

Comprehensive Review of Recent Advances in Chiral A-Ring Flavonoid Containing Compounds: Structure, Bioactivities, and Synthesis

Abstract
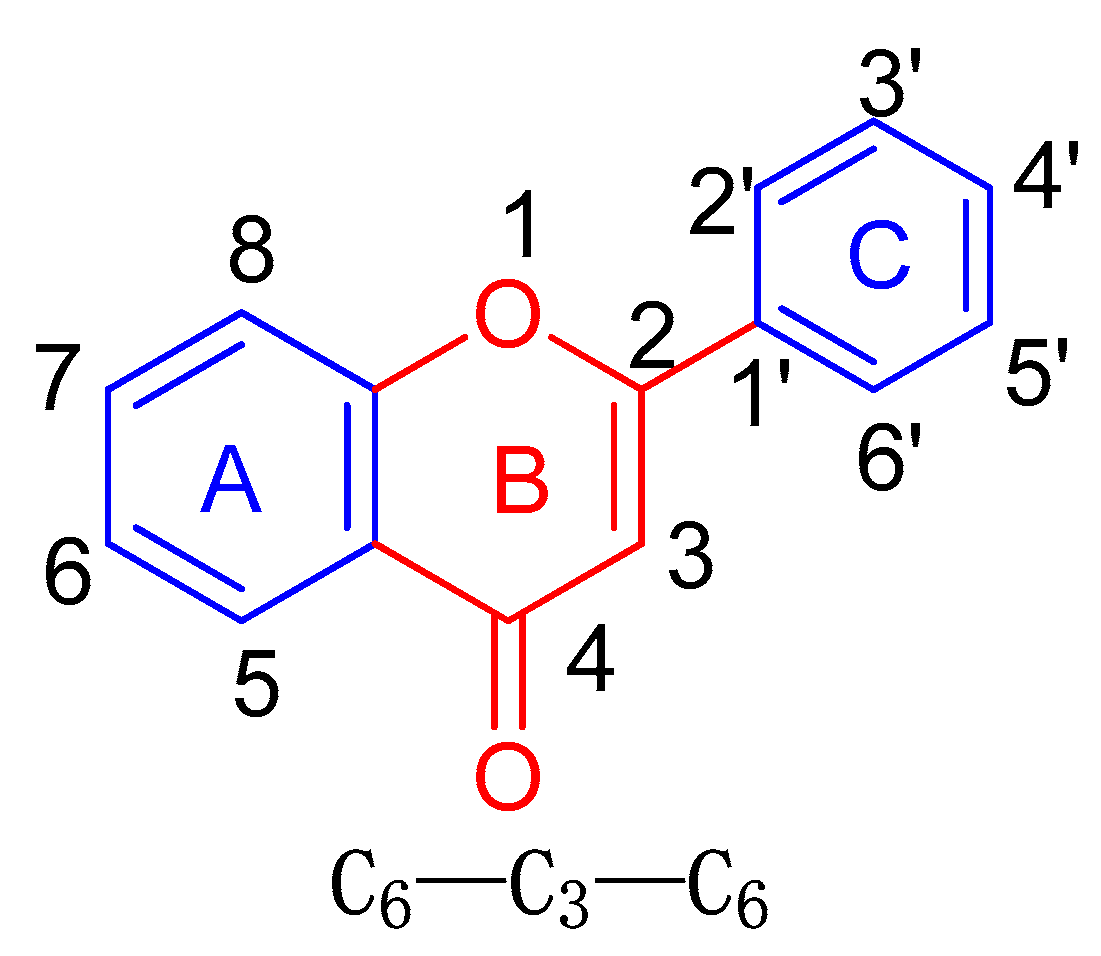
:1. Introduction
2. Isolation and Structural Elucidation of Chiral A-Ring Flavonoid-Containing Compounds
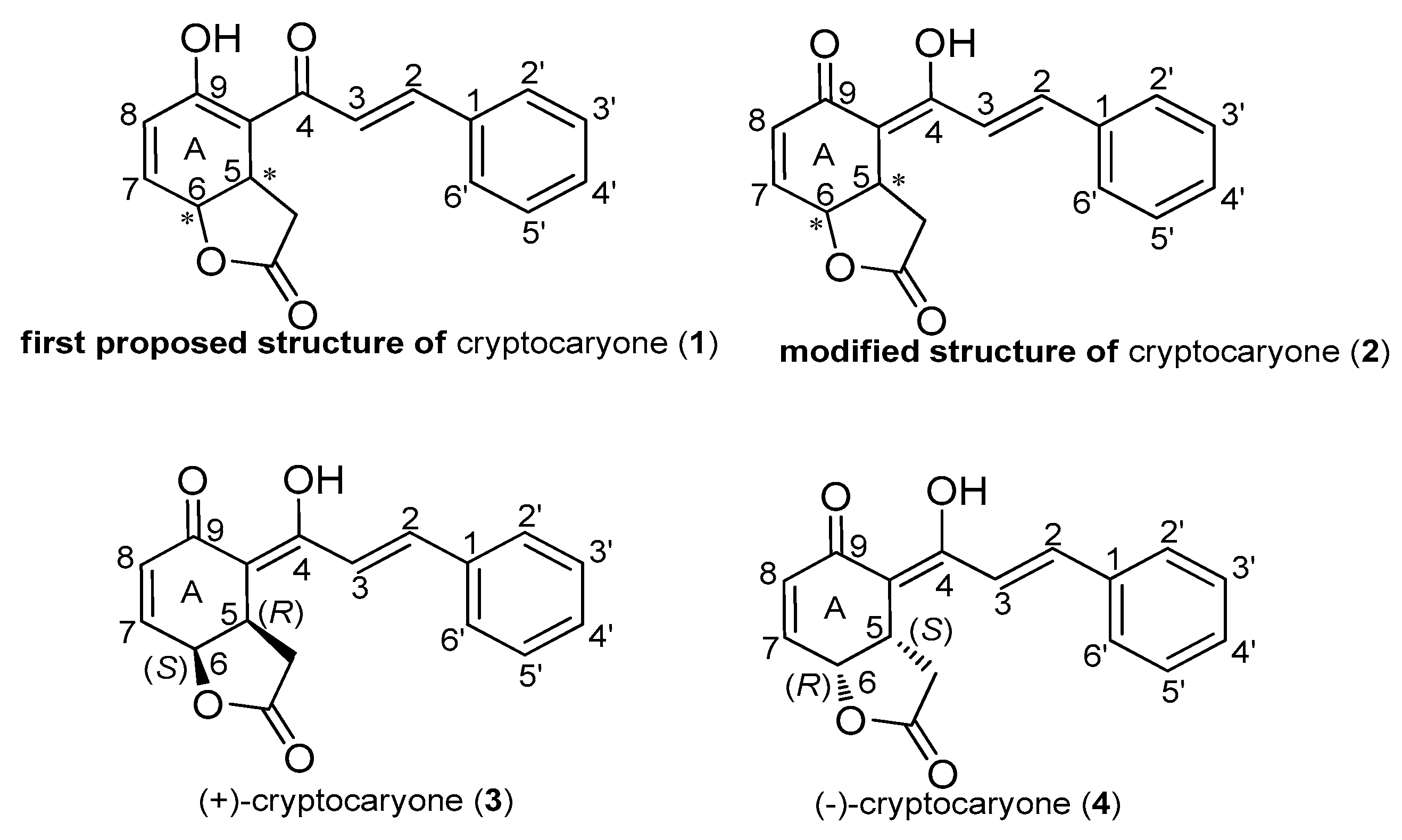
2.1. Cryptocaryone
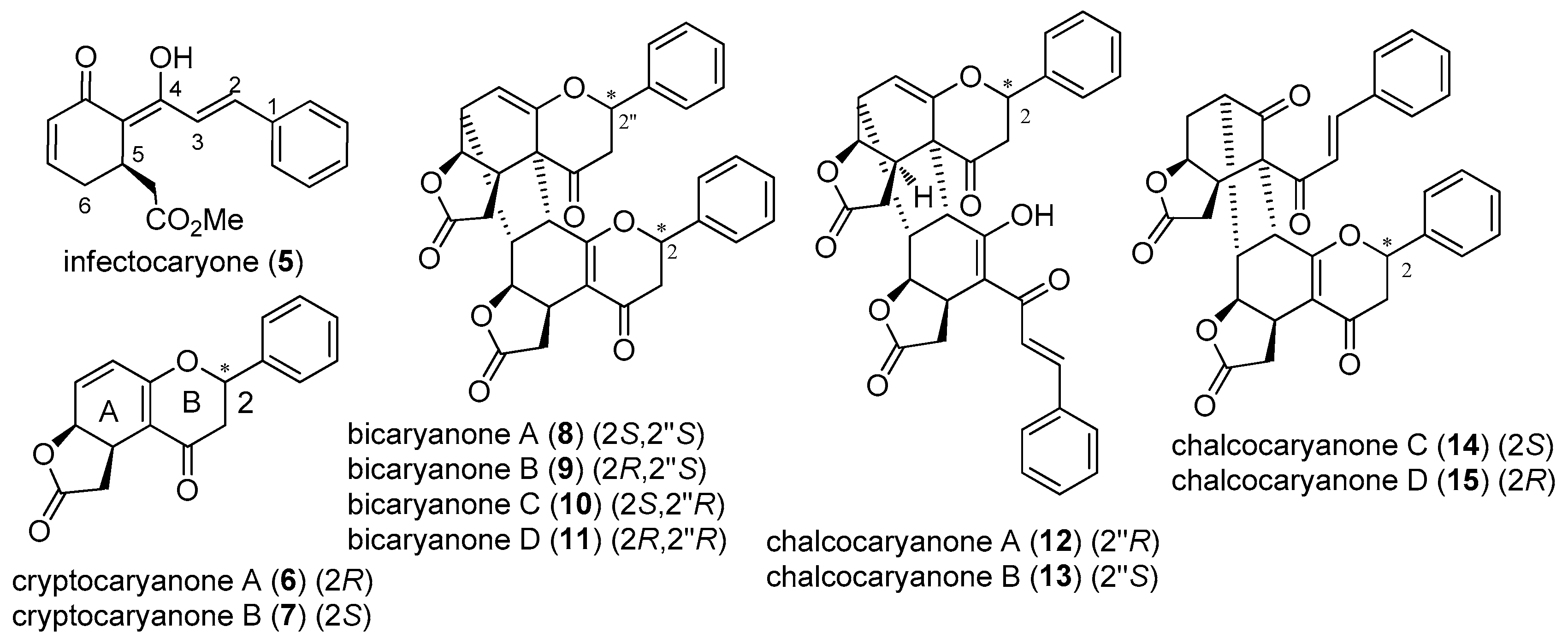
2.2. Infectocaryone, Cryptocaryanones, Bicaryanones, and Chalcocaryanones
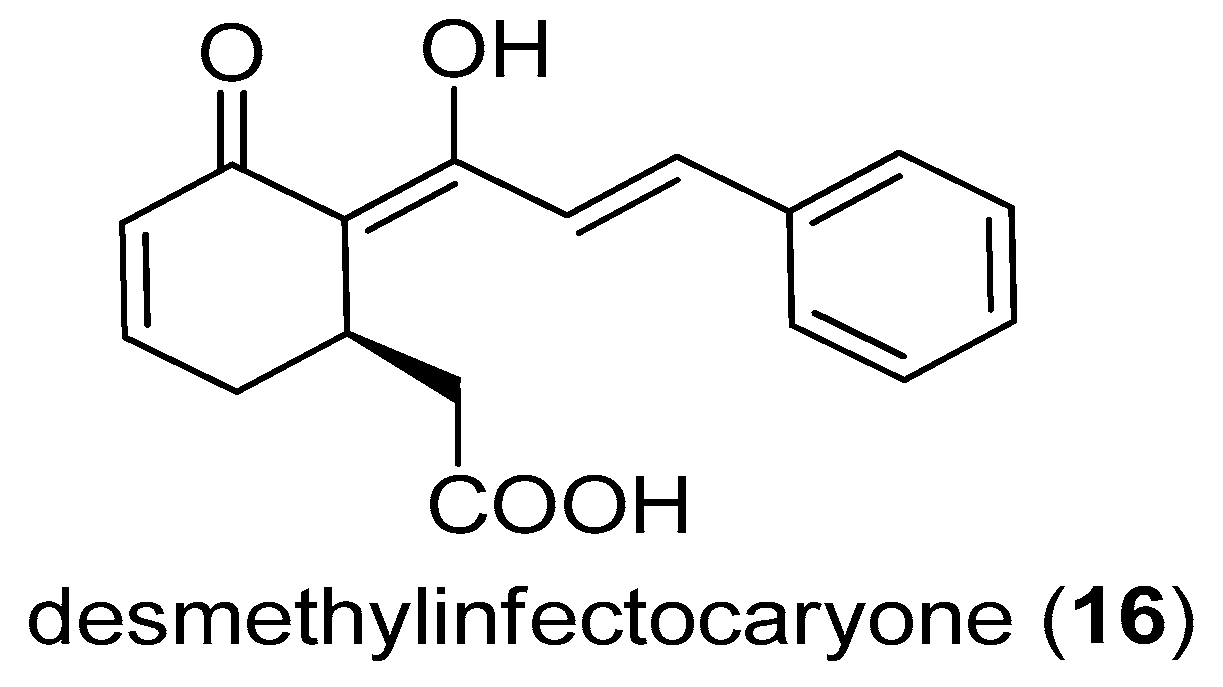
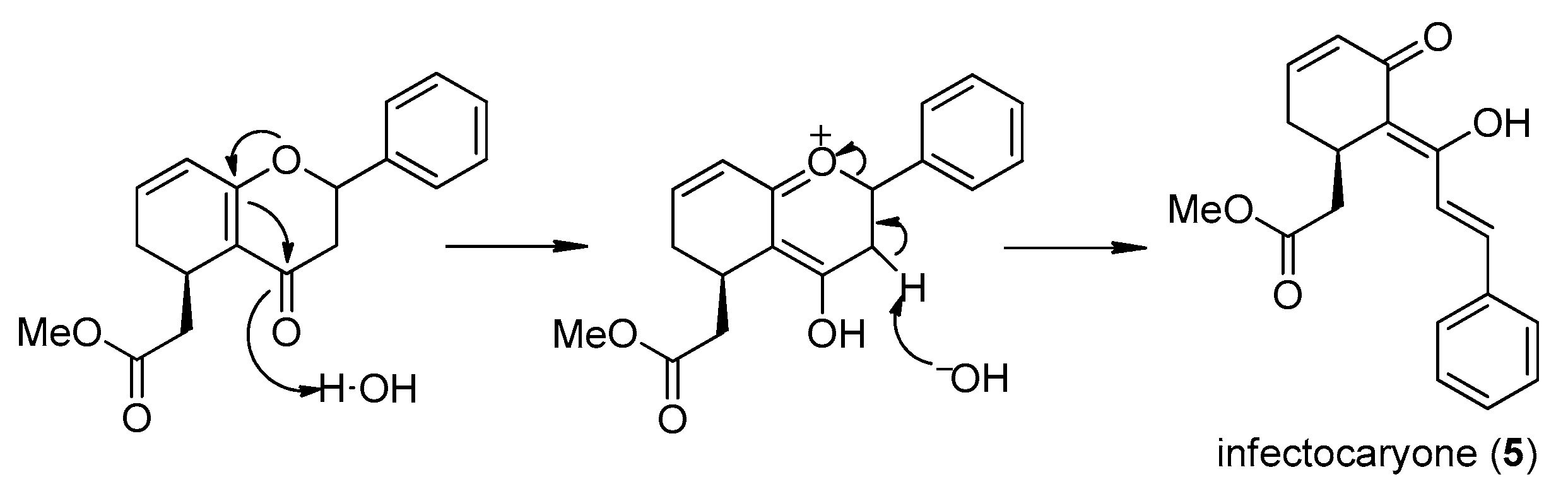
2.3. Desmethylinfectocaryone
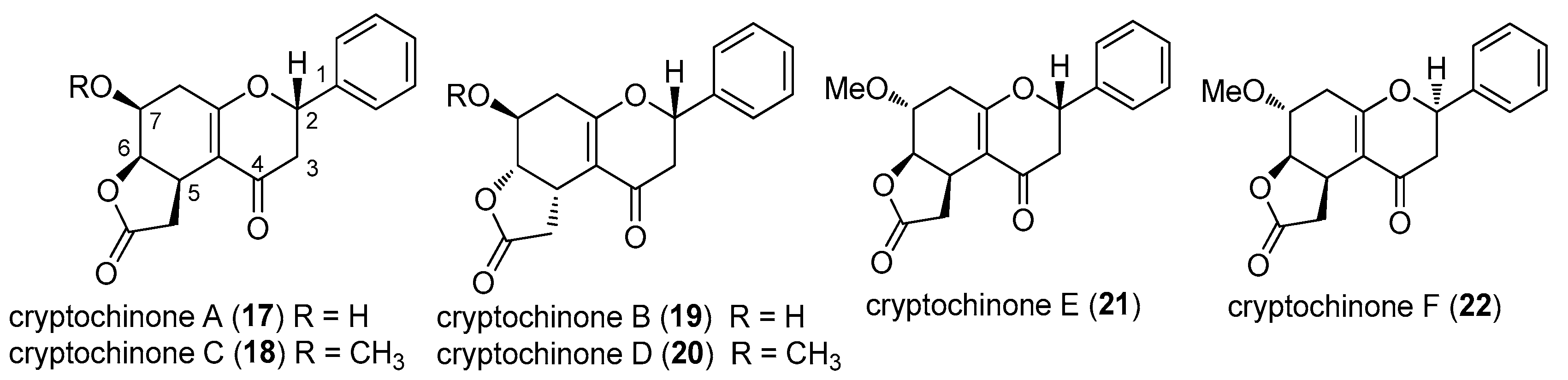
2.4. Cryptochinones
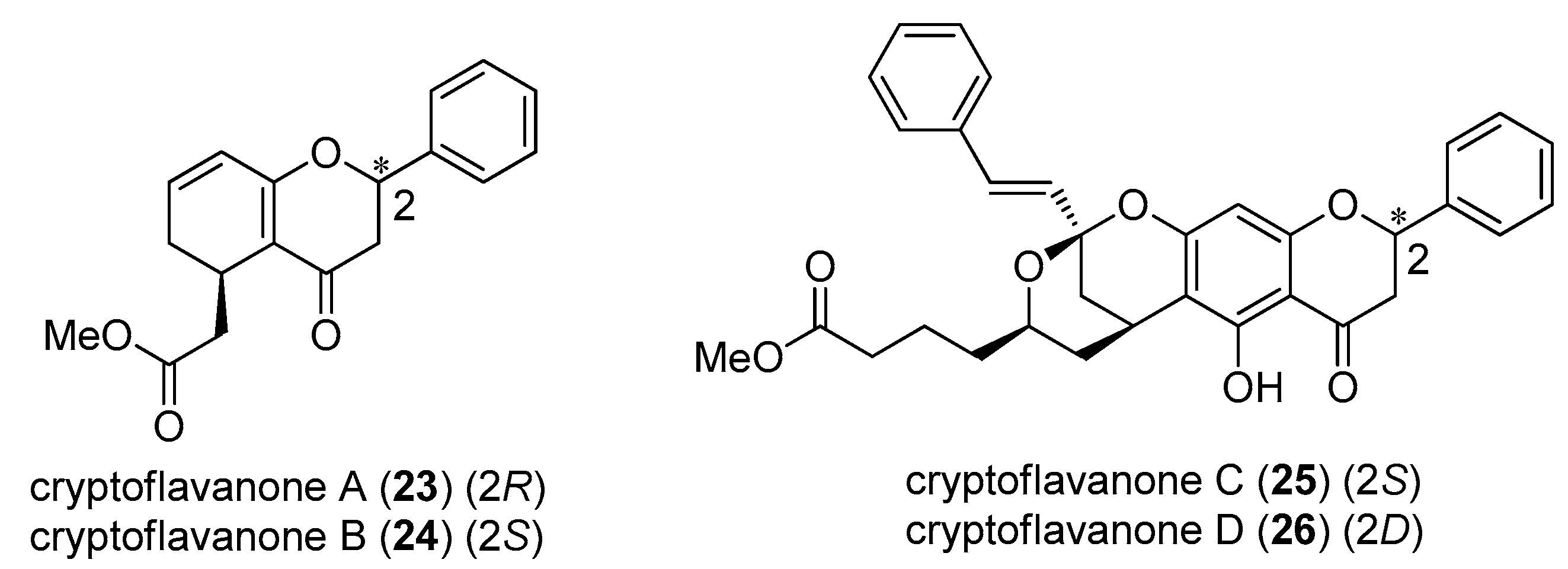
2.5. Cryptoflavanones
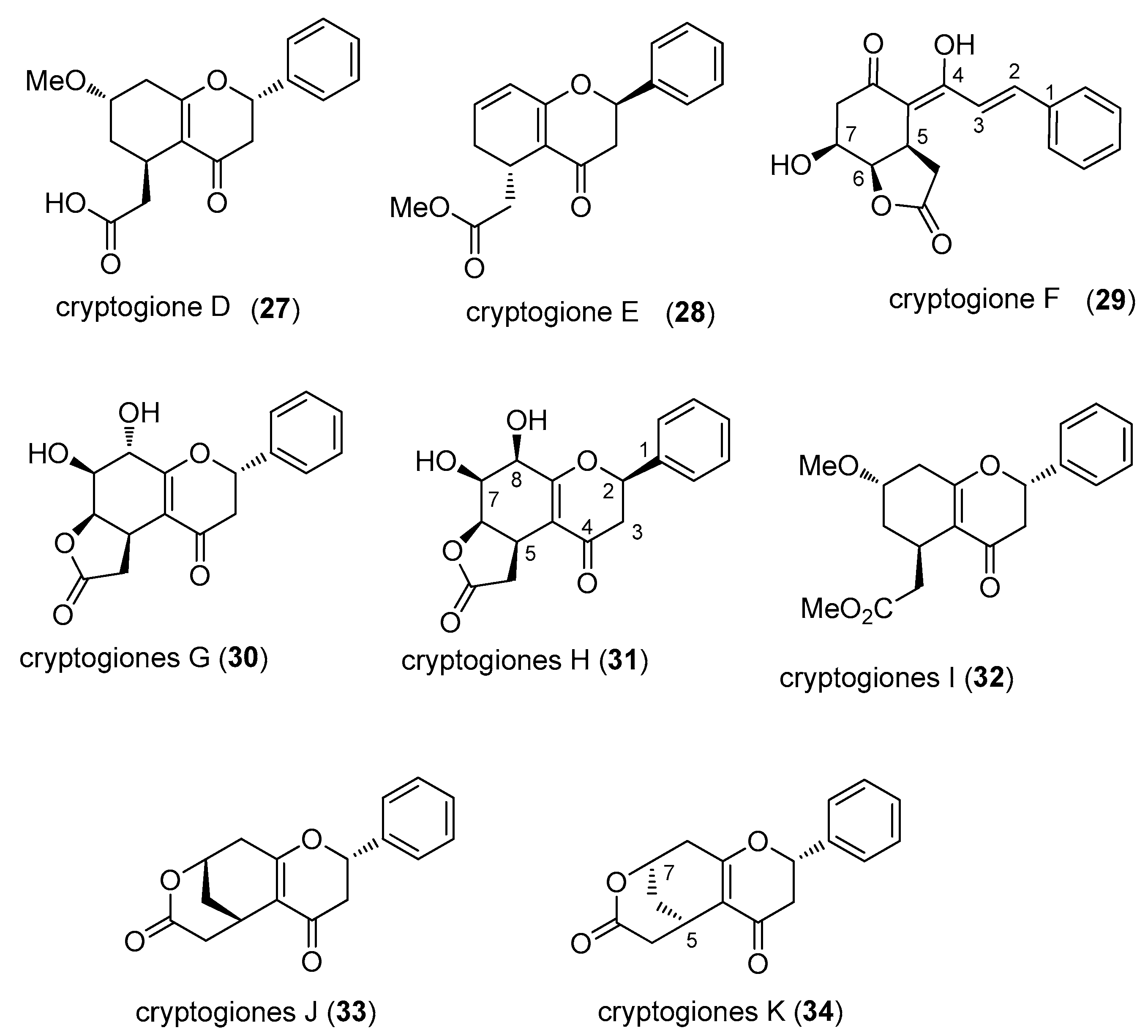
2.6. Cryptogiones
2.7. Cryptoconones
3. Biological Activity of Chiral A-Ring Flavonoid-Containing Compounds
4. Progress in the Synthesis of Chiral A-Ring-Containing Flavonoids
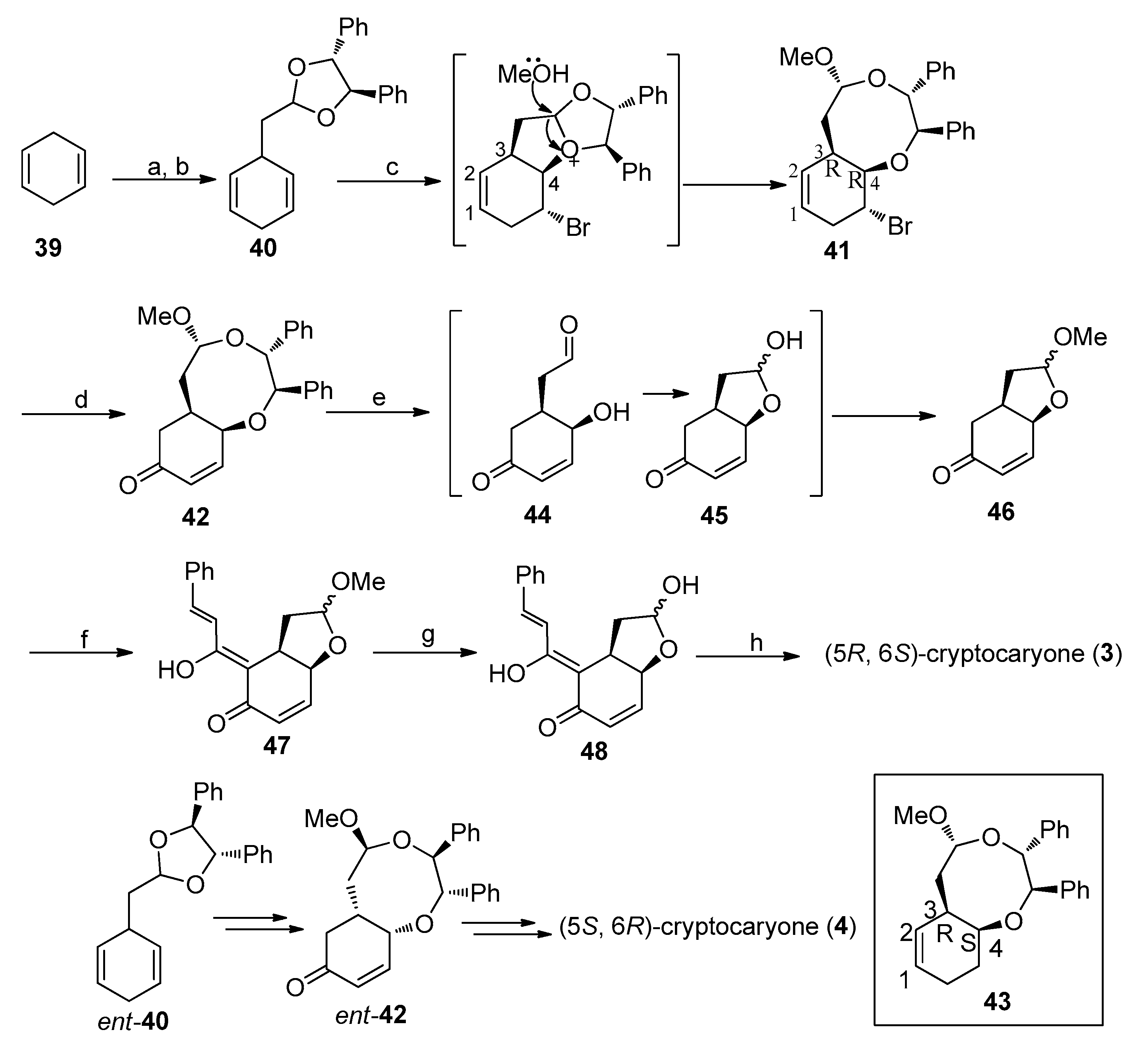
4.1. Synthesis of the Natural Product Cryptocaryone
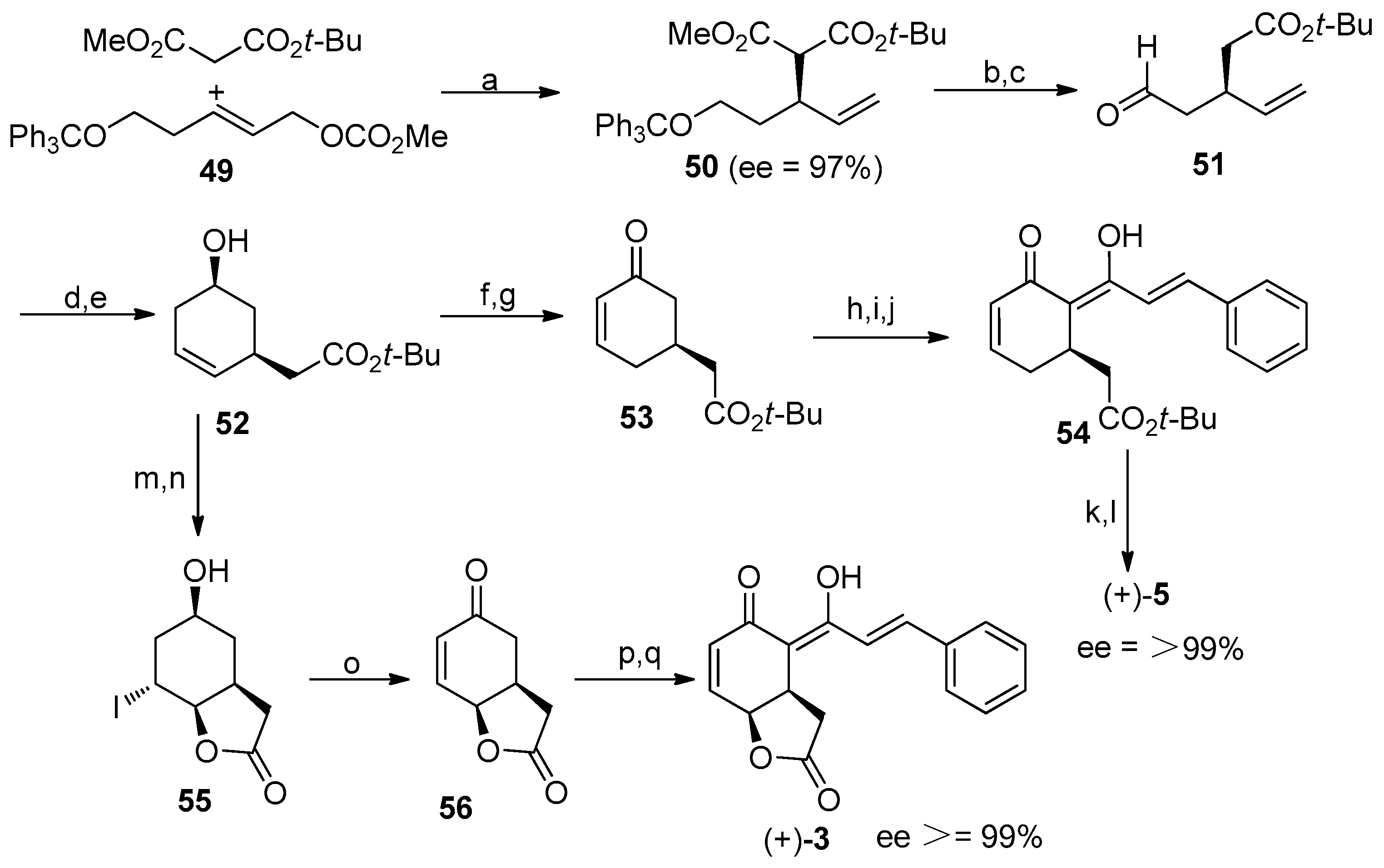
4.2. Synthesis of the Natural Products (+)-Cryptocaryone and (+)-Infectocaryone
4.3. Asymmetric Synthesis of the Natural Product Cryptocaryanone A
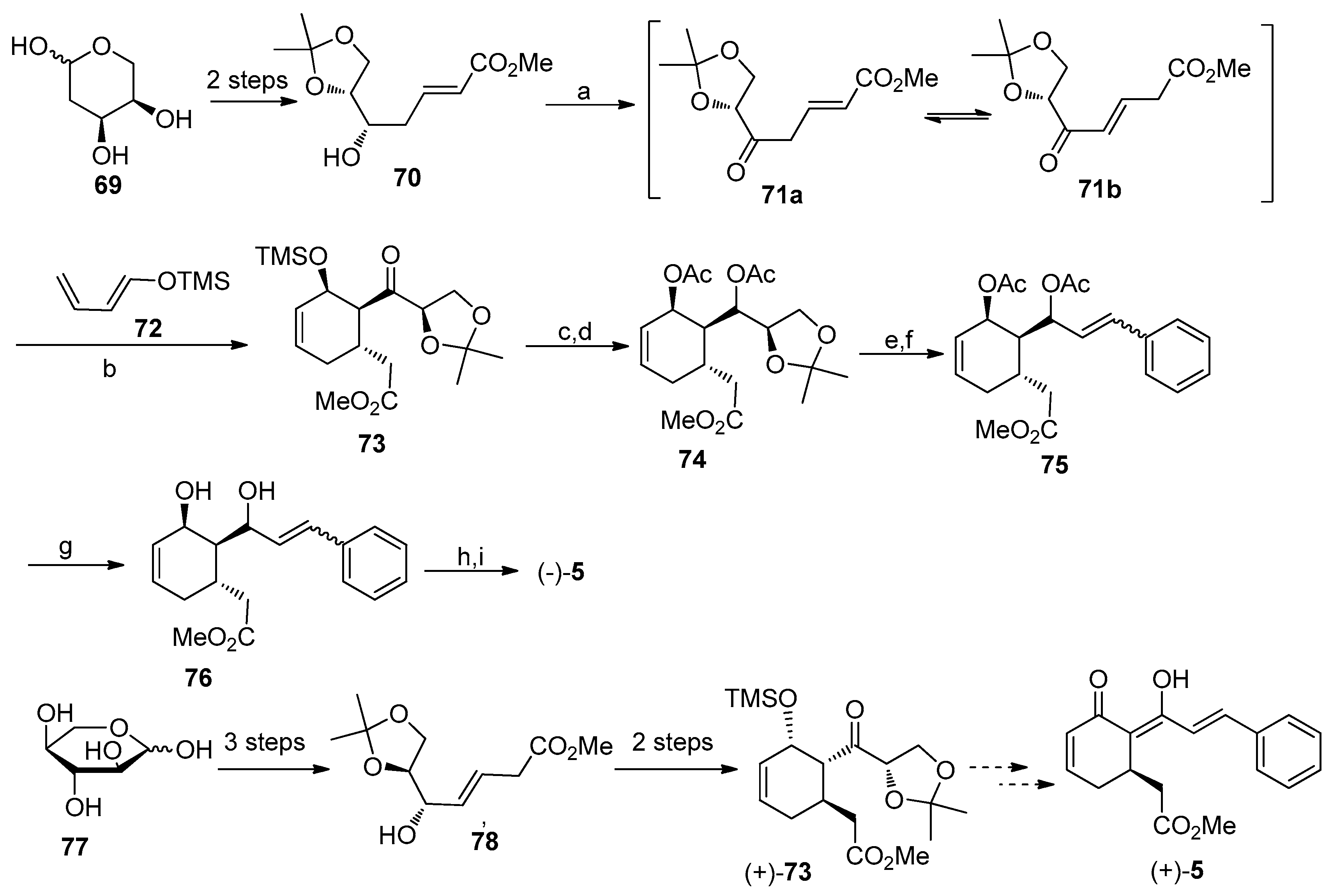
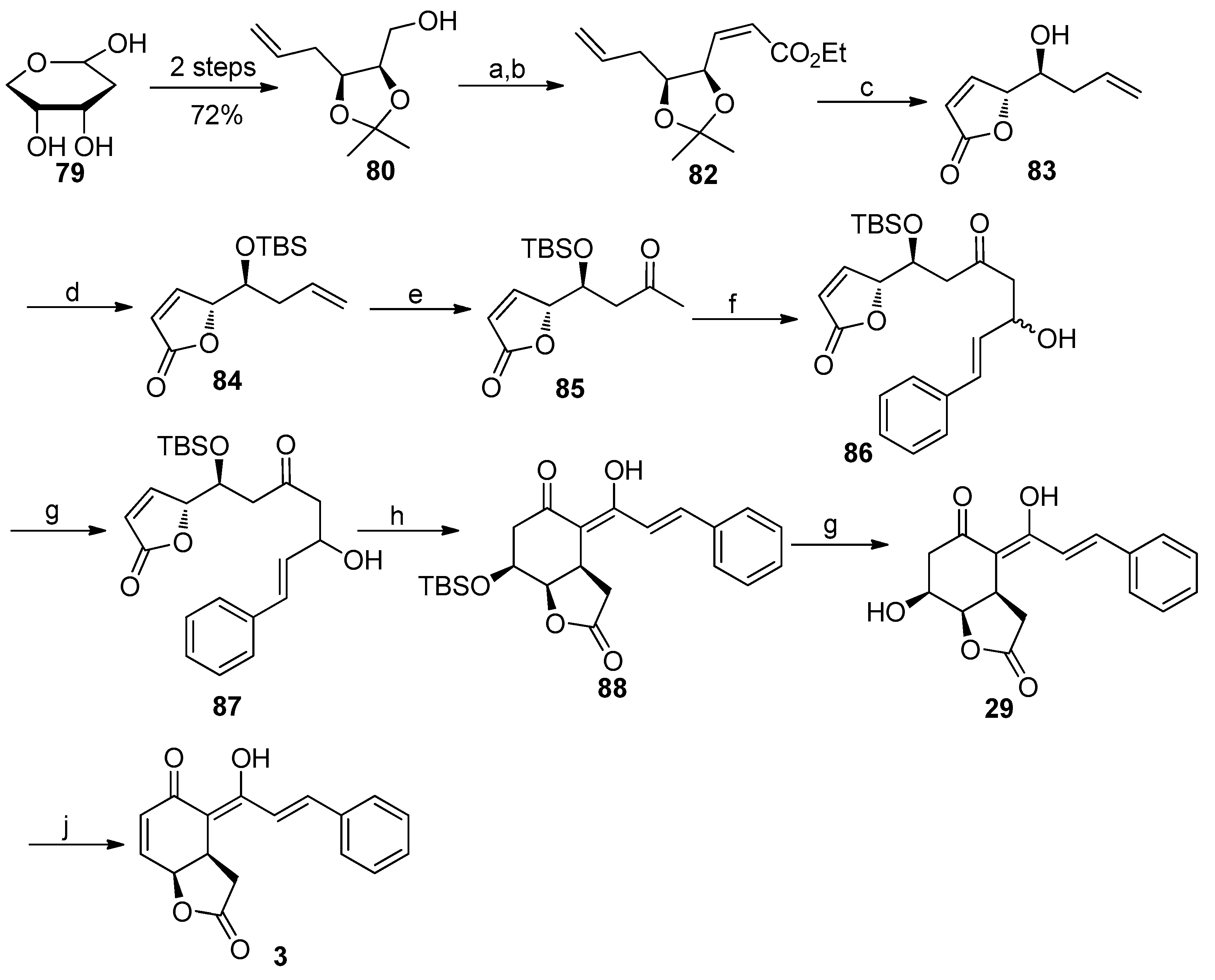
4.4. Asymmetric Synthesis of Infectocaryone
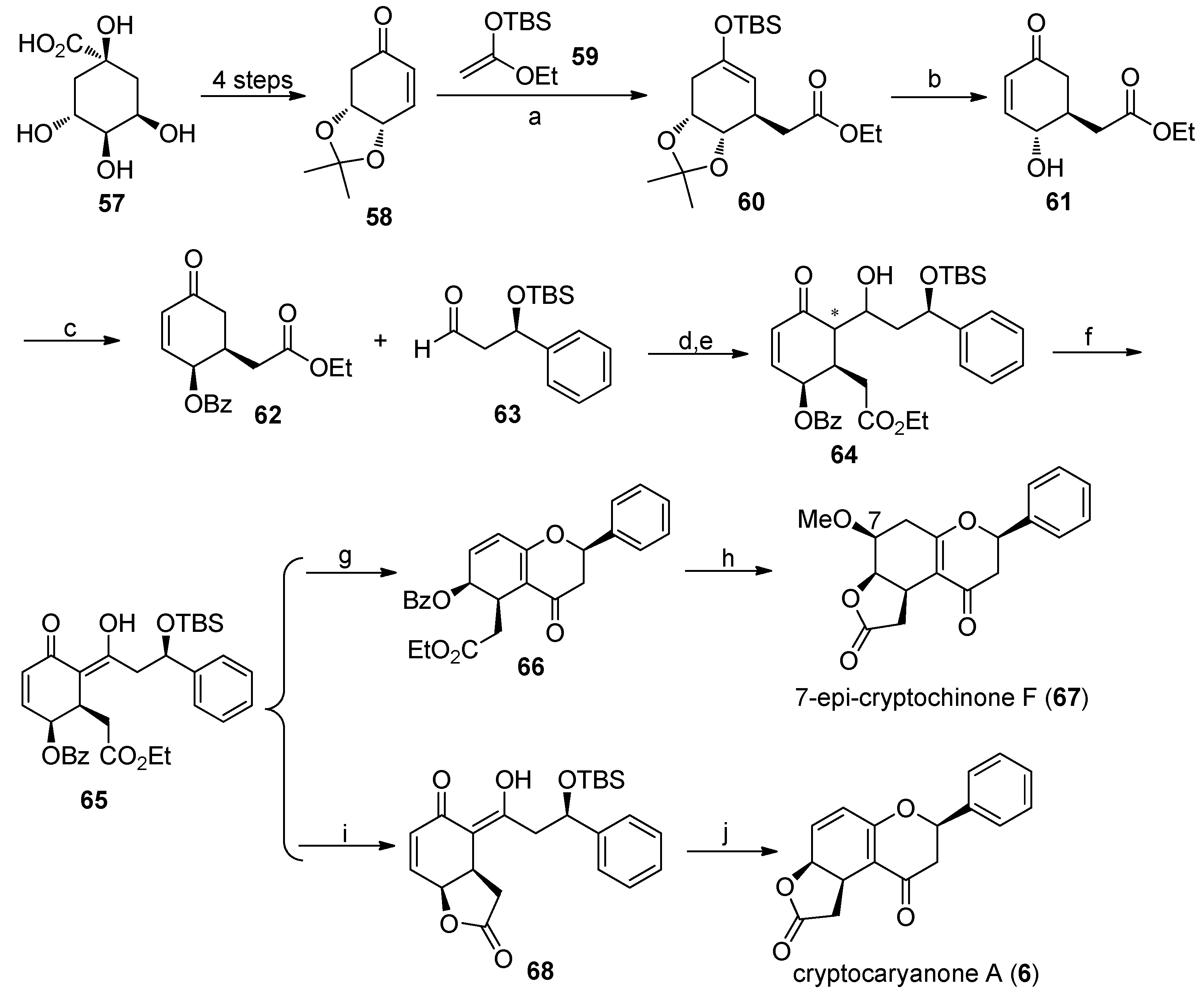
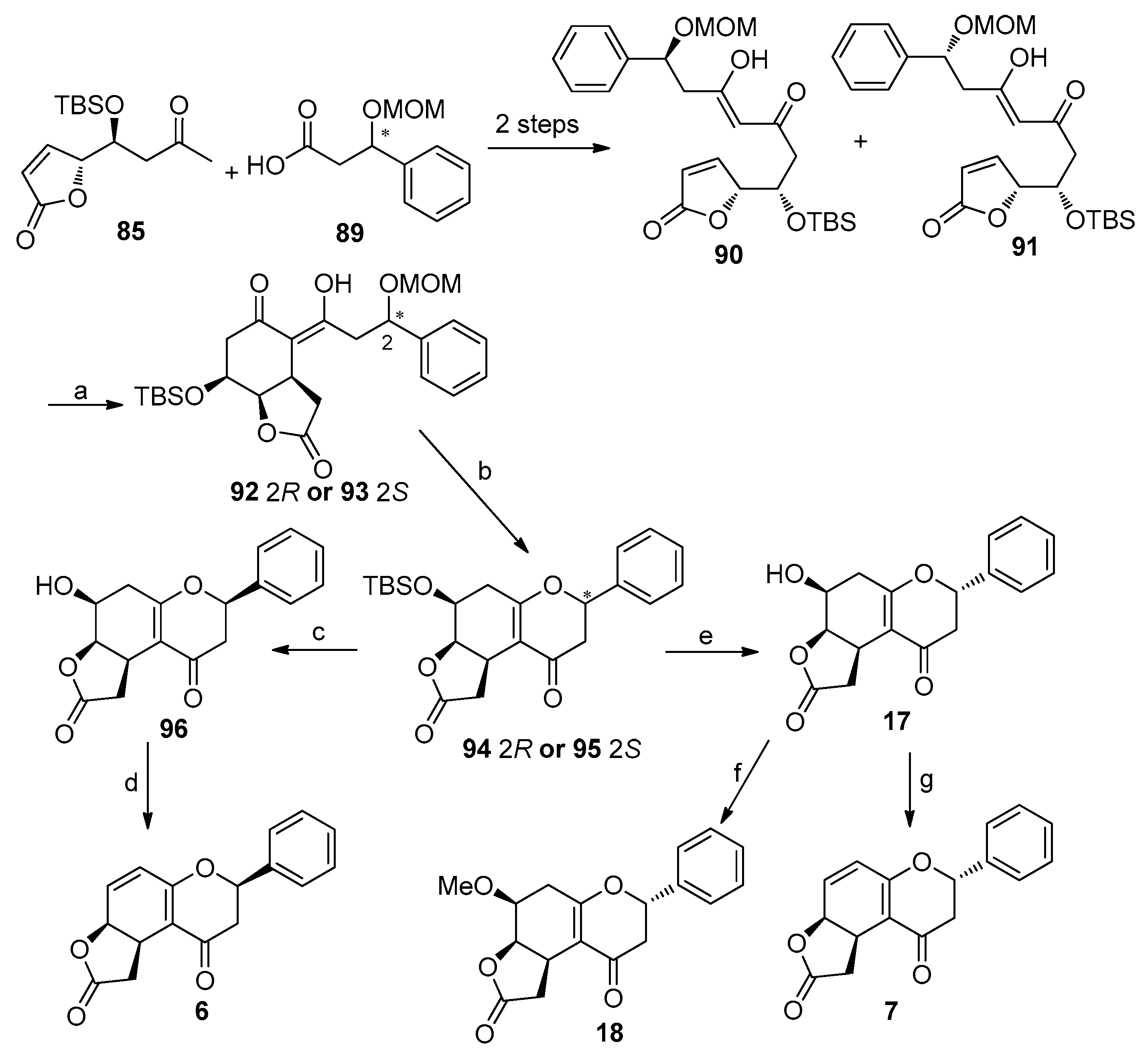
4.5. Asymmetric Synthesis of Cryptogione F, Cryptocaryanone B, Cryptochinones A and C, Cryptocaryone, and Cryptocaryanone A
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAN | Ceric ammonium nitrate |
| DBU | 1,8-Diazabicyclo [5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DMAP | 4-Dimethylamino-pyridine |
| DMF | Dimethyl formamide |
| DMP | Dess-Martin periodinane |
| IBX | 2-Iodoxybenzoic acid |
| IC50 | Half maximal inhibitory concentration |
| LiHMDS | Lithium bis(trimethylsilyl)amide |
| NBS | N-Bromosuccinimide |
| NIS | N-iodosuccinimide |
| PDC | Pyridinium dichromate |
| RCM | Ring-closing metathesis |
| TBAI | Tetrabutylammonium Iodide |
| TFAA | Trifluoroacetic anhydride |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
References
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Filippi, A.; Braidot, E.; Petrussa, E.; Fabro, M.; Vuerich, M.; Boscutti, F. Plant growth shapes the effects of elevation on the content and variability of flavonoids in subalpine bilberry stands. Plant Biol. 2021, 23, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, F.; Lian, Y.; Xiao, H.; Zheng, J. Biosynthesis of citrus flavonoids and their health effects. Crit. Rev. Food Sci. Nutr. 2020, 60, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef] [PubMed]
- Rakha, A.; Umar, N.; Rabail, R.; Butt, M.S.; Kieliszek, M.; Hassoun, A.; Aadil, R.M. Anti-inflammatory and anti-allergic potential of dietary flavonoids: A review. Biomed. Pharmacother. 2022, 156, 113945. [Google Scholar] [CrossRef]
- Wen, K.; Fang, X.; Yang, J.; Yao, Y.; Nandakumar, K.S.; Salem, M.L.; Cheng, K. Recent Research on Flavonoids and their Biomedical Applications. Curr. Med. Chem. 2021, 28, 1042–1066. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Hassan, A.R.; Amer, K.F.; El-Toumy, S.A.; Nielsen, J.; Christensen, S.B. A new flavonol glycoside and other flavonoids from the aerial parts of Taverniera aegyptiaca. Nat. Prod. Res. 2019, 33, 1135–1139. [Google Scholar] [CrossRef]
- Sun, Z.G.; Li, Z.N.; Zhang, J.M.; Hou, X.Y.; Yeh, S.M.; Ming, X. Recent Developments of Flavonoids with Various Activities. Curr. Top. Med. Chem. 2022, 22, 305–329. [Google Scholar] [CrossRef]
- Kocic, B.; Kitic, D.; Brankovic, S. Dietary flavonoid intake and colorectal cancer risk: Evidence from human population studies. J. BUON. 2013, 18, 34–43. [Google Scholar]
- Speisky, H.; Shahidi, F.; Costa de Camargo, A.; Fuentes, J. Revisiting the Oxidation of Flavonoids: Loss, Conservation or Enhancement of Their Antioxidant Properties. Antioxidants 2022, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.P.; Chen, Q.X.; Huang, H.; Liu, X.D.; Chen, H.T.; Zhang, R.Q. Inhibitory effects of cupferron on the monophenolase and diphenolase activity of mushroom tyrosinase. Int. J. Biochem. Cell Biol. 2003, 35, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Delomenède, M.; Bedos-Belval, F.; Duran, H.; Vindis, C.; Baltas, M.; Nègre-Salvayre, A. Development of novel antiatherogenic biaryls: Design, synthesis, and reactivity. J. Med. Chem. 2008, 51, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Nishida, J.; Saito, S.; Kawabata, J. Inhibitory effects of 5,6,7-trihydroxyflavones on tyrosinase. Molecules 2007, 12, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Xie, L.; Liu, K.; Liang, Y.; Dai, X.; Wang, X.; Lu, J.; Zhang, X.; Li, X. The antihypertensive potential of flavonoids from Chinese Herbal Medicine: A review. Pharmacol. Res. 2021, 174, 105919. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.P.; Chen, Q.X.; Huang, H.; Wang, H.Z.; Zhang, R.Q. Inhibitory effects of some flavonoids on the activity of mushroom tyrosinase. Biochemistry 2003, 68, 487–491. [Google Scholar] [PubMed]
- Ren, L.; Perera, C.; Hemar, Y. Antitumor activity of mushroom polysaccharides: A review. Food Funct. 2012, 3, 1118–1130. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, M.; Lu, L.; Sun, L.; Xu, M. Nitric oxide fumigation stimulates flavonoid and phenolic accumulation and enhances antioxidant activity of mushroom. Food Chem. 2012, 135, 1220–1225. [Google Scholar] [CrossRef]
- Liu, H.; Huang, B.; Qin, X.; Zhang, X.; Wang, Z. Fabrication and photocatalytic activity of mushroom-like ZnO microcrystals via a solvothermal route. Rare Metals 2011, 30, 173–176. [Google Scholar] [CrossRef]
- Gupta, T.; Kataria, R.; Sardana, S. A Comprehensive Review on Current Perspectives of Flavonoids as Antimicrobial Agent. Curr. Top. Med. Chem. 2022, 22, 425–434. [Google Scholar] [CrossRef]
- Tan, Z.; Deng, J.; Ye, Q.; Zhang, Z. The Antibacterial Activity of Natural-derived Flavonoids. Curr. Top. Med. Chem. 2022, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Overview of antibacterial, antitoxin, antiviral, and antifungal activities of tea flavonoids and teas. Mol. Nutr. Food Res. 2007, 51, 116–134. [Google Scholar] [CrossRef] [PubMed]
- Orhan, D.D.; Ozçelik, B.; Ozgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010, 165, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Liao, J.; Zhou, H.; Wang, C.C.; Wang, L.; Fan, Y. Flavonoids from Lycium barbarum Leaves Exhibit Anti-Aging Effects through the Redox-Modulation. Molecules 2022, 27, 4952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, A.; Yang, X. Effect of lemon seed flavonoids on the anti-fatigue and antioxidant effects of exhausted running exercise mice. J. Food Biochem. 2021, 45, e13620. [Google Scholar] [CrossRef]
- Chen, X.; Liang, D.; Huang, Z.; Jia, G.; Zhao, H.; Liu, G. Anti-fatigue effect of quercetin on enhancing muscle function and antioxidant capacity. J. Food Biochem. 2021, 45, e13968. [Google Scholar] [CrossRef]
- Kujumgiev, A.; Tsvetkova, I.; Serkedjieva, Y.; Bankova, V.; Christov, R.; Popov, S. Antibacterial, antifungal and antiviral activity of propolis of different geographic origin. J. Ethnopharmacol. 1999, 64, 235–240. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Li, X.; Zhang, L.; Ren, L. Flavonoids as Potential Antiviral Agents for Porcine Viruses. Pharmaceutics 2022, 14, 1793. [Google Scholar] [CrossRef]
- Cataneo, A.H.D.; Ávila, E.P.; Mendes, L.A.O.; de Oliveira, V.G.; Ferraz, C.R.; de Almeida, M.V.; Frabasile, S.; Duarte Dos Santos, C.N.; Verri, W.A.; Bordignon, J.; et al. Flavonoids as Molecules with Anti-Zika virus Activity. Front. Microbiol. 2021, 12, 710359. [Google Scholar] [CrossRef]
- Badshah, S.L.; Faisal, S.; Muhammad, A.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Antiviral activities of flavonoids. Biomed. Pharmacother. 2021, 140, 111596. [Google Scholar] [CrossRef]
- Zhou, Z.G.; Li, D.D.; Chen, Y.; Chen, X.; Man, R.J. Discussion on the Structural Modification and Anti-tumor Activity of Flavonoids. Curr. Top. Med. Chem. 2022, 22, 561–577. [Google Scholar] [CrossRef]
- Ferdous, U.T.; Balia Yusof, Z.N. Insight into Potential Anticancer Activity of Algal Flavonoids: Current Status and Challenges. Molecules 2021, 26, 6844. [Google Scholar] [CrossRef]
- Ponte, L.G.S.; Pavan, I.C.B.; Mancini, M.C.S.; da Silva, L.G.S.; Morelli, A.P.; Severino, M.B.; Bezerra, R.M.N.; Simabuco, F.M. The Hallmarks of Flavonoids in Cancer. Molecules 2021, 26, 2029. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.; Abdellatif, B.; Zhai, K.; Liskova, A.; Kubatka, P.; Büsselberg, D. Flavonoids Alleviate Peripheral Neuropathy Induced by Anticancer Drugs. Cancers 2021, 13, 1576. [Google Scholar] [CrossRef] [PubMed]
- Hozzein, W.N.; Mohany, M.; Alhawsawi, S.M.M.; Zaky, M.Y.; Al-Rejaie, S.S.; Alkhalifah, D.H.M. Flavonoids from Marine-Derived Actinobacteria as Anticancer Drugs. Curr. Pharm. Des. 2021, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.X. Chemical composition, characterization, and differentiation of honey botanical and geographical origins. Adv. Food Nutr. Res. 2011, 62, 89–137. [Google Scholar]
- Kurniadewi, F.; Juliawaty, L.D.; Syah, Y.M.; Achmad, S.A.; Hakim, E.H.; Koyama, K.; Kinoshita, K.; Takahashi, K. Phenolic compounds from Cryptocarya konishii: Their cytotoxic and tyrosine kinase inhibitory properties. J. Nat. Med. 2010, 64, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Chen, J.J.; Peng, C.F.; Cheng, M.J.; Chen, I.S. New flavanones from the leaves of Cryptocarya chinensis and their antituberculosis activity. Chem. Biodivers. 2011, 8, 2015–2024. [Google Scholar] [CrossRef]
- Ren, Y.; Yuan, C.; Qian, Y.; Chai, H.B.; Chen, X.; Goetz, M.; Kinghorn, A.D. Constituents of an extract of Cryptocarya rubra housed in a repository with cytotoxic and glucose transport inhibitory effects. J. Nat. Prod. 2014, 77, 550–556. [Google Scholar] [CrossRef] [Green Version]
- Govindachari, T.; Parthasarathy, P. Cryptocaryone, a novel 5′,6′-dihydrochalcone, from Cryptocaryabourdilloni gamb. Tetrahedron Lett. 1972, 13, 3419–3420. [Google Scholar] [CrossRef]
- Maddry, J.A.; Joshi, B.S.; Gary Newton, M.; William Pelletier, S.; Parthasarathy, P. Crytocaryone: A revised structure. Tetrahedron Lett. 1985, 26, 5491–5492. [Google Scholar] [CrossRef]
- Dumontet, V.; Gaspard, C.; Van Hung, N.; Fahy, J.; Tchertanov, L.; Sévenet, T.; Guéritte, F. New cytotoxic flavonoids from Cryptocarya infectoria. Tetrahedron 2001, 57, 6189–6196. [Google Scholar] [CrossRef]
- Fujioka, H.; Nakahara, K.; Oki, T.; Hirano, K.; Hayashi, T.; Kita, Y. The first asymmetric total syntheses of both enantiomers of cryptocaryone. Tetrahedron Lett. 2010, 51, 1945–1946. [Google Scholar] [CrossRef]
- Chou, T.H.; Chen, J.J.; Lee, S.J.; Chiang, M.Y.; Yang, C.W.; Chen, I.S. Cytotoxic flavonoids from the leaves of Cryptocarya chinensis. J. Nat. Prod. 2010, 73, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Guo, Z.K.; Yan, C.M.; Li, E.G.; Tan, R.X.; Ge, H.M. Anti-inflammatory flavonoids from Cryptocarya chingii. Phytochemistry 2012, 76, 98–105. [Google Scholar] [CrossRef]
- Feng, R.; Wang, T.; Wei, W.; Tan, R.X.; Ge, H.M. Cytotoxic constitutents from Cryptocarya maclurei. Phytochemistry 2013, 90, 147–153. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, W.J.; Cheng, Y.Q.; Jiang, R.; Wei, W.; Chen, C.J.; Wang, G.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Cytotoxic and antimicrobial flavonoids from Cryptocarya concinna. Planta. Med. 2014, 80, 925–930. [Google Scholar] [CrossRef]
- Chen, Y.C.; Kung, F.L.; Tsai, I.L.; Chou, T.H.; Chen, I.S.; Guh, J.H. Cryptocaryone, a natural dihydrochalcone, induces apoptosis in human androgen independent prostate cancer cells by death receptor clustering in lipid raft and nonraft compartments. J. Urol. 2010, 183, 2409–2418. [Google Scholar] [CrossRef]
- Chen, Y.C.; Yang, C.W.; Chan, T.F.; Farooqi, A.A.; Chang, H.S.; Yen, C.H.; Huang, M.Y.; Chang, H.W. Cryptocaryone Promotes ROS-Dependent Antiproliferation and Apoptosis in Ovarian Cancer Cells. Cells 2022, 11, 641. [Google Scholar] [CrossRef]
- Meragelman, T.L.; Scudiero, D.A.; Davis, R.E.; Staudt, L.M.; McCloud, T.G.; Cardellina, J.H.; Shoemaker, R.H. Inhibitors of the NF-kappaB activation pathway from Cryptocarya rugulosa. J. Nat. Prod. 2009, 72, 336–339. [Google Scholar] [CrossRef]
- Hexum, J.K.; Tello-Aburto, R.; Struntz, N.B.; Harned, A.M.; Harki, D.A. Bicyclic Cyclohexenones as Inhibitors of NF-κB Signaling. ACS Med. Chem. Lett. 2012, 3, 459–464. [Google Scholar] [CrossRef]
- Lin, H.R.; Chou, T.H.; Huang, D.W.; Chen, I.S. Cryptochinones from Cryptocarya chinensis act as farnesoid X receptor agonists. Bioorg. Med. Chem. Lett. 2014, 24, 4181–4186. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.S.; Tang, J.Y.; Yen, C.Y.; Huang, H.W.; Wu, C.Y.; Chung, Y.A.; Wang, H.R.; Chen, I.S.; Huang, M.Y.; Chang, H.W. Antiproliferation of Cryptocarya concinna-derived cryptocaryone against oral cancer cells involving apoptosis, oxidative stress, and DNA damage. BMC Complementary Altern. Med. 2016, 16, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.C.; Chang, H.S.; Tang, J.Y.; Farooqi, A.A.; Kuo, Y.T.; Hsuuw, Y.D.; Lee, J.W.; Chang, H.W. Combined Treatment with Cryptocaryone and Ultraviolet C Promotes Antiproliferation and Apoptosis of Oral Cancer Cells. Int. J. Mol. Sci. 2022, 23, 2981. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, H.; Kotoku, N.; Sawama, Y.; Nagatomi, Y.; Kita, Y. Concise asymmetric synthesis of a model compound, (4S,5S,6S)-6-(2,2-dimethoxy)ethyl-4,5-epoxy-6-hydroxy-2-cyclohexenone, for the cyclohexenone core of scyphostatin. Tetrahedron Lett. 2002, 43, 4825–4828. [Google Scholar] [CrossRef]
- Ates, A.; Gautier, A.; Leroy, B.; Plancher, J.M.; Quesnel, Y.; Markó, I.E. Remarkably efficient deprotection of cyclic acetals and ketals. Tetrahedron Lett. 1999, 40, 1799–1802. [Google Scholar] [CrossRef]
- Markó, I.E.; Ates, A.; Gautier, A.; Leroy, B.; Plancher, J.M.; Quesnel, Y.; Vanherck, J.C. Cerium(IV)-Catalyzed Deprotection of Acetals and Ketals under Mildly Basic Conditions. Angew. Chem. Int. Ed. 1999, 38, 3207–3209. [Google Scholar] [CrossRef]
- Ates, A.; Gautier, A.; Leroy, B.; Plancher, J.M.; Quesnel, Y.; Vanherck, J.C.; Markó, I.E. Mild and chemoselective catalytic deprotection of ketals and acetals using cerium(IV) ammonium nitrate. Tetrahedron 2003, 59, 8989–8999. [Google Scholar] [CrossRef]
- Fujioka, H.O.Y.; Hirose, H.; Murai, K.; Kita, Y. Mild and efficient removal of hydroxyethyl unit from 2-hydroxyethyl ether derivatives leading to alcohols. Org. Lett. 2005, 7, 3303–3306. [Google Scholar] [CrossRef]
- Fujioka, H.; Hirose, H.; Ohba, Y.; Murai, K.N.K.; Kita, Y. Cerium ammonium nitrate (CAN) for mild and efficient reagent to remove hydroxyethyl units from 2-hydroxyethyl ethers and 2-hydroxyethyl amines. Tetrahedron 2007, 63, 625–637. [Google Scholar] [CrossRef]
- Hanessian, S.; Wong, D.H.; Therien, M. Oxidation of Alcohols with N-Halosuccinimides-New and Efficient Variants. Synthesis 1981, 1981, 394–396. [Google Scholar] [CrossRef]
- Franck, G.; Brödner, K.; Helmchen, G. Enantioselective modular synthesis of cyclohexenones: Total syntheses of (+)-crypto- and (+)-infectocaryone. Org. Lett. 2010, 12, 3886–3889. [Google Scholar] [CrossRef] [PubMed]
- Spiess, S.W.C.; Franck, G.; Taquet, J.P.; Helmchen, G. Iridium-catalyzed asymmetric allylic substitutions—Very high regioselectivity and air stability with a catalyst derived from dibenzo[a,e]cyclooctatetraene and a phosphoramidite. Angew. Chem. Int. Ed. 2008, 47, 7652–7655. [Google Scholar] [CrossRef] [PubMed]
- Alexakis, A.; Polet, D. Very efficient phosphoramidite ligand for asymmetric iridium-catalyzed allylic alkylation. Org. Lett. 2004, 6, 3529–3532. [Google Scholar] [CrossRef] [PubMed]
- Streiff, S.W.C.; Schelwies, M.; Lipowsky, G.; Miller, N.; Helmchen, G. Carbocycles via enantioselective inter- and intramolecular iridium-catalysed allylic alkylations. Chem. Commun. 2005, 2005, 2957–2959. [Google Scholar] [CrossRef]
- Cowden, C.J.; Paterson, I. Asymmetric aldol reactions using boron enolates. Org. React. 1997, 51, 1. [Google Scholar]
- Kurasaki, H.; Okamoto, I.; Morita, N.; Tamura, O. A flexible approach to grandisine alkaloids: Total synthesis of grandisines B., D., and F. Chem. Eur. J. 2009, 15, 12754–12763. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, Z.; Yang, J.; Zhao, G.; Ma, B.; Xie, X.; She, X. Total Synthesis of (+)-Cryptocaryanone A. Synlett 2012, 23, 2129–2131. [Google Scholar]
- Trost, B.M.; Romero, A.G. Synthesis of optically active isoquinuclidines utilizing a diastereoselectivity control element. J. Org. Chem. 1986, 51, 2332–2342. [Google Scholar] [CrossRef]
- Audia, J.E.; Boisvert, L.; Patten, A.D.; Villalobos, A.; Danishefsky, S.J. Synthesis of two useful, enantiomerically pure derivatives of (S)-4-hydroxy-2-cyclohexenone. J. Org. Chem. 1989, 54, 3738–3740. [Google Scholar] [CrossRef]
- Jeroncic, L.O.; Cabal, M.P.; Danishefsky, S.J.; Shulte, G.M. On the diastereofacial selectivity of Lewis acid-catalyzed carbon-carbon bond forming reactions of conjugated cyclic enones bearing electron-withdrawing substituents at the.gamma.-position. J. Org. Chem. 1991, 56, 387–395. [Google Scholar] [CrossRef]
- Zhang, W.; Baranczak, A.; Sulikowski, G.A. Stereocontrolled assembly of the C3/C3’ dideoxy core of lomaiviticin A/B and congeners. Org. Lett. 2008, 10, 1939–1941. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, J.D.; Godfrey, A.A.; Taylor, R.J.K. The preparation of (−)-grandisine B from (+)-grandisine D; a biomimetic total synthesis or formation of an isolation artefact? Org. Lett. 2011, 13, 3976–3979. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, L.; Liu, X.; Jia, J.; Jiang, L.; Lin, J.; Chen, X. A new approach to asymmetric synthesis of infectocaryone. Org. Biomol. Chem. 2014, 12, 7603–7611. [Google Scholar] [CrossRef] [PubMed]
- Raban, M.; Mislow, K. Modern methods for the determination of optical purity. Top. Stereochem. 1967, 2, 199–230. [Google Scholar]
- Trost, B.M.; Belletire, J.L.; Godleski, S.; McDougal, P.G.; Balkovec, J.M. On the use of the O-methylmandelate ester for establishment of absolute configuration of secondary alcohols. J. Org. Chem. 1986, 51, 2370–2374. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and .alpha.-methoxy-.alpha.- trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Liu, X.; Jia, J.; Jia, Y.; Gu, H.; Luo, J.; Chen, X. A Flexible and Divergent Strategy to Flavonoids with a Chiral A-Ring Featuring Intramolecular Michael Addition: Stereoselective Synthesis of (+)-Cryptocaryone, (+)-Cryptogione F, and (+)-Cryptocaryanones A and B, as Well as (+)-Cryptochinones A and C. Org Lett. 2018, 20, 1945–1948. [Google Scholar] [CrossRef]
- Takahashi, K.; Morita, H.; Honda, T. Formal synthesis of (−)-oleocanthal by means of a SmI2-promoted intramolecular coupling of bromoalkyne with α,β-unsaturated ester. Tetrahedron Lett. 2012, 53, 3342–3345. [Google Scholar] [CrossRef]
- Wood, J.M.; Furkert, D.P.; Brimble, M.A. Synthesis of the 2-formylpyrrole spiroketal pollenopyrroside A and structural elucidation of xylapyrroside A, shensongine A and capparisine B. Org. Biomol. Chem. 2016, 14, 7659–7664. [Google Scholar] [CrossRef]
- Liu, X.; Chen, R.; Duan, F.; Jia, J.; Zhou, Y.; Chen, X. A concise synthesis of (+)-botryolide-E and its C-7 epimer. Tetrahedron Lett. 2017, 58, 3947–3950. [Google Scholar] [CrossRef]
- Burgess, E.M.; Penton, H.R., Jr.; Taylor, E.A. Thermal reactions of alkyl N-carbomethoxysulfamate esters. J. Org. Chem. 1973, 38, 26–31. [Google Scholar] [CrossRef]
- Yuan, P.; Liu, X.; Yang, X.; Zhang, Y.; Chen, X. Total Syntheses of (+)-Gabosine P, (+)-Gabosine Q, (+)-Gabosine E, (−)-Gabosine G, (−)-Gabosine I, (−)-Gabosine K, (+)-Streptol, and (−)-Uvamalol A by a Diversity-Oriented Approach Featuring Tunable Deprotection Manipulation. J. Org. Chem. 2017, 82, 3692–3701. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cytotoxicity (IC50) µM [Cell Line] | Ref. |
|---|---|---|
| 3 | 1.8 [KB], 2.0 [K562], 2.0 [K565-DOX], 0.04 [P-388], 1.6 [PC-3], 2.3 [DU-145], 3.4 [LNCaP], 9.35 [HCT116], 7.23 [HT-29], 6.52 [SW480], 9.03 [MDA-MB-231], 0.32 [HT-29], 1.5 [TOV-21G], 3 [SKOV3], 9.5 [TOV-112D] | [37,39,42,47,48,49] |
| 5 | 1.7 [KB], 0.08 [P-388], 24.3 [MCF-7], 11.0 [NCI-H460], 3.7 [SF-268] | [37,42,44] |
| 6 | 2.5 [KB], 10.9 [HT-29], 5.1 [MCF-7], 4.3 [NCI-H460], 5.0 [SF-268] | [39,42] |
| 7 | 2.1 [KB] | [42] |
| 16 | 2.17 [P-388] | [37] |
| Antimicrobial activity (MIC; µg/mL) | ||
| 3 | 25 [H37Rv] | [44] |
| 6 | 10 [Phytophthora capsici], 5 [Fusarium moniliforme] | [47] |
| Anti-inflammatory activity (IC50) µM | ||
| 2 | 2.0 [TNFa] | [46] |
| 7 | 2.0 [TNFa] | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Liu, Y.; Liu, X.; Chen, X.; Chen, R. Comprehensive Review of Recent Advances in Chiral A-Ring Flavonoid Containing Compounds: Structure, Bioactivities, and Synthesis. Molecules 2023, 28, 365. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28010365
Zhang C, Liu Y, Liu X, Chen X, Chen R. Comprehensive Review of Recent Advances in Chiral A-Ring Flavonoid Containing Compounds: Structure, Bioactivities, and Synthesis. Molecules. 2023; 28(1):365. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28010365
Chicago/Turabian StyleZhang, Changyue, Yanzhi Liu, Xiaojing Liu, Xiaochuan Chen, and Ruijiao Chen. 2023. "Comprehensive Review of Recent Advances in Chiral A-Ring Flavonoid Containing Compounds: Structure, Bioactivities, and Synthesis" Molecules 28, no. 1: 365. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28010365