
Bioactive Terphenyls Isolated from the Antarctic Lichen Stereocaulon alpinum

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
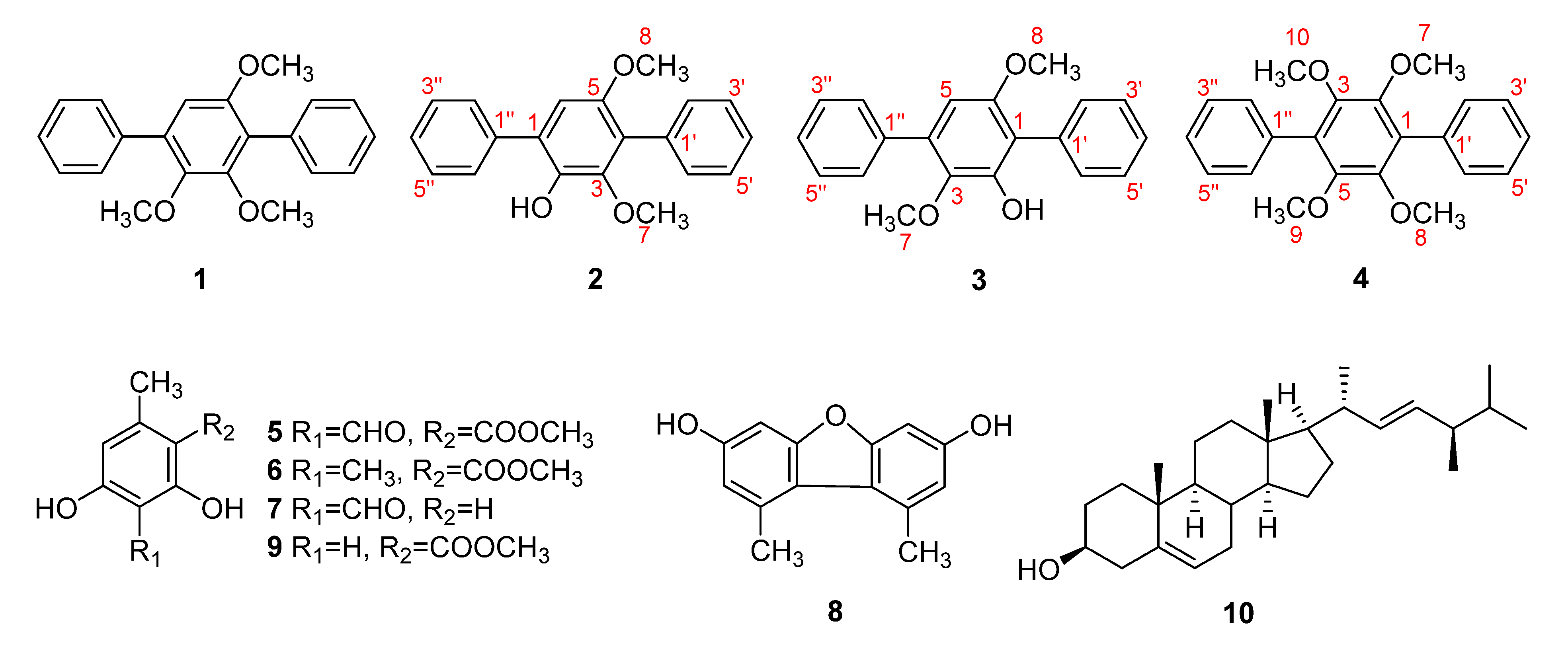
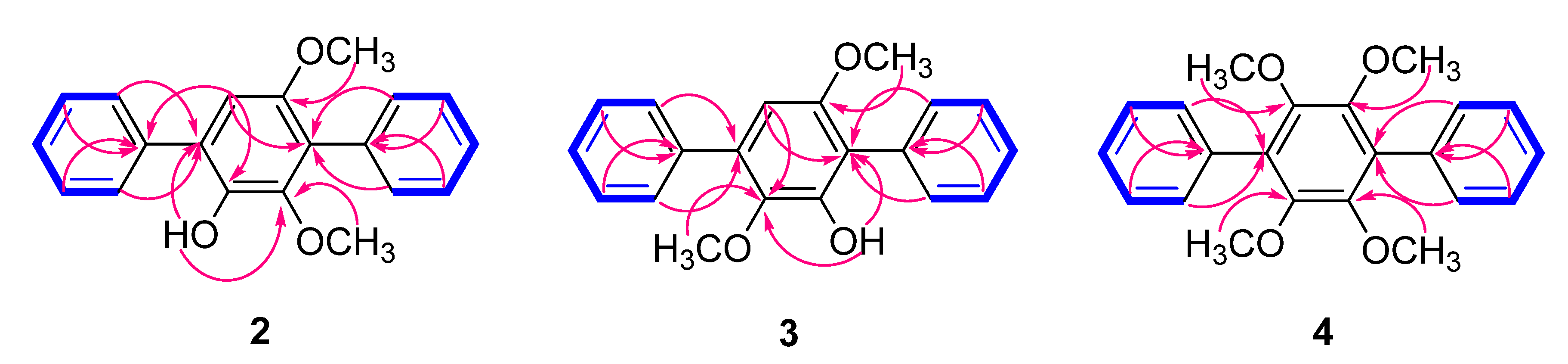
2.1. Structure Elucidation of the Compounds
2.2. Biological Evaluation
3. Materials and Methods
3.1. General Procedures
3.2. Lichen Material
3.3. Extraction and Isolation
3.4. Cell Culture
3.5. MTS Assay
3.6. Measurement of Nitric Oxide (NO) Production
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branislav, R.; Marijana, K. Lichens as a Potential Source of Bioactive Secondary Metabolites. In Lichen Secondary Metabolites; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 1–30. [Google Scholar]
- Crawford, S.D. Lichens Used in Traditional Medicinal. In Lichen Secondary Metabolites; Springer International Publishing: Cham, Switzerland, 2015; pp. 31–97. [Google Scholar]
- Ismed, F.; Lohézic-Le Dévéhat, F.; Guiller, A.; Corlay, N.; Bakhtiar, A.; Boustie, J. Phytochemical Review of the Lichen Genus Stereocaulon (Fam. Stereocaulaceae) and Related Pharmacological Activities Highlighted by a Focus on Nine Species. Phytochem. Rev. 2018, 17, 1165–1178. [Google Scholar] [CrossRef]
- Sharma, G.K. Ethnomedicinal Flora: Ayurvedic System of Medicine in a Remote Part of the Indo-Tibetan Himalayas. J. Tennessee Acad. Sci. 1997, 72, 53–55. [Google Scholar]
- Fraser, M.H.; Cuerrier, A.; Haddad, P.S.; Arnason, J.T.; Owen, P.L.; Johns, T. Medicinal Plants of Cree Communities (Québec, Canada): Antioxidant Activity of Plants Used to Treat Type 2 Diabetes Symptoms. Can. J. Physiol. Pharmacol. 2007, 85, 1200–1214. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Tsuchiya, T.; Kishibe, K.; Noya, S.; Shiro, M.; Hirasawa, Y. Antimitotic Activity of Lobaric Acid and a New Benzofuran, Sakisacaulon A from Stereocaulon Sasakii. Bioorganic Med. Chem. Lett. 2009, 19, 3679–3681. [Google Scholar] [CrossRef]
- Bucar, F.; Schneider, I.; Ögmundsdóttir, H.; Ingólfsdóttir, K. Anti-Proliferative Lichen Compounds with Inhibitory Activity on 12(S)-HETE Production in Human Platelets. Phytomedicine 2004, 11, 602–606. [Google Scholar] [CrossRef]
- Bhattarai, H.D.; Kim, T.; Oh, H.; Yim, J.H. A New Pseudodepsidone from the Antarctic Lichen Stereocaulon Alpinum and Its Antioxidant, Antibacterial Activity. J. Antibiot. 2013, 66, 559–561. [Google Scholar] [CrossRef]
- Seo, C.; Yim, J.H.; Lee, H.K.; Park, S.M.; Sohn, J.H.; Oh, H. Stereocalpin A, a Bioactive Cyclic Depsipeptide from the Antarctic Lichen Stereocaulon Alpinum. Tetrahedron Lett. 2008, 49, 29–31. [Google Scholar] [CrossRef]
- Seo, C.; Sohn, J.H.; Ahn, J.S.; Yim, J.H.; Lee, H.K.; Oh, H. Protein Tyrosine Phosphatase 1B Inhibitory Effects of Depsidone and Pseudodepsidone Metabolites from the Antarctic Lichen Stereocaulon Alpinum. Bioorganic Med. Chem. Lett. 2009, 19, 2801–2803. [Google Scholar] [CrossRef]
- Valeria, C.; Carmela, S.; Corrado, T. Polyhyroxy-p-Terphenyls and Related p-Terphenylquinones from Fungi: Overview and Biological Properties. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V: Amsterdam, The Netherlands, 2003; pp. 263–308. [Google Scholar]
- Shukla, V.; Joshi, G.P.; Rawat, M.S.M. Lichen as a Potential Natural Source of Bioactive Compounds: A Review. Phytochem. Rev. 2010, 9, 303–314. [Google Scholar] [CrossRef]
- Xu, K.; Gao, Y.; Li, Y.L.; Xie, F.; Zh, Z.T.; Lou, H.X. Cytotoxic p-Terphenyls from the Endolichenic Fungus Floricola striata. J. Nat. Prod. 2018, 81, 2041–2049. [Google Scholar] [CrossRef]
- Li, W.; Li, X.B.; Lou, H.X. Structural and biological diversity of natural p-terphenyls. J Asian Nat Prod Res. 2017, 20, 1–13. [Google Scholar] [CrossRef]
- Biggins, J.B.; Liu, X.; Feng, Z.; Brady, S.F. Metabolites from the Induced Expression of Cryptic Single Operons Found in the Genome of Burkholderia Pseudomallei. J. Am. Chem. Soc. 2011, 133, 1638–1641. [Google Scholar] [CrossRef] [Green Version]
- Blatchly, J.M.; Green, R.J.S.; Mcomie, J.F.W.; Saleh, S.A. Thiele-Winter Acetoxylation of Quinone. Part IV. Methoxy- and Hydroxy-(Phenyl)-1,4-Benzoquinones and (4-Substituted Phenyl)-1,4-Benzoquinones. J. Chem. Soc. Perkin Trans. 1 1975, 309–314. [Google Scholar] [CrossRef]
- Briggs, L.H.; Cambie, R.C.; Dean, I.C.; Dromgoole, S.H.; Fergus, B.J.; Ingram, W.B.; Lewis, K.G.; Small, C.W.; Thomas, R.; Walker, D.A. Chemistry of fungi. 10. Metabolites of some fungal species. N. Zeal. J. Sci. 1975, 18, 565–576. [Google Scholar]
- Kouam, S.F.; Ngadjui, B.T.; Krohn, K.; Wafo, P.; Ajaz, A.; Choudhary, M.I. Prenylated Anthronoid Antioxidants from the Stem Bark of Harungana Madagascariensis. Phytochemistry 2005, 66, 1174–1179. [Google Scholar] [CrossRef]
- Wang, X.; Yu, W.; Lou, H. Antifungal Constituents from the Chinese Moss Homalia Trichomanoides. Chem. Biodivers. 2005, 2, 139–145. [Google Scholar] [CrossRef]
- Banwell, M.G.; Chand, S. Exploitation of Co-Operative Directed Ortho-Metallation (Dom) by 1, 3-Related-Omom Groups in the Development of a Fully Regio-Controlled Synthesis of Atranol from Orcinol. Org. Prep. Proced. Int. 2005, 37, 275–279. [Google Scholar] [CrossRef]
- Tanahashi, T.; Takenaka, Y.; Nagakura, N.; Hamada, N. Dibenzofurans from the Cultured Lichen Mycobionts of Lecanora cinereocarnea. Phytochemistry 2001, 58, 1129–1134. [Google Scholar] [CrossRef]
- Lopes, T.I.B.; Coelho, R.G.; Yoshida, N.C.; Honda, N.K. Radical-Scavenging Activity of Orsellinates. Chem. Pharm. Bull. 2008, 56, 1551–1554. [Google Scholar] [CrossRef] [Green Version]
- Shukla, V.; Negi, S.; Rawat, M.S.M.; Pant, G.; Nagatsu, A. Chemical Study of Ramalina africana (Ramalinaceae) from the Garhwal Himalayas. Biochem. Syst. Ecol. 2004, 32, 449–453. [Google Scholar] [CrossRef]
- Schröder, H.; Haslinger, E. Long-Range Proton Spin-Spin Coupling in Rigid Cyclic Structures by 2D NMR/‘Through-Space Coupling’. Magn. Reson. Chem. 1994, 32, 12–15. [Google Scholar] [CrossRef]
- Sveshnikov, N.N.; Fomichov, A.A.; Vystorop, V.G. Inter-ring Long-range Spin-Spin Proton Coupling in some 8-Hydroxyquinoline Derivatives. Mendeleev Commun. 1993, 3, 107–108. [Google Scholar] [CrossRef]
- Alvarez-Cisneros, C.; Muñoz, M.A.; Suárez-Castillo, O.R.; Pérez-Hernández, N.; Cerda-García-Rojas, C.M.; Morales-Ríos, M.S.; Joseph-Nathan, P. Stereospecific 5JHortho,OMe couplings in methoxyindoles, methoxycoumarins, and methoxyflavones. Magn. Reson. Chem. 2014, 52, 491–499. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No. | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 126.9 | 117.1 | 130.3 | |||
| 2 | 140.6 | 147.3 | 147.2 | |||
| 3 | 145.5 | 139.0 | 147.2 | |||
| 4 | 123.3 | 133.1 | 130.3 | |||
| 5 | 150.4 | 6.51, s | 104.1 | 147.2 | ||
| 6 | 6.75, s | 108.8 | 153.6 | 147.2 | ||
| 7 | 3.37, s | 60.8 | 3.45, s | 61.1 | 3.59, s | 61.0 |
| 8 | 3.72, s | 56.6 | 3.75, s | 56.1 | 3.59, s | 61.0 |
| 9 | 3.59, s | 61.0 | ||||
| 10 | 3.59, s | 61.0 | ||||
| 1′ | 133.5 | 133.3 | 134.0 | |||
| 2′/6′ | 7.49, m | 128.2 | 7.47, m | 130.9 | 7.44, m | 130.4 |
| 3′/5′ | 7.45, m | 130.7 | 7.47 m | 128.2 | 7.44, m | 128.0 |
| 4′ | 7.37, td (7.5, 1.2) | 127.5 | 7.38, m | 127.5 | 7.38, m | 127.3 |
| 1″ | 137.9 | 138.2 | 134.0 | |||
| 2″/6″ | 7.67, dd (8.3, 1.2) | 128.5 | 7.66, dd (8.3, 1.3) | 128.9 | 7.44, m | 130.4 |
| 3″/5″ | 7.47 | 129.2 | 7.47, m | 128.7 | 7.44, m | 128.0 |
| 4″ | 7.37, td (7.5, 1.2) | 127.5 | 7.38 m | 127.7 | 7.38, m | 127.3 |
| 2-OH | 5.74, s | 5.95, s | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phi, K.-H.; Shin, M.-J.; Lee, S.; So, J.E.; Kim, J.H.; Suh, S.-S.; Koo, M.H.; Shin, S.C.; Kim, J.-H.; Lee, J.H.; et al. Bioactive Terphenyls Isolated from the Antarctic Lichen Stereocaulon alpinum. Molecules 2022, 27, 2363. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072363
Phi K-H, Shin M-J, Lee S, So JE, Kim JH, Suh S-S, Koo MH, Shin SC, Kim J-H, Lee JH, et al. Bioactive Terphenyls Isolated from the Antarctic Lichen Stereocaulon alpinum. Molecules. 2022; 27(7):2363. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072363
Chicago/Turabian StylePhi, Kim-Hoa, Min-Ji Shin, Seulah Lee, Jae Eun So, Ji Hee Kim, Sung-Suk Suh, Man Hyung Koo, Seung Chul Shin, Jin-Hyoung Kim, Jun Hyuck Lee, and et al. 2022. "Bioactive Terphenyls Isolated from the Antarctic Lichen Stereocaulon alpinum" Molecules 27, no. 7: 2363. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072363