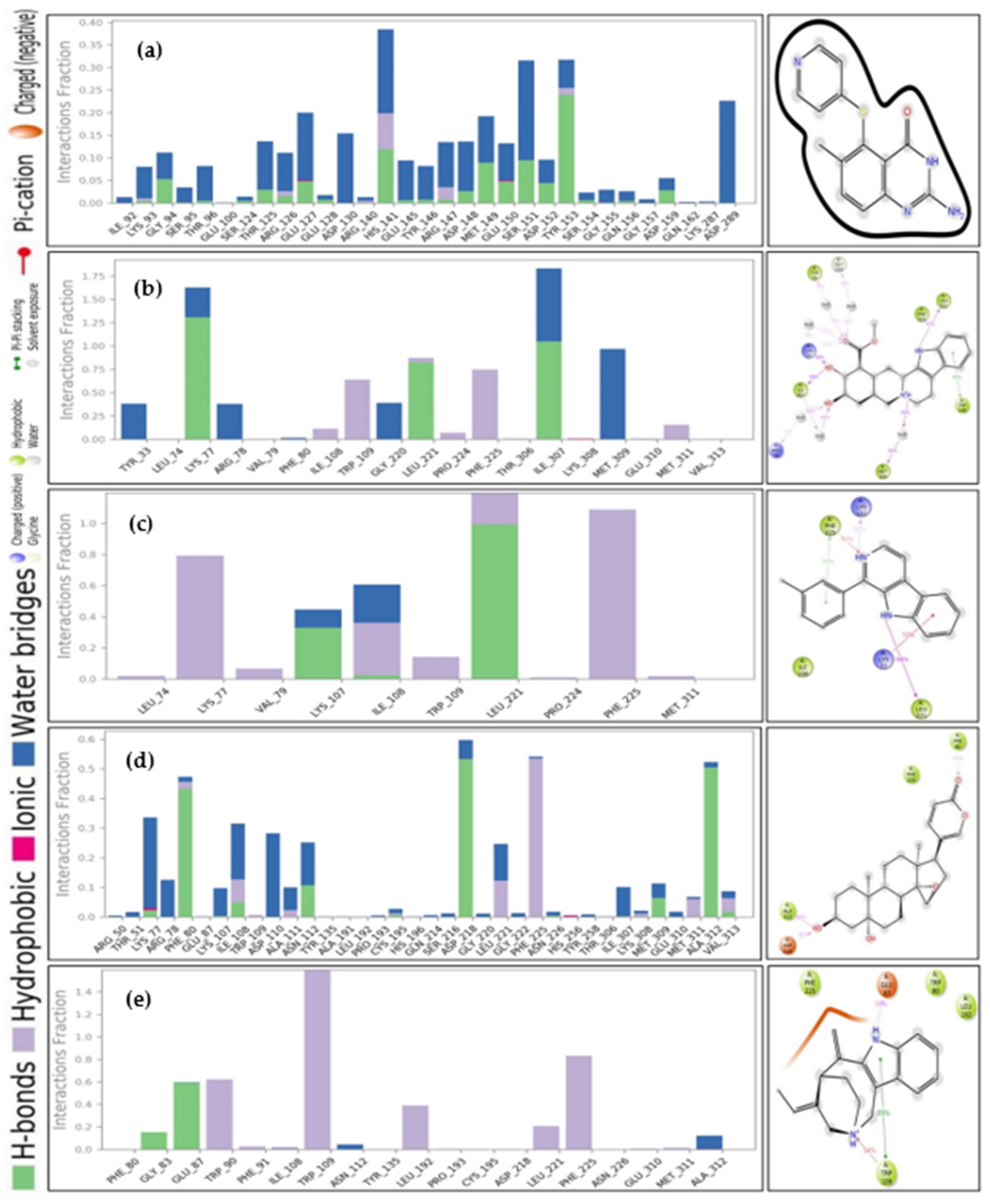{kind=link}
| No | Compound ID | Chemical Formula | 2D Structure | Score (Kcal/mol) |
|---|---|---|---|---|
| 1 | CID:102004710 | 18-Beta-hydroxy-3-epi-alpha-yohimbine |  | −8.8 |
| 2 | CID:198912 | 1-(3-Methylphenyl)-2,3,4,9-tetrahydro-1H-β-carboline |  | −8.7 |
| 3 | CID:11969465 | Marinobufagenin |  | −8.7 |
| 4 | CID:5281349 | Apparicine |  | −8.6 |
| 5 | PubChem CID:135400184 | Nolatrexed |  | −7.4 |
| Properties | CID:102004710 | CID:198912 | CID:11969465 | CID:5281349 | |
|---|---|---|---|---|---|
| Physicochemical properties | MW (g/mol) < 500 | 370.44 | 262.35 | 400.51 | 264.36 |
| Heavy atoms | 27 | 20 | 29 | 20 | |
| Arom. heavy atoms | 9 | 15 | 26 | 9 | |
| Rotatable bonds | 2 | 1 | 1 | 0 | |
| H-bond acceptors < 10 | 5 | 1 | 5 | 1 | |
| H-bond donors < 5 | 3 | 2 | 2 | 1 | |
| TPSA ≤ 140 (A2) | 85.79 | 27.82 | 83.2 | 19.03 | |
| Lipophilicity | Log Po/w ≤ 5 | 1.71 | 3.40 | 3.14 | 3.30 |
| Plasma protein binding | 100% | 100% | 100% | 100% | 100% |
| Water solubility | Log S (ESOL) | −3.49 | −4.19 | −3.99 | −3.54 |
| Pharmacokinetics | GI absorption | High | High | High | High |
| Drug-likeness | Lipinski | Yes | Yes | Yes | Yes |
| Medi. Chemistry | Synth. accessibility | Easy | Easy | Easy | Easy |
| Parameters | Compounds | |||
|---|---|---|---|---|
| CID: 102004710 | CID: 198912 | CID: 11969465 | CID: 5281349 | |
| Ames toxicity | No | Yes | No | Yes |
| hERG I inhibition | No | No | No | No |
| Carcinogens | NC | NC | NC | NC |
| Rat acute toxicity | 2.853 | 2.82 | 2.665 | 2.973 |
| TP toxicity | 0.316 | 0.433 | 0.352 | 0.888 |
| HB toxicity | No | Yes | Yes | No |
| Fish toxicity | No | No | Yes | Yes |
| Skin sensitization | No | No | No | Yes |
| ID | Residues | Distance (Å) | Bond Category | Bond Type |
|---|---|---|---|---|
| CID:102004710 | PHE80 | 2.86 | Hydrogen Bond | Conventional Hydrogen Bond |
| ILE307 | 3.39 | Hydrogen Bond | Carbon Hydrogen Bond | |
| ILE108 | 3.69 | Hydrophobic | Pi-Sigma | |
| ILE108 | 3.65 | Hydrophobic | Pi-Sigma | |
| LEU221 | 3.84 | Hydrophobic | Pi-Sigma | |
| PHE225 | 4.98 | Hydrophobic | Pi-Pi T-shaped | |
| PHE225 | 5.29 | Hydrophobic | Pi-Pi T-shaped | |
| CID:198912 | LEU221 | 2.77 | Hydrogen Bond | Conventional Hydrogen Bond |
| LYS77 | 4.17 | Hydrogen Bond | Pi-Cation | |
| PHE80 | 4.04 | Hydrogen Bond | Pi-Donor Hydrogen Bond | |
| ILE108 | 4.38 | Hydrophobic | Alkyl | |
| PHE225 | 4.20 | Hydrophobic | Pi-Alkyl | |
| LEU221 | 5.15 | Hydrophobic | Pi-Alkyl | |
| VAL79 | 4.91 | Hydrophobic | Pi-Alkyl | |
| ILE108 | 5.00 | Hydrophobic | Pi-Alkyl | |
| LEU221 | 5.23 | Hydrophobic | Pi-Alkyl | |
| CID:11969465 | ILE108 | 2.35 | Hydrogen Bond | Conventional Hydrogen Bond |
| PHE225 | 3.56 | Hydrophobic | Pi-Sigma | |
| PHE80 | 5.48 | Hydrophobic | Pi-Pi Stacked | |
| VAL79, PHE80 | 5.71 | Hydrophobic | Amide-Pi Stacked | |
| ILE108 | 5.28 | Hydrophobic | Alkyl | |
| PHE225 | 4.94 | Hydrophobic | Pi-Alkyl | |
| CID:5281349 | ASN226 | 4.09 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| CYS195 | 4.93 | Other | Pi-Sulfur | |
| TRP109 | 5.32 | Hydrophobic | Pi-Pi T-shaped | |
| TRP109 | 4.76 | Hydrophobic | Pi-Pi T-shaped | |
| ILE108 | 4.73 | Hydrophobic | Alkyl | |
| LEU221 | 5.18 | Hydrophobic | Alkyl |
| PubChem CID | Energy (a.u) | Dipole Moment (Debye) |
|---|---|---|
| 102004710 | −1225.94832 | 3.878234 |
| 198912 | −806.07028 | 2.516531 |
| 11969465 | −1309.527491 | 9.732138 |
| 5281349 | −807.213195 | 2.127067 |
| PubChem CID | IP (eV) | EA (eV) | η | S | µ | χ | ω |
|---|---|---|---|---|---|---|---|
| 102004710 | 5.296 | 0.868 | 2.214 | 0.452 | −3.082 | 3.082 | 2.145 |
| 198912 | 5.126 | 0.157 | 2.485 | 0.402 | −2.642 | 2.642 | 1.404 |
| 11969465 | 6.202 | 1.659 | 2.272 | 0.44 | −3.931 | 3.931 | 3.401 |
| 5281349 | 5.095 | 0.796 | 2.149 | 0.465 | −2.946 | 2.946 | 2.019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.R.; Awal, M.A.; Khames, A.; Abourehab, M.A.S.; Samad, A.; Hassan, W.M.I.; Alam, R.; Osman, O.I.; Nur, S.M.; Molla, M.H.R.; et al. Computational Identification of Druggable Bioactive Compounds from Catharanthus roseus and Avicennia marina against Colorectal Cancer by Targeting Thymidylate Synthase. Molecules 2022, 27, 2089. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072089
Islam MR, Awal MA, Khames A, Abourehab MAS, Samad A, Hassan WMI, Alam R, Osman OI, Nur SM, Molla MHR, et al. Computational Identification of Druggable Bioactive Compounds from Catharanthus roseus and Avicennia marina against Colorectal Cancer by Targeting Thymidylate Synthase. Molecules. 2022; 27(7):2089. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072089
Chicago/Turabian StyleIslam, Md Rashedul, Md Abdul Awal, Ahmed Khames, Mohammad A. S. Abourehab, Abdus Samad, Walid M. I. Hassan, Rahat Alam, Osman I. Osman, Suza Mohammad Nur, Mohammad Habibur Rahman Molla, and et al. 2022. "Computational Identification of Druggable Bioactive Compounds from Catharanthus roseus and Avicennia marina against Colorectal Cancer by Targeting Thymidylate Synthase" Molecules 27, no. 7: 2089. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072089