
Charge-Shift Bonding Propensity in Halogen-Bonded BXY (B Is a Small Lewis Base H2O or NH3; X and Y Are Halogen Atoms) Complexes: An NBO/NRT/AIM Investigation
 ,
,
Abstract
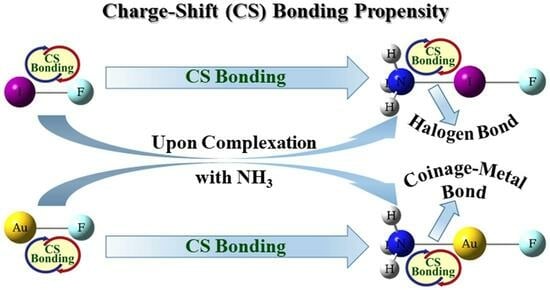
:
1. Introduction
2. Results and Discussion
2.1. Geometries and Dissociation Energies
2.2. 3c/4e ω-Bonding in a Halogen-Bonded Complex
2.3. Charge-Shift Bonding in a Halogen-Bonded Complex
2.4. CS Bonding Propensity in a Halogen-Bonded Complex
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shaik, S.; Maitre, P.; Sini, G.; Hiberty, P.C. The Charge-Shift Bonding Concept. Electron-Pair Bonds with Very Large Ionic-Covalent Resonance Energies. J. Am. Chem. Soc. 1992, 114, 7861–7866. [Google Scholar] [CrossRef]
- Shaik, S.; Danovich, D.; Wu, W.; Hiberty, P.C. Charge-Shift Bonding and Its Manifestations in Chemistry. Nat. Chem. 2009, 1, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Zhang, L.; Ying, F.; Wu, W.; Hiberty, P.C.; Shaik, S. Topology of Electron Charge Density for Chemical Bonds from Valence Bond Theory: A Probe of Bonding Types. Chem.-A Eur. J. 2009, 15, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Danovich, D.; Braida, B.; Wu, W.; Hiberty, P.C. New Landscape of Electron-Pair Bonding: Covalent, Ionic, and Charge-Shift Bonds. In Chemical Bond II: 100 Years Old and Getting Stronger; Mingos, D.M.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 170, pp. 169–212. [Google Scholar]
- Shaik, S.; Danovich, D.; Galbraith, J.M.; Braida, B.; Wu, W.; Hiberty, P.C. Charge-Shift Bonding: A New and Unique Form of Bonding. Angew. Chem.-Int. Ed. 2020, 59, 984–1001. [Google Scholar] [CrossRef]
- Radenkovic, S.; Danovich, D.; Shaik, S.; Hiberty, P.C.; Braida, B. The Nature of Bonding in Metal-Metal Singly Bonded Coinage Metal Dimers: Cu2, Ag2 and Au2. Comput. Theor. Chem. 2017, 1116, 195–201. [Google Scholar] [CrossRef]
- Dordevic, S.; Radenkovic, S.; Shaik, S.; Braida, B. On the Nature of the Bonding in Coinage Metal Halides. Molecules 2022, 27, 490. [Google Scholar] [CrossRef]
- Shaik, S.; Danovich, D.; Silvi, B.; Lauvergnat, D.L.; Hiberty, P.C. Charge-Shift Bonding-A Class of Electron-Pair Bonds That Emerges from Valence Bond Theory and Is Supported by the Electron Localization Function Approach. Chem.-A Eur. J. 2005, 11, 6358–6371. [Google Scholar] [CrossRef]
- Levine, D.S.; Head-Gordon, M. Energy Decomposition Analysis of Single Bonds within Kohn-Sham Density Functional Theory. Proc. Natl. Acad. Sci. USA 2017, 114, 12649–12656. [Google Scholar] [CrossRef]
- Joy, J.; Danovich, D.; Kaupp, M.; Shaik, S. Covalent vs Charge-Shift Nature of the Metal-Metal Bond in Transition Metal Complexes: A Unified Understanding. J. Am. Chem. Soc. 2020, 142, 12277–12287. [Google Scholar] [CrossRef]
- Braida, B.; Hiberty, P.C. The Essential Role of Charge-Shift Bonding in Hypervalent Prototype XeF2. Nat. Chem. 2013, 5, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Braida, B.; Ribeyre, T.; Hiberty, P.C. A Valence Bond Model for Electron-Rich Hypervalent Species: Application to SFn (n = 1, 2, 4), PF5, and ClF3. Chem.-A Eur. J. 2014, 20, 9643–9649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Su, Y.; Zou, X.; Fu, L.; Song, J.; Chen, D.; Sun, C. Charge-Shift Bonding in Xenon Hydrides: An NBO/NRT Investigation on HXeY∙∙∙HX (Y = Cl, Br, I.; X = OH, Cl, Br, I, CCH, CN) via H-Xe Blue-Shift Phenomena. Front. Chem. 2020, 8, 277. [Google Scholar] [CrossRef]
- Legon, A.C.; Walker, N.R. What’s in a Name? ‘Coinage-Metal’ Non-Covalent Bonds and Their Definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef]
- Song, J.; Fu, L.; Li, H.; Su, Y.; Wang, Q.; Zhang, G.; Wang, J. Equivalent and Complement of the omega-Bonding Model and Charge-Shift Bonding Model: A Natural Bond Orbital/Natural Resonance Theory/Atoms in Molecules Investigation via Cu/Ag/Au Bonding in BMX (B = H2O, H2S, NH3, and PH3; M = Cu, Ag, and Au; and X. = F, Cl, Br, and I). J. Phys. Chem. Lett. 2023, 14, 5226–5233. [Google Scholar] [PubMed]
- Gershoni-Poranne, R.; Chen, P. The Carbon-Nitrogen Bonds in Ammonium Compounds Are Charge Shift Bonds. Chem.-A Eur. J. 2017, 23, 4659–4668. [Google Scholar] [CrossRef]
- Joy, J.; Danovich, D.; Shaik, S. Nature of the Trigger Linkage in Explosive Materials Is a Charge-Shift Bond. J. Org. Chem. 2021, 86, 15588–15596. [Google Scholar] [CrossRef]
- Sarr, S.; Graton, J.; Montavon, G.; Pilme, J.; Galland, N. On the Interplay between Charge-Shift Bonding and Halogen Bonding. ChemPhysChem 2020, 21, 240–250. [Google Scholar] [CrossRef]
- Sarr, S.; Pilme, J.; Montavon, G.; Le Questel, J.-Y.; Galland, N. Astatine Facing Janus: Halogen Bonding vs. Charge-Shift Bonding. Molecules 2021, 26, 4568. [Google Scholar] [CrossRef]
- Weinhold, F.; Clark, R.L. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005; pp. 281–289. [Google Scholar]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Legon, A.C. A Reduced Radial Potential Energy Function for the Halogen Bond and the Hydrogen Bond in Complexes B∙∙∙XY and B∙∙∙HX, Where X and Y Are Halogen Atoms. Phys. Chem. Chem. Phys. 2014, 16, 12415–12421. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Legon, A.C. Systematic Behaviour of Electron Redistribution on Formation of Halogen-Bonded Complexes BXY, as Determined via XY Halogen Nuclear Quadrupole Coupling Constants. Phys. Chem. Chem. Phys. 2019, 21, 16914–16922. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Fundamentals and Applications. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2008; Volume 126, pp. 1–221. [Google Scholar]
- Legon, A.C. An Assessment of Radial Potential Functions for the Halogen Bond: Pseudo-Diatomic Models for Axially Symmetric Complexes B∙∙∙ClF (B=N2, CO, PH3, HCN, and NH3). Chempluschem 2021, 86, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Rezac, J. Introduction: Noncovalent Interactions. Chem. Rev. 2016, 116, 4911–4912. [Google Scholar] [CrossRef] [PubMed]
- Sachs, L. Applied Statistics: A Handbook of Techniques; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Bloemink, H.I.; Hinds, K.; Holloway, J.H.; Legon, A.C. Characterisation of A Pre-Reactive Intermediate in Gas-Phase Mixtures of Fluorine and Ammonia: The Rotational Spectrum of the H3N∙∙∙F2 Complex. Chem. Phys. Lett. 1995, 245, 598–604. [Google Scholar] [CrossRef]
- Karpfen, A. Charge-Transfer Complexes Between NH3 and the Halogens F2, ClF, and Cl2: An ab initio Study on the Intermolecular Interaction. J. Phys. Chem. A 2000, 104, 6871–6879. [Google Scholar] [CrossRef]
- Cooke, S.A.; Cotti, G.; Holloway, J.H.; Legon, A.C. Detection and Characterization of a Pre-Reactive Complex in a Mixture of Water and Fluorine: Rotational Spectrum of H2O∙∙∙F2. Angew. Chem.-Int. Ed. 1997, 36, 129–130. [Google Scholar] [CrossRef]
- Bloemink, H.I.; Evans, C.M.; Holloway, J.H.; Legon, A.C. Is the Gas-Phase Complex of Ammonia and Chlorine Monofluoride H3N∙∙∙ClF or [H3NCl]+ ∙∙∙F−? Evidence from Rotational Spectroscopy. Chem. Phys. Lett. 1996, 248, 260–268. [Google Scholar] [CrossRef]
- Davey, J.B.; Legon, A.C.; Thumwood, J.M.A. Interaction of Water and Dichlorine in the Gas Phase: An Investigation of H2O∙∙∙Cl2 by Rotational Spectroscopy and ab initio Calculations. J. Chem. Phys. 2001, 114, 6190–6202. [Google Scholar] [CrossRef]
- Legon, A.C.; Lister, D.G.; Thorn, J.C. Nonreactive Interaction of Ammonia and Molecular Chlorine-Rotational Spectrum of the Charge-Transfer Complex H3N∙∙∙Cl2. J. Chem. Soc.-Faraday Trans. 1994, 90, 3205–3212. [Google Scholar] [CrossRef]
- Legon, A.C.; Thumwood, J.M.A.; Waclawik, E.R. The Interaction of Water and Dibromine in the Gas Phase: An Investigation of the Complex H2O∙∙∙Br2 by Rotational Spectroscopy and ab initio Calculations. Chem.-A Eur. J. 2002, 8, 940–950. [Google Scholar] [CrossRef]
- Bloemink, H.I.; Legon, A.C.; Thorn, J. ‘Charge-Transfer’Complexes of Ammonia with Halogens. Nature of the Binding in H3N∙∙∙BrCl from its Rotational Spectrum. J. Chem. Soc.-Faraday Trans. 1995, 91, 781–787. [Google Scholar] [CrossRef]
- Cooke, S.A.; Cotti, G.; Evans, C.M.; Holloway, J.H.; Legon, A.C. The Pre-Reactive Complex H2O∙∙∙ClF Identified in Mixtures of Water Vapour and Chlorine Monofluoride by Rotational Spectroscopy. Chem. Commun. 1996, 20, 2327–2328. [Google Scholar] [CrossRef]
- Bloemink, H.I.; Legon, A.C. The Complex H3N∙∙∙Br2 Characterized in the Gas Phase by Rotational Spectroscopy. J. Chem. Phys. 1995, 103, 876–882. [Google Scholar] [CrossRef]
- Davey, J.B.; Legon, A.C. Rotational Spectroscopy of the Gas Phase Complex of Water and Bromine Monochloride in the Microwave Region: Geometry, Binding Strength and Charge Transfer. Phys. Chem. Chem. Phys. 2001, 3, 3006–3011. [Google Scholar] [CrossRef]
- Davey, J.B.; Legon, A.C.; Waclawik, E.R. An Investigation of the Gas-Phase Complex of Water and Iodine Monochloride by Microwave Spectroscopy: Geometry, Binding Strength and Electron Redistribution. Phys. Chem. Chem. Phys. 2000, 2, 1659–1665. [Google Scholar] [CrossRef]
- Guichemerre, M.; Chambaud, G.; Stoll, H. Electronic Structure and Spectroscopy of Monohalides of Metals of Group I-B. Chem. Phys. 2002, 280, 71–102. [Google Scholar] [CrossRef]
- Reynard, L.M.; Evans, C.J.; Gerry, M.C.L. The Pure Rotational Spectrum of AuI. J. Mol. Spectrosc. 2001, 205, 344–346. [Google Scholar] [CrossRef]
- Weinhold, F. Natural Bond Orbital Analysis: A Critical Overview of Relationships to Alternative Bonding Perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Weinhold, F. Natural Resonance Theory: III. Chemical Applications. J. Comput. Chem. 1998, 19, 628–646. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory: I. General Formalism. J. Comput. Chem. 1998, 19, 593–609. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory: II. Natural Bond Order and Valency. J. Comput. Chem. 1998, 19, 610–627. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The Characterization of Atomic Interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Silvi, B.; Gillespie, R.J.; Gatti, C. Electron Density Analysis. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 187–226. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 Energy Evaluation by Direct Nethods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-Optimized Gaussian Basis Sets for Molecular Response Calculations. J. Chem. Phys. 2010, 133, 169–211. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Headgordon, M.A. 5th-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations III. The Atoms Aluminum Through Argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. I. Correlation Consistent Basis Sets for the Post-D Group 13–15 Elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-D Group 16-18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Weinhold, F. NBOPro 6.0; Theoretical Chemistry Institute: Madison, WI, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B | NH3 | H2O | Free | |||
|---|---|---|---|---|---|---|
| XY | rB–X (Å) | rX–Y (Å) | rB–X (Å) | rX–Y (Å) | rX–Y (Å) | |
| FF | 2.595 (2.71) [29] | 1.416 (1.43) [30] | 2.649 (2.70) [31] | 1.407 | 1.401 (1.41) [30] | |
| ClF | 2.232 (2.37) [32] | 1.714 (1.70) [30] | 2.515 (2.54) [33] | 1.656 | 1.638 (1.63) [30] | |
| ClCl | 2.592 (2.73) [34] | 2.034 (2.00) [34] | 2.771 (2.81) [35] | 2.008 (2.01) [24] | 1.999 (1.99) [30] | |
| BrF | 2.292 (2.34) [25] | 1.829 (1.82) [25] | 2.493 | 1.780 | 1.758 (1.76) [25] | |
| BrCl | 2.469 (2.63) [36] | 2.203 (2.19) [36] | 2.698 (2.74) [37] | 2.155 (2.15) [24] | 2.138 (2.14) [25] | |
| BrBr | 2.538 (2.72) [38] | 2.332 (2.34) [38] | 2.757 (2.80) [39] | 2.292 (2.29) [24] | 2.279 (2.28) [25] | |
| IF | 2.443 | 1.976 | 2.585 | 1.941 | 1.920 | |
| ICl | 2.540 (2.54) [24] | 2.390 (2.39) [24] | 2.731 (2.78) [40] | 2.343 (2.34) [24] | 2.321 (2.32) [24] | |
| IBr | 2.583 (2.58) [24] | 2.530 (2.53) [24] | 2.781 (2.78) [24] | 2.484 (2.48) [24] | 2.465 (2.47) [24] | |
| II | 2.672 (2.67) [24] | 2.717 (2.72) [24] | 2.868 (2.87) [24] | 2.678 (2.68) [24] | 2.663 (2.66) [24] | |
| AuF | 1.995 | 1.909 | 2.038 | 1.893 | 1.902 (1.92) [41] | |
| AuCl | 2.029 | 2.199 | 2.079 | 2.178 | 2.174 (2.20) [41] | |
| AuBr | 2.040 | 2.310 | 2.094 | 2.289 | 2.284 (2.32) [41] | |
| AuI | 2.058 | 2.466 | 2.121 | 2.444 | 2.436 (2.47) [42] | |
| B | NH3 | H2O | |||
|---|---|---|---|---|---|
| XY | wI | wII | wI | wII | |
| FF | 98.88% | 0.85% | 99.85% | 0.15% | |
| ClF | 75.74% | 22.81% | 94.48% | 4.94% | |
| ClCl | 94.94% | 3.99% | 98.86% | 0.88% | |
| BrF | 70.47% | 28.80% | 90.55% | 8.67% | |
| BrCl | 87.69% | 10.60% | 97.23% | 2.26% | |
| BrBr | 91.06% | 7.26% | 98.06% | 1.51% | |
| IF | 67.37% | 32.27% | 86.53% | 12.82% | |
| ICl | 82.30% | 17.16% | 94.74% | 4.55% | |
| IBr | 85.87% | 13.41% | 96.16% | 3.20% | |
| II | 90.57% | 7.76% | 97.67% | 1.82% | |
| AuF | 49.59% | 49.26% | 65.68% | 33.22% | |
| AuCl | 62.87% | 36.27% | 76.69% | 22.00% | |
| AuBr | 66.17% | 32.85% | 79.05% | 19.51% | |
| AuI | 69.96% | 29.12% | 82.29% | 16.12% | |
| B | NH3 | H2O | Free | |||
|---|---|---|---|---|---|---|
| XY | DIN–X | DIX–Y | DIO–X | DIX–Y | DIX–Y | |
| FF | 0.151 | 1.229 | 0.077 | 1.269 | 1.286 | |
| ClF | 0.533 | 1.051 | 0.250 | 1.184 | 1.253 | |
| ClCl | 0.283 | 1.340 | 0.144 | 1.414 | 1.446 | |
| BrF | 0.563 | 0.976 | 0.316 | 1.087 | 1.175 | |
| BrCl | 0.414 | 1.221 | 0.208 | 1.350 | 1.415 | |
| BrBr | 0.368 | 1.265 | 0.186 | 1.371 | 1.419 | |
| IF | 0.549 | 0.902 | 0.344 | 0.982 | 1.074 | |
| ICl | 0.464 | 1.126 | 0.260 | 1.247 | 1.340 | |
| IBr | 0.432 | 1.186 | 0.237 | 1.305 | 1.386 | |
| II | 0.374 | 1.258 | 0.202 | 1.354 | 1.408 | |
| AuF | 0.913 | 0.919 | 0.720 | 0.976 | 1.098 | |
| AuCl | 0.849 | 1.244 | 0.652 | 1.318 | 1.464 | |
| AuBr | 0.831 | 1.311 | 0.632 | 1.390 | 1.533 | |
| AuI | 0.803 | 1.403 | 0.600 | 1.479 | 1.608 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Wang, M.; Xu, X.; Shao, Q.; Zhao, Y.; Zhang, G.; Sun, N. Charge-Shift Bonding Propensity in Halogen-Bonded BXY (B Is a Small Lewis Base H2O or NH3; X and Y Are Halogen Atoms) Complexes: An NBO/NRT/AIM Investigation. Molecules 2023, 28, 6212. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28176212
Song J, Wang M, Xu X, Shao Q, Zhao Y, Zhang G, Sun N. Charge-Shift Bonding Propensity in Halogen-Bonded BXY (B Is a Small Lewis Base H2O or NH3; X and Y Are Halogen Atoms) Complexes: An NBO/NRT/AIM Investigation. Molecules. 2023; 28(17):6212. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28176212
Chicago/Turabian StyleSong, Junjie, Mengyang Wang, Xiaocheng Xu, Qinghao Shao, Ying Zhao, Guiqiu Zhang, and Nan Sun. 2023. "Charge-Shift Bonding Propensity in Halogen-Bonded BXY (B Is a Small Lewis Base H2O or NH3; X and Y Are Halogen Atoms) Complexes: An NBO/NRT/AIM Investigation" Molecules 28, no. 17: 6212. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28176212