

The Microbial Degradation of Natural and Anthropogenic Phosphonates
 , , ,
, , ,
Abstract
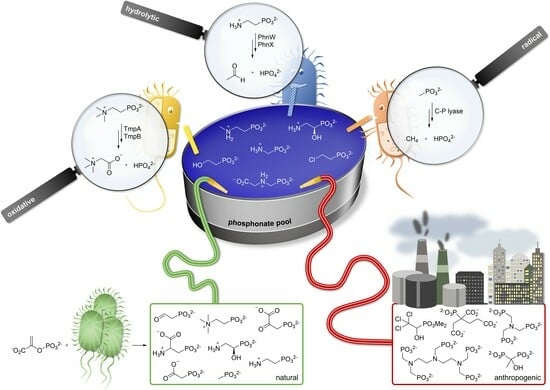
:
1. Introduction
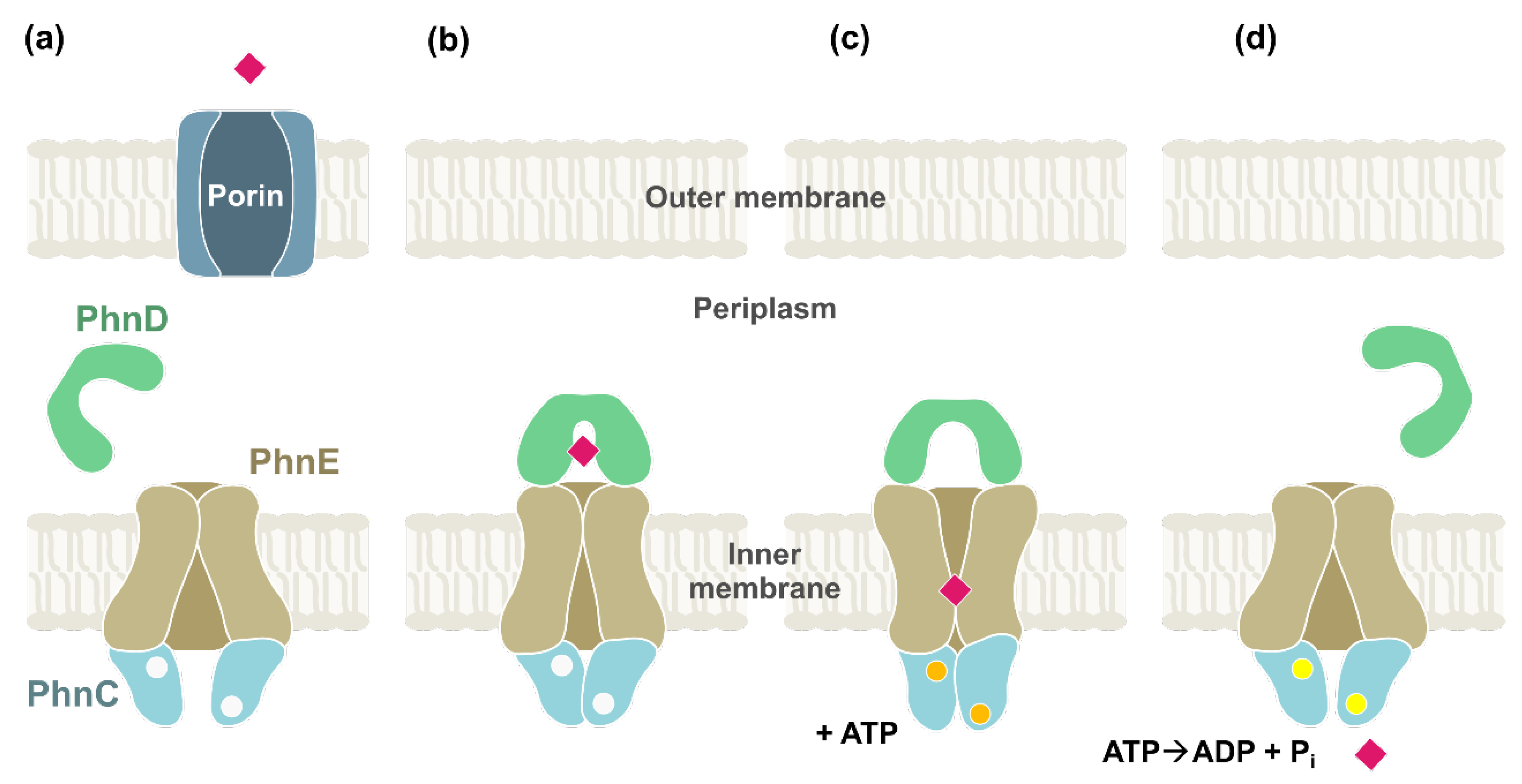
2. Bacterial Phosphonate Transporters
2.1. ABC Transporters
2.2. Symporters/Antiporters (Facilitators)
3. Hydrolytic Pathways for the Catabolism of Phosphonates
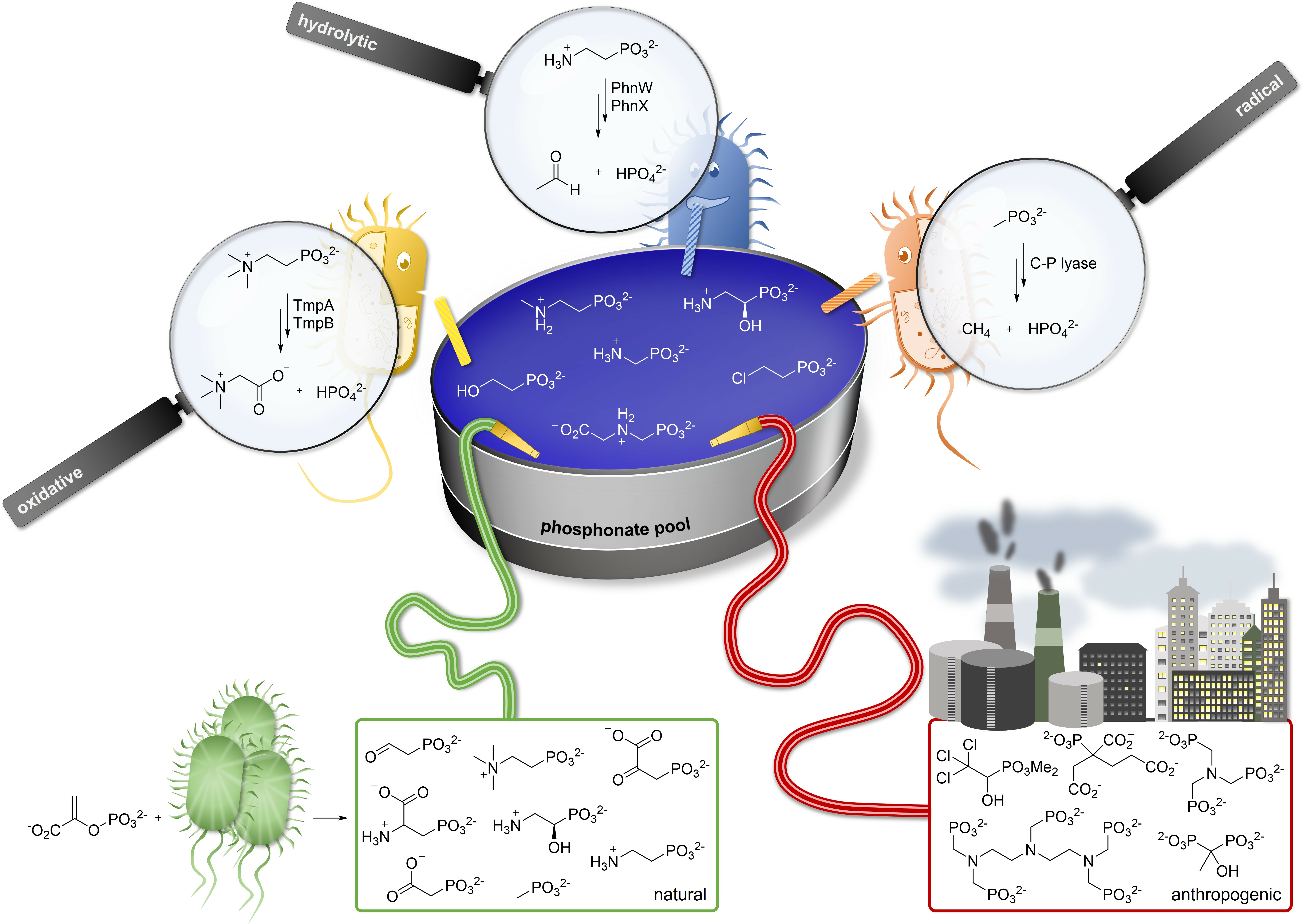
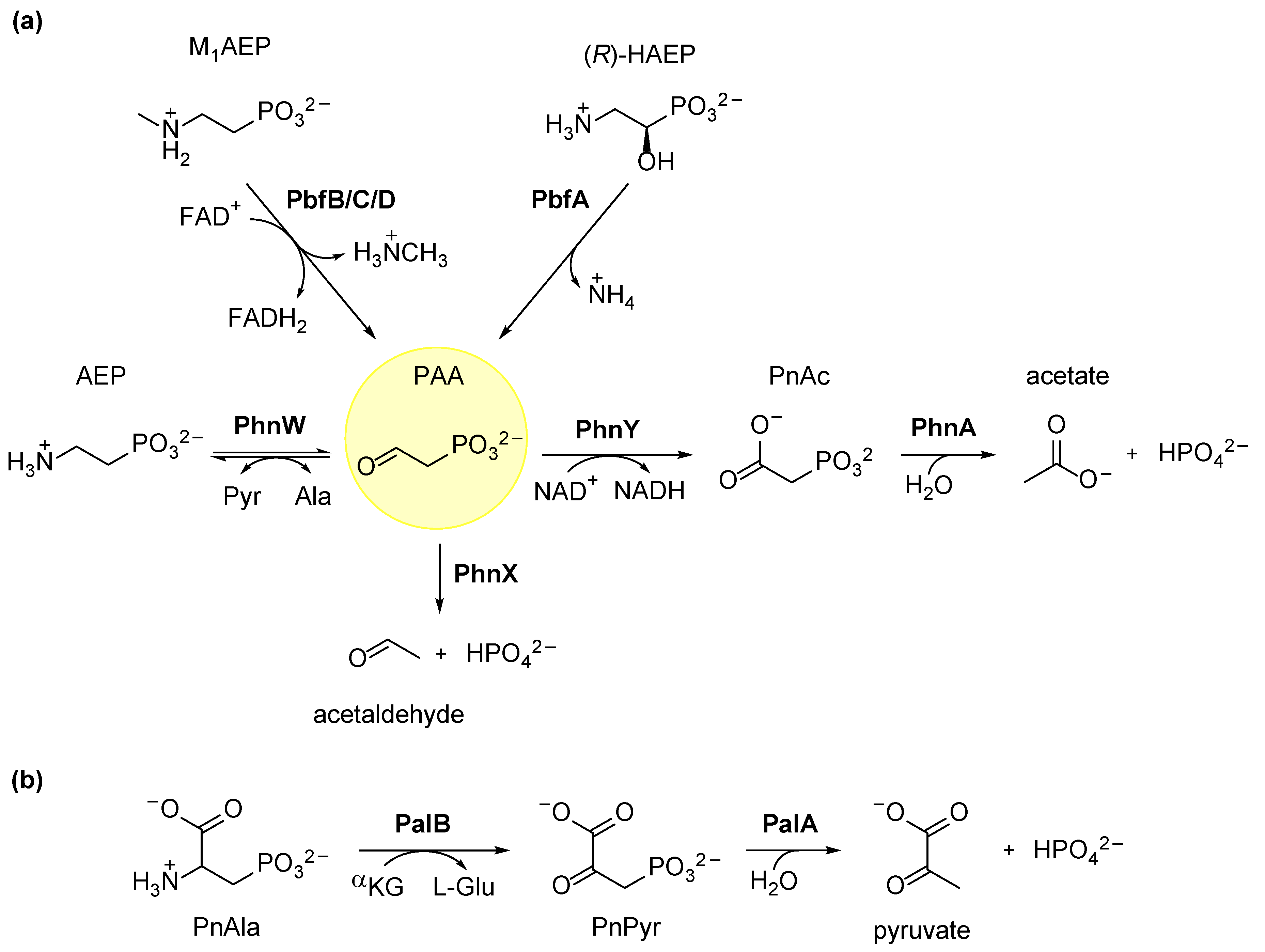
3.1. Degradation of AEP through the PhnWX and PhnWAY Pathways
3.2. Ancillary Enzymes Can Expand the Usefulness of AEP Degradation Pathways
3.3. Hydrolytic Degradation of Phosphonoalanine (PnAla)
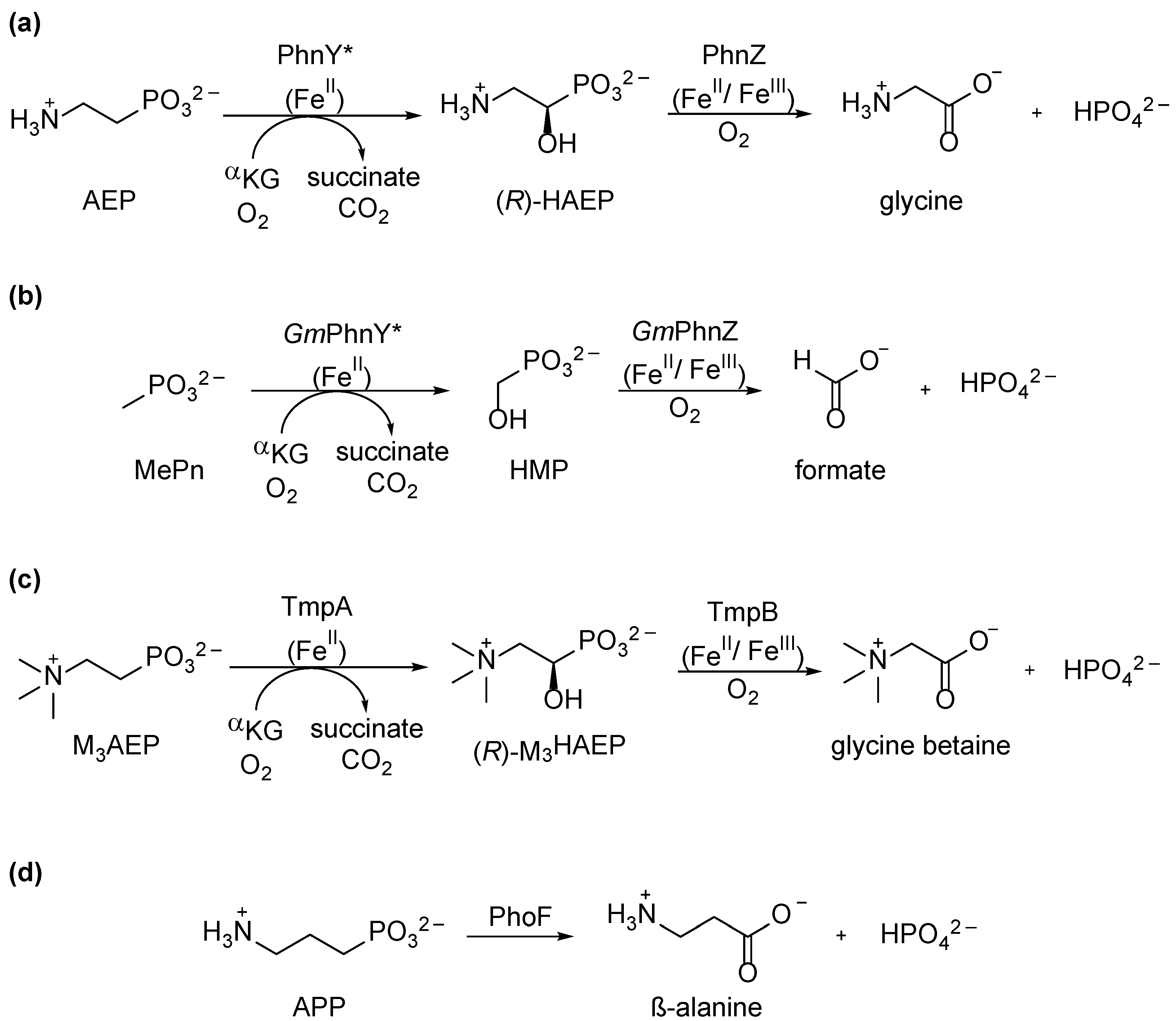
4. Oxidative Pathways for Phosphonate Catabolism
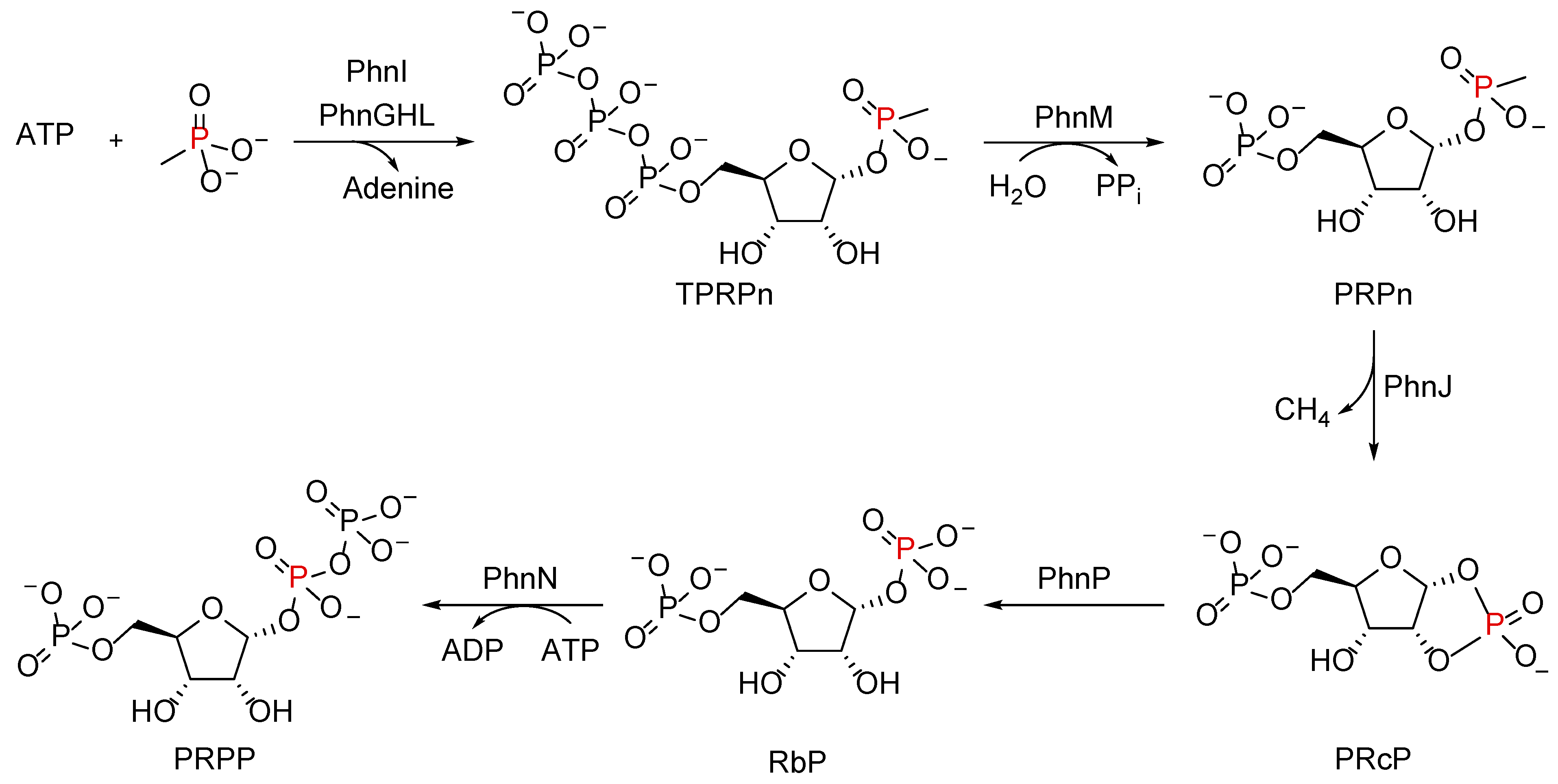
5. The C–P–Lyase Complex
5.1. Organization of the C–P Lyase Complex
5.2. Reaction Mechanism by the ‘Central’ C–P Lyase Subunits
5.3. Role of Nonessential Subunits in the C–P Lyase Function
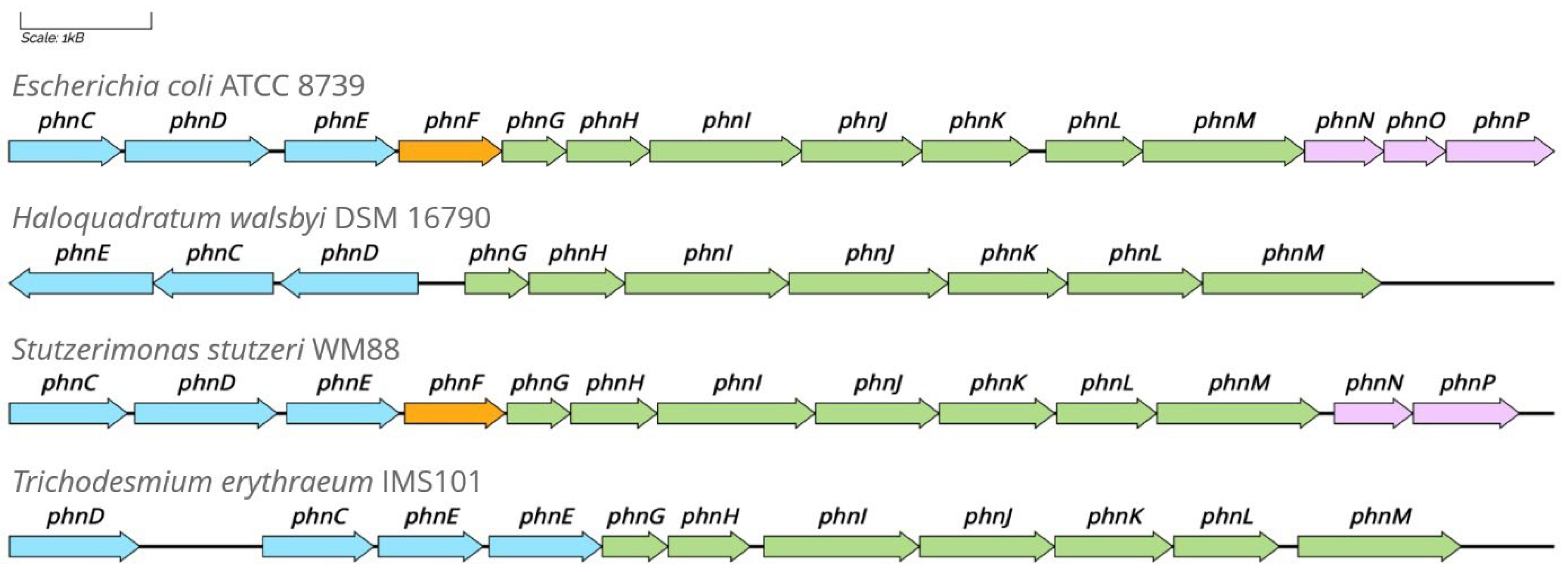
6. Genomic and Environmental Distribution of the Known Phosphonate Degradation Pathways
6.1. Studies in Marine Systems
6.2. Studies in Freshwater and Soil Systems
7. Regulation of Phosphonate Degradation Pathways
7.1. Phosphonate Catabolism Is Usually under the Pho Regulon Control
7.2. Substrate-Specific Hydrolytic Pathways Expression Can Be Phosphate-Independent and Substrate-Inducible
7.3. LysR-Like Transcriptional Regulator Proteins Are Recurrent in Operons for C–P Hydrolysis
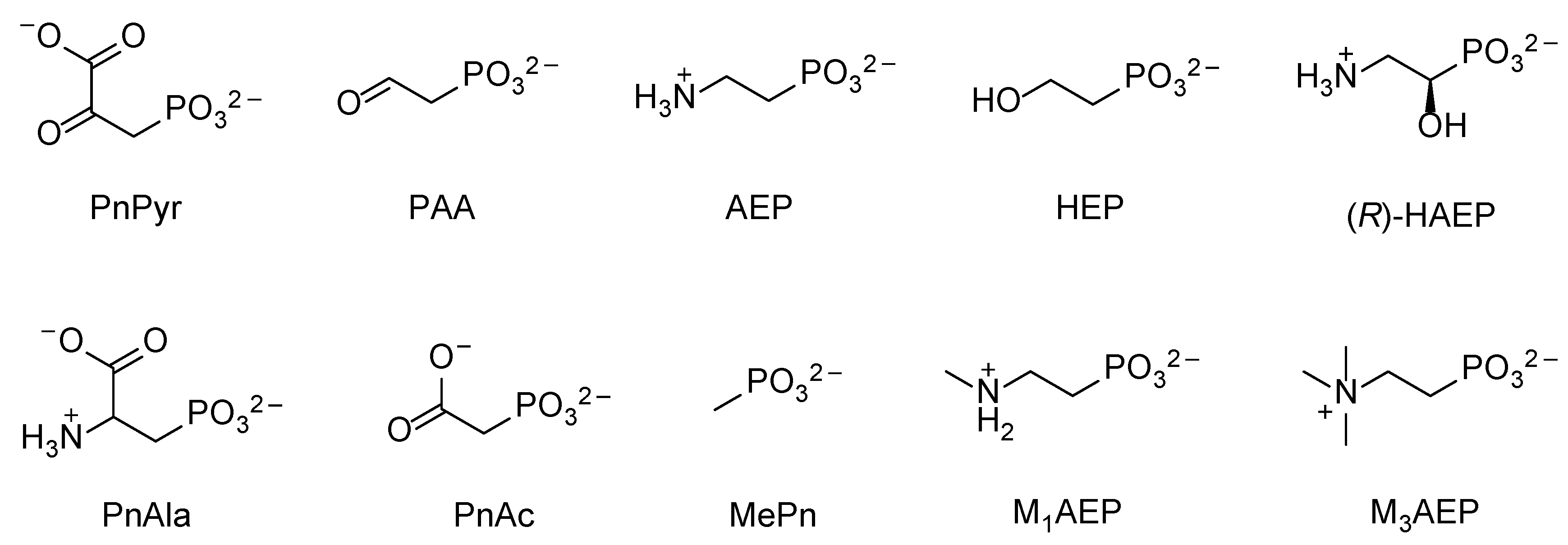
8. The Microbial Degradation of Man-Made Phosphonates
8.1. Polyphosphonates (PPns)
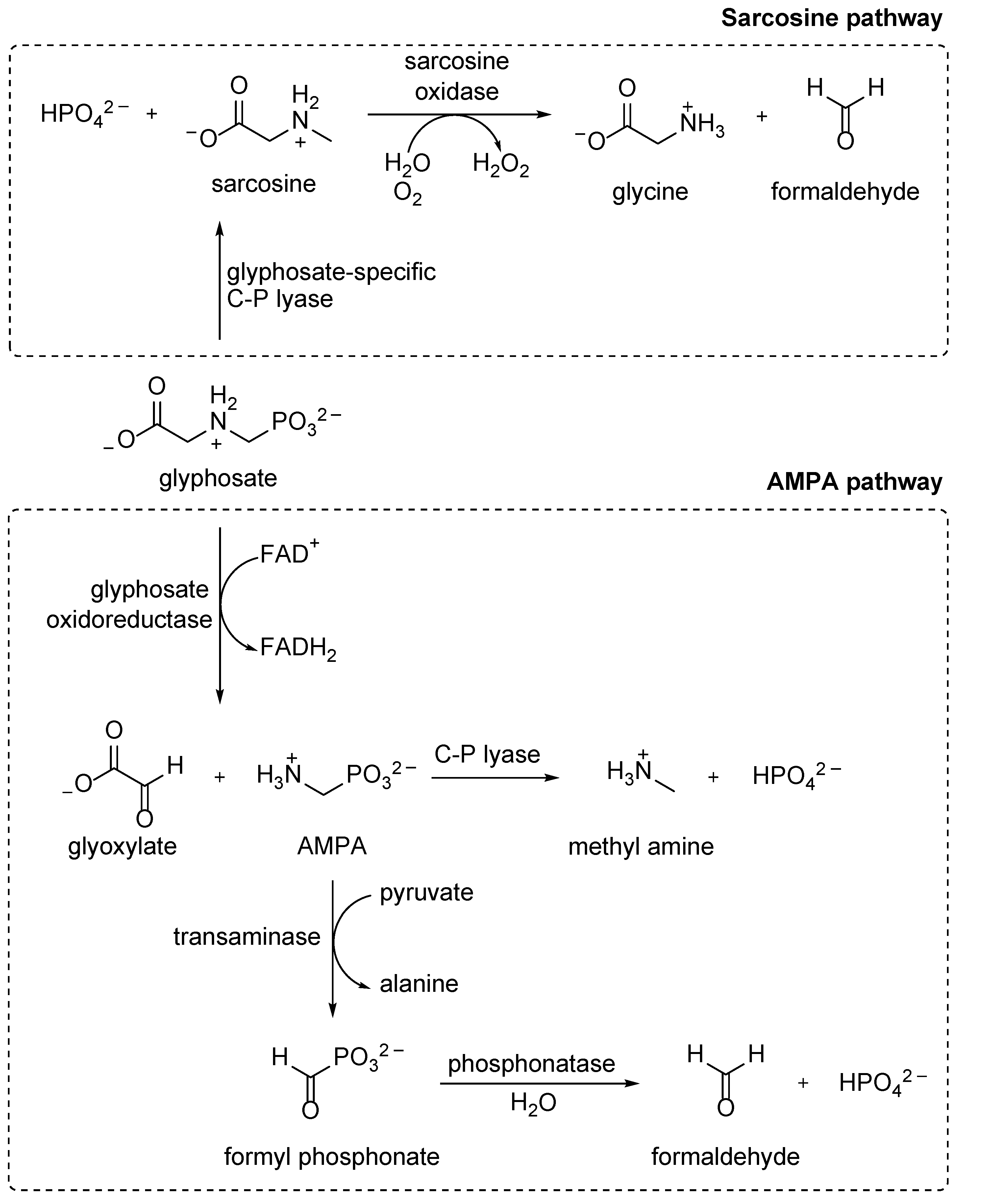
8.2. Glyphosate
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef]
- Black, S.; Morel, B.; Zapf, P. Verification of the Chemical Convention. Nature 1991, 351, 515–516. [Google Scholar] [CrossRef]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The Genes and Enzymes of Phosphonate Metabolism by Bacteria, and Their Distribution in the Marine Environment. Front. Microbiol. 2012, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Doroghazi, J.R.; Janga, S.C.; Zhang, J.K.; Circello, B.; Griffin, B.M.; Labeda, D.P.; Metcalf, W.W. Diversity and Abundance of Phosphonate Biosynthetic Genes in Nature. Proc. Natl. Acad. Sci. USA 2013, 110, 20759–20764. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Horsman, G.P. An Inventory of Early Branch Points in Microbial Phosphonate Biosynthesis. Microb. Genom. 2022, 8, 000781. [Google Scholar] [CrossRef]
- Wieczorek, D.; Żyszka-haberecht, B.; Kafka, A.; Lipok, J. Phosphonates as Unique Components of Plant Seeds—A Promising Approach to Use Phosphorus Profiles in Plant Chemotaxonomy. Int. J. Mol. Sci. 2021, 22, 11501. [Google Scholar] [CrossRef] [PubMed]
- Kilby, P.M.; Radda, G.K.; Wegener, G. 2-Aminoethylphosphonic Acid Is the Main Phosphorus Compound in Locust Hemolymph. Naturwissenschaften 1991, 78, 514–517. [Google Scholar] [CrossRef]
- Martinez, A.; Tyson, G.W.; DeLong, E.F. Widespread Known and Novel Phosphonate Utilization Pathways in Marine Bacteria Revealed by Functional Screening and Metagenomic Analyses. Environ. Microbiol. 2010, 12, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.L.; Ingall, E.D.; Benner, R. Marine Phosphorus Is Selectively Remineralized. Nature 1998, 393, 426. [Google Scholar] [CrossRef]
- Kolowith, L.C.; Ingall, E.D.; Benner, R. Composition and Cycling of Marine Organic Phosphorus. Limnol. Oceanogr. 2001, 46, 309–320. [Google Scholar] [CrossRef]
- Newman, R.H.; Tate, K.R. Soil Phosphorus Characterisation By 31P Nuclear Magnetic Resonance. Commun. Soil. Sci. Plant Anal. 1980, 11, 835–842. [Google Scholar] [CrossRef]
- Vestergren, J.; Vincent, A.G.; Jansson, M.; Persson, P.; Ilstedt, U.; Gröbner, G.; Giesler, R.; Schleucher, J. High-Resolution Characterization of Organic Phosphorus in Soil Extracts Using 2D 1H-31P NMR Correlation Spectroscopy. Environ. Sci. Technol. 2012, 46, 3950–3956. [Google Scholar] [CrossRef] [PubMed]
- Rott, E.; Steinmetz, H.; Metzger, J.W. Organophosphonates: A Review on Environmental Relevance, Biodegradability and Removal in Wastewater Treatment Plants. Sci. Total Environ. 2018, 615, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Kononova, S.V.; Nesmeyanova, M.A. Phosphonates and Their Degradation by Microorganisms. Biochemistry 2002, 67, 184–195. [Google Scholar] [PubMed]
- Fox, E.M.; Mendz, G.L. Phosphonate Degradation in Microorganisms. Enzyme Microb. Technol. 2006, 40, 145–150. [Google Scholar] [CrossRef]
- Sosa, O.A.; Repeta, D.J.; Ferrón, S.; Bryant, J.A.; Mende, D.R.; Karl, D.M.; DeLong, E.F. Isolation and Characterization of Bacteria That Degrade Phosphonates in Marine Dissolved Organic Matter. Front. Microbiol. 2017, 8, 1786. [Google Scholar] [CrossRef]
- Krzyśko-Lupicka, T.; Strof, W.; Kubsâ, K.; Skorupa, M.; Wieczorek, P.; Lejczak, B.; Kafarski, P. The Ability of Soil-Borne Fungi to Degrade Organophosphonate Carbon-to-Phosphorus Bonds. Appl. Microbiol. Biotechnol. 1997, 48, 549–552. [Google Scholar] [CrossRef]
- Klimek-Ochab, M. Phosphate-Independent Utilization of Phosphonoacetic Acid as Sole Phosphorus Source by a Psychrophilic Strain of Geomyces pannorum P15. Folia Microbiol. 2014, 59, 375–380. [Google Scholar] [CrossRef]
- Bolhuis, H.; Palm, P.; Wende, A.; Falb, M.; Rampp, M.; Rodriguez-Valera, F.; Pfeiffer, F.; Oesterhelt, D. The Genome of the Square Archaeon Haloquadratum walsbyi: Life at the Limits of Water Activity. BMC Genom. 2006, 7, 169. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Roth, J.R. Selfish Operons: Horizontal Transfer May Drive the Evolution of Gene Clusters. Genetics 1996, 143, 184–1860. [Google Scholar] [CrossRef]
- Manav, M.C.; Sofos, N.; Hove-Jensen, B.; Brodersen, D.E. The Abc of Phosphonate Breakdown: A Mechanism for Bacterial Survival. BioEssays 2018, 40, e1800091. [Google Scholar] [CrossRef] [PubMed]
- Stosiek, N.; Klimek-Ochab, M. Carbon-Phosphorus Lyase—The State of the Art. Appl. Biochem. Biotechnol. 2020, 190, 1525–1552. [Google Scholar] [CrossRef] [PubMed]
- Pallitsch, K.; Zechel, D.L. The Functional Importance of Bacterial Oxidative Phosphonate Pathways. Biochem. Soc. Trans. 2023, 51, 487–499. [Google Scholar] [CrossRef]
- Ternan, N.G.; Grath, J.W.M.; Mullan, G.M.; Quinn, J.P. Organophosphonates: Occurrence, Synthesis and Biodegradation by Microorganisms. World J. Microbiol. Biotechnol. 1998, 14, 635–647. [Google Scholar] [CrossRef]
- McGrath, J.W.; Chin, J.P.; Quinn, J.P. Organophosphonates Revealed: New Insights into the Microbial Metabolism of Ancient Molecules. Nat. Rev. Microbiol. 2013, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.F.; Liras, P. Molecular Mechanisms of Phosphate Sensing, Transport and Signalling in Streptomyces and Related Actinobacteria. Int. J. Mol. Sci. 2021, 22, 1129. [Google Scholar] [CrossRef]
- Mulligan, C.; Fischer, M.; Thomas, G.H. Tripartite ATP-Independent Periplasmic (TRAP) Transporters in Bacteria and Archaea. FEMS Microbiol. Rev. 2011, 35, 68–86. [Google Scholar] [CrossRef]
- Rugg, S.J. Characterisation of Bacterial Phosphonate Transporters and Their Application as Glyphosate Biosensors. Ph.D. Thesis, University of York, York, UK, 2018. [Google Scholar]
- Alicea, I.; Marvin, J.S.; Miklos, A.E.; Ellington, A.D.; Looger, L.L.; Schreiter, E.R. Structure of the Escherichia coli Phosphonate Binding Protein PhnD and Rationally Optimized Phosphonate Biosensors. J. Mol. Biol. 2011, 414, 356–369. [Google Scholar] [CrossRef]
- Stasi, R.; Neves, H.I.; Spira, B. Phosphate Uptake by the Phosphonate Transport System PhnCDE. BMC Microbiol. 2019, 19, 79. [Google Scholar] [CrossRef]
- Rao, N.N.; Torriani, A. Molecular Aspects of Phosphate Transport in Escherichia coli. Mol. Microbiol. 1990, 4, 1083–1090. [Google Scholar] [CrossRef]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal Structures Explain Functional Properties of Two E. coli Porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Rizk, S.S.; Cuneo, M.J.; Hellinga, H.W. Identification of Cognate Ligands for the Escherichia coli phnD Protein Product and Engineering of a Reagentless Fluorescent Biosensor for Phosphonates. Protein Sci. 2006, 15, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, M.; Diefenbach, M.; Biesalski, M.; Laube, B. Electrical Sensing of Phosphonates by Functional Coupling of Phosphonate Binding Protein PhnD to Solid-State Nanopores. ACS Sens. 2020, 5, 234–241. [Google Scholar] [CrossRef]
- Hilderbrand, R.L.; Henderson, T. Phosphonic Acids in Nature. In The Role of Phosphonates in Living Systems; Hilderbrand, R.L., Ed.; CRC Press: Boca Raton, FL, USA, 1983; pp. 5–28. [Google Scholar]
- Lockwood, S.; Greening, C.; Baltar, F.; Morales, S.E. Global and Seasonal Variation of Marine Phosphonate Metabolism. ISME J. 2022, 16, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Metcalf, W.W.; Lee, K.-S.; Wanner, B.L. Molecular Cloning, Mapping, and Regulation of Pho Regulon Genes for Phosphonate Breakdown by the Phosphonatase Pathway of Salmonella typhimurium LT2. J. Bacteriol. 1995, 177, 6411–6421. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.D.; Baker, A.S.; Dunaway-Mariano, D.; Metcalf, W.W.; Wanner, B.L.; Martin, B.M. The 2-Aminoethylphosphonate-Specific Transaminase of the 2-Aminoethylphosphonate Degradation Pathway. J. Bacteriol. 2002, 184, 4134–4140. [Google Scholar] [CrossRef]
- Murphy, A.R.J.; Scanlan, D.J.; Chen, Y.; Adams, N.B.P.; Cadman, W.A.; Bottrill, A.; Bending, G.; Hammond, J.P.; Hitchcock, A.; Wellington, E.M.H.; et al. Transporter Characterisation Reveals Aminoethylphosphonate Mineralisation as a Key Step in the Marine Phosphorus Redox Cycle. Nat. Commun. 2021, 12, 4554. [Google Scholar] [CrossRef]
- Borisova, S.A.; Christman, H.D.; Mourey Metcalf, M.E.; Zulkepli, N.A.; Zhang, J.K.; Van Der Donk, W.A.; Metcalf, W.W. Genetic and Biochemical Characterization of a Pathway for the Degradation of 2-Aminoethylphosphonate in Sinorhizobium meliloti 1021. J. Biol. Chem. 2011, 286, 22283–22290. [Google Scholar] [CrossRef]
- Voegele, R.T.; Bardin, S.; Finan, T.M. Characterization of the Rhizobium (Sinorhizobium) meliloti High- and Low-Affinity Phosphate Uptake Systems. J. Bacteriol. 1997, 179, 7226–7232. [Google Scholar] [CrossRef]
- Wicke, D.; Schulz, L.M.; Lentes, S.; Scholz, P.; Poehlein, A.; Gibhardt, J.; Daniel, R.; Ischebeck, T.; Commichau, F.M. Identification of the First Glyphosate Transporter by Genomic Adaptation. Environ. Microbiol. 2019, 21, 1287–1305. [Google Scholar] [CrossRef]
- Chen, C.C.H.; Zhang, H.; Kim, A.D.; Howard, A.; Sheldrick, G.M.; Mariano-Dunaway, D.; Herzberg, O. Degradation Pathway of the Phosphonate Ciliatine: Crystal Structure of 2-Aminoethylphosphonate Transaminase. Biochemistry 2002, 41, 13162–13169. [Google Scholar] [CrossRef]
- Jia, H.; Chen, Y.; Chen, Y.; Liu, R.; Zhang, Q.; Bartlam, M. Structural Characterization of a 2-Aminoethylphosphonate:Pyruvate Aminotransferase from Pseudomonas aeruginosa PAO1. Biochem. Biophys. Res. Commun. 2021, 552, 114–119. [Google Scholar] [CrossRef]
- Zangelmi, E.; Stanković, T.; Malatesta, M.; Acquotti, D.; Pallitsch, K.; Peracchi, A. Discovery of a New, Recurrent Enzyme in Bacterial Phosphonate Degradation: (R)-1-Hydroxy-2-Aminoethylphosphonate Ammonia-Lyase. Biochemistry 2021, 60, 1214–1225. [Google Scholar] [CrossRef]
- Zangelmi, E.; Ruffolo, F.; Dinhof, T.; Gerdol, M.; Malatesta, M.; Chin, J.P.; Rivetti, C.; Secchi, A.; Pallitsch, K.; Peracchi, A. Deciphering the Role of Recurrent FAD-Dependent Enzymes in Bacterial Phosphonate Catabolism. iScience, 2023; accepted for publication. [Google Scholar] [CrossRef]
- La Nauze, J.M.; Rosenberg, H. The Breakdown of Aminoethylphosphonate by Cell-Free Extracts of Bacillus cereus. Biochim. Biophys. Acta 1967, 148, 811–813. [Google Scholar] [CrossRef]
- La Nauze, J.M.; Rosenberg, H.; Shaw, D.C. The Enzymic Cleavage of the Carbon-Phosphorus Bond: Purification and Properties of Phosphonatase. Biochim. Biophys. Acta 1970, 212, 332–350. [Google Scholar] [CrossRef] [PubMed]
- Dumora, C.; Lacoste, A.-M.; Cassaigne, A. Phosphonoacetaldehyde Hydrolase from Pseudomonas aeuginosa: Purification Properties and Comparison with Bacillus cereus Enzyme. Biochim. Biophys. Acta 1989, 997, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.S.; Ciocci, M.J.; Metcalf, W.W.; Kim, J.; Babbitt, P.C.; Wanner, B.L.; Martin, B.M.; Dunaway-Mariano, D. Insights into the Mechanism of Catalysis by the P-C Bond-Cleaving Enzyme Phosphonoacetaldehyde Hydrolase Derived from Gene Sequence Analysis and Mutagenesis. Biochemistry 1998, 37, 9305–9315. [Google Scholar] [CrossRef]
- Morais, M.C.; Zhang, W.; Baker, A.S.; Zhang, G.; Dunaway-Mariano, D.; Allen, K.N. The Crystal Structure of Bacillus cereus Phosphonoacetaldehyde Hydrolase: Insight into Catalysis of Phosphorus Bond Cleavage and Catalytic Diversification within the HAD Enzyme Superfamily. Biochemistry 2000, 39, 10385–10396. [Google Scholar] [CrossRef]
- Cui, Y.; Lin, X.; Zhang, H.; Lin, L.; Lin, S. PhnW-PhnX Pathway in Dinoflagellates Not Functional to Utilize Extracellular Phosphonates. Front. Mar. Sci. 2016, 2, 120. [Google Scholar] [CrossRef]
- Kulakova, A.N.; Kulakov, L.A.; Akulenko, N.V.; Ksenzenko, V.N.; Hamilton, J.T.G.; Quinn, J.P. Structural and Functional Analysis of the Phosphonoacetate Hydrolase (phnA) Gene Region in Pseudomonas fluorescens 23F. J. Bacteriol. 2001, 183, 3268–3275. [Google Scholar] [CrossRef]
- Cooley, N.A.; Kulakova, A.N.; Villarreal-Chiu, J.F.; Gilbert, J.A.; McGrath, J.W.; Quinn, J.P. Phosphonoacetate Biosynthesis: In Vitro Detection of a Novel NADP+-Dependent Phosphonoacetaldehyde-Oxidizing Activity in Cell-Extracts of a Marine Roseobacter. Microbiology 2011, 80, 335–340. [Google Scholar] [CrossRef]
- Kim, A.; Benning, M.M.; OkLee, S.; Quinn, J.; Martin, B.M.; Holden, H.M.; Dunaway-Mariano, D. Divergence of Chemical Function in the Alkaline Phosphatase Superfamily: Structure and Mechanism of the P-C Bond Cleaving Enzyme Phosphonoacetate Hydrolase. Biochemistry 2011, 50, 3481–3494. [Google Scholar] [CrossRef] [PubMed]
- Mcmullan, G.; Quinn, J.P. Detection of a Novel Carbon-Phosphorus Bond Cleavage Activity in Cell-Free Extracts of an Environmental Pseudomonas fluorescens Isolate. Biochem. Biophys. Res. Commun. 1992, 184, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Mcmullan, G.; Harrington, F.; Quinn, J.P. Metabolism of Phosphonoacetate as the Sole Carbon and Phosphorus Source by an Environmental Bacterial Isolate. Appl. Environ. Microbiol. 1992, 58, 1364–1366. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.W.; Wisdom, G.B.; McMullan, G.; Larkin, M.J.; Quinn, J.P. The Purification and Properties of Phosphonoacetate Hydrolase, a Novel Carbon-Phosphorus Bond-Cleavage Enzyme from Pseudomonas fluorescens 23F. Eur. J. Biochem. 1995, 234, 225–230. [Google Scholar] [CrossRef]
- Panas, P.; Ternan, N.G.; Dooley, J.S.G.; McMullan, G. Detection of Phosphonoacetate Degradation and phnA Genes in Soil Bacteria from Distinct Geographical Origins Suggest Its Possible Biogenic Origin. Environ. Microbiol. 2006, 8, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Burdett, H.; Temperton, B.; Wick, R.; Snelling, D.; McGrath, J.W.; Quinn, J.P.; Munn, C.; Gilbert, J.A. Evidence for Phosphonate Usage in the Coral Holobiont. ISME J. 2010, 4, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Caescu, C.I.; Vidal, O.; Krzewinski, F.; Artenie, V.; Bouquelet, S. Bifidobacterium longum Requires a Fructokinase (Frk; ATP:D-Fructose 6-Phosphotransferase, EC 2.7.1.4) for Fructose Catabolism. J. Bacteriol. 2004, 186, 6515–6525. [Google Scholar] [CrossRef]
- Kentache, T.; Thabault, L.; Peracchi, A.; Frédérick, R.; Bommer, G.T.; Van Schaftingen, E. The Putative Escherichia coli Dehydrogenase YjhC Metabolises Two Dehydrated Forms of N-Acetylneuraminate Produced by Some Sialidases. Biosci. Rep. 2020, 40, BSR20200927. [Google Scholar] [CrossRef]
- Quin, L.D.; Quin, G.S. Screening for Carbon-Bound Phosphorus in Marine Animals by High-Resolution 31P-NMR Spectroscopy: Coastal and Hydrothermal Vent Invertebrates. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 128, 173–185. [Google Scholar] [CrossRef]
- Ternan, N.G.; Hamilton, J.; Quinn, J.P. Initial in Vitro Characterisation of Phosphonopyruvate Hydrolase, a Novel Phosphate Starvation-Independent, Carbon-Phosphorus Bond Cleavage Enzyme in Burkholderia cepacia Pal6. Arch. Microbiol. 2000, 173, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kulakova, A.N.; Wisdom, G.B.; Kulakov, L.A.; Quinn, J.P. The Purification and Characterization of Phosphonopyruvate Hydrolase, a Novel Carbon-Phosphorus Bond Cleavage Enzyme from Variovorax sp. Pal2. J. Biol. Chem. 2003, 278, 23426–23431. [Google Scholar] [CrossRef] [PubMed]
- Kulakova, A.N.; Kulakov, L.A.; Villarreal-Chiu, J.F.; Gilbert, J.A.; McGrath, J.W.; Quinn, J.P. Expression of the Phosphonoalanine-Degradative Gene Cluster from Variovorax sp. Pal2 Is Induced by Growth on Phosphonoalanine and Phosphonopyruvate. FEMS Microbiol. Lett. 2009, 292, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Van Staalduinen, L.M.; McSorley, F.R.; Schiessl, K.; Séguin, J.; Wyatt, P.B.; Hammerschmidt, F.; Zechel, D.L.; Jia, Z. Crystal Structure of PhnZ in Complex with Substrate Reveals a Di-Iron Oxygenase Mechanism for Catabolism of Organophosphonates. Proc. Natl. Acad. Sci. USA 2014, 111, 5171–5176. [Google Scholar] [CrossRef]
- McSorley, F.R.; Wyatt, P.B.; Martinez, A.; DeLong, E.F.; Hove-Jensen, B.; Zechel, D.L. PhnY and PhnZ Comprise a New Oxidative Pathway for Enzymatic Cleavage of a Carbon—Phosphorus Bond. J. Am. Chem. Soc. 2012, 134, 8364–8367. [Google Scholar] [CrossRef]
- Wörsdörfer, B.; Lingaraju, M.; Yennawar, N.H.; Boal, A.K.; Krebs, C.; Bollinger, J.M.; Pandelia, M.E. Organophosphonate-Degrading PhnZ Reveals an Emerging Family of HD Domain Mixed-Valent Diiron Oxygenases. Proc. Natl. Acad. Sci. USA 2013, 110, 18874–18879. [Google Scholar] [CrossRef]
- Langton, M.; Sun, S.; Ueda, C.; Markey, M.; Chen, J.; Paddy, I.; Jiang, P.; Chin, N.; Milne, A.; Pandelia, M.E. The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries. Catalysts 2020, 10, 1191. [Google Scholar] [CrossRef]
- Bollinger, J.M.; Diao, Y.; Matthews, M.L.; Xing, G.; Krebs, C. Myo-Inositol Oxygenase: A Radical New Pathway for O2 and C–H Activation at a Nonheme Diiron Cluster. Dalton Trans. 2009, 905–914. [Google Scholar] [CrossRef]
- Gama, S.R.; Vogt, M.; Kalina, T.; Hupp, K.; Hammerschmidt, F.; Pallitsch, K.; Zechel, D.L. An Oxidative Pathway for Microbial Utilization of Methylphosphonic Acid as a Phosphate Source. ACS Chem. Biol. 2019, 14, 735–741. [Google Scholar] [CrossRef]
- Sosa, O.A.; Casey, J.R.; Karl, D.M. Methylphosphonate Oxidation in Prochlorococcus Strain MIT9301 Supports Phosphate Acquisition, Formate Excretion, and Carbon Assimilation into Purines. Appl. Environ. Microbiol. 2019, 85, e00289-19. [Google Scholar] [CrossRef]
- Rajakovich, L.J.; Pandelia, M.E.; Mitchell, A.J.; Chang, W.C.; Zhang, B.; Boal, A.K.; Krebs, C.; Bollinger, J.M. A New Microbial Pathway for Organophosphonate Degradation Catalyzed by Two Previously Misannotated Non-Heme-Iron Oxygenases. Biochemistry 2019, 58, 1627–1647. [Google Scholar] [CrossRef] [PubMed]
- Rajakovich, L.J.; Zhang, B.; McBride, M.J.; Boal, A.K.; Krebs, C.; Martin Bollinger, J. Emerging Structural and Functional Diversity in Proteins With Dioxygen-Reactive Dinuclear Transition Metal Cofactors. In Comprehensive Natural Products III; Liu, H.-W., Begley, T.P., Eds.; Elsevier: Oxford, UK, 2020; pp. 215–250. [Google Scholar]
- Langton, M.; Appell, M.; Koob, J.; Pandelia, M.-E. Domain Fusion of Two Oxygenases Affords Organophosphonate Degradation in Pathogenic Fungi. Biochemistry 2022, 61, 956–962. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Zechel, D.L.; Jochimsen, B. Utilization of Glyphosate as Phosphate Source: Biochemistry and Genetics of Bacterial Carbon-Phosphorus Lyase. Microbiol. Mol. Biol. Rev. 2014, 78, 176–197. [Google Scholar] [CrossRef] [PubMed]
- Nowack, B. Environmental Chemistry of Phosphonates. Water Res. 2003, 37, 2533–2546. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, V.; Gill, J.P.K.; Datta, S.; Singh, S.; Dhaka, V.; Kapoor, D.; Wani, A.B.; Dhanjal, D.S.; Kumar, M.; et al. Herbicide Glyphosate: Toxicity and Microbial Degradation. Int. J. Environ. Res. Public. Health 2020, 17, 7519. [Google Scholar] [CrossRef] [PubMed]
- Ingall, E.D. Making Methane. Nat. Geosci. 2008, 1, 420. [Google Scholar] [CrossRef]
- Chin, J.P.; McGrath, J.W.; Quinn, J.P. Microbial Transformations in Phosphonate Biosynthesis and Catabolism, and Their Importance in Nutrient Cycling. Curr. Opin. Chem. Biol. 2016, 31, 50–57. [Google Scholar] [CrossRef]
- Wackett, L.P.; Shames, S.L.; Venditti, C.P.; Walsh, C.T. Bacterial Carbon-Phosphorus Lyase: Products, Rates, and Regulation of Phosphonic and Phosphinic Acid Metabolism. J. Bacteriol. 1987, 169, 710–717. [Google Scholar] [CrossRef]
- Seweryn, P.; Van, L.B.; Kjeldgaard, M.; Russo, C.J.; Passmore, L.A.; Hove-Jensen, B.; Jochimsen, B.; Brodersen, D.E. Structural Insights into the Bacterial Carbon-Phosphorus Lyase Machinery. Nature 2015, 525, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Amstrup, S.K.; Ong, S.C.; Sofos, N.; Karlsen, J.L.; Skjerning, R.B.; Boesen, T.; Enghild, J.J.; Hove-Jensen, B.; Brodersen, D.E. Structural Remodelling of the Carbon–Phosphorus Lyase Machinery by a Dual ABC ATPase. Nat. Commun. 2023, 14, 1001. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; Wanner, B.L. Evidence for a Fourteen-Gene, PhnC to PhnP Locus for Phosphonate Metabolism in Escherichia coli. Gene 1993, 129, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Elgorriaga, A.; Amrhein, N. Evidence for Two Distinct Phosphonate-Degrading Enzymes (C-P Lyases) in Arthrobacter sp. GLP-1. Biodegradation 1991, 2, 53–59. [Google Scholar] [CrossRef]
- Sviridov, A.V.; Shushkova, T.V.; Zelenkova, N.F.; Vinokurova, N.G.; Morgunov, I.G.; Ermakova, I.T.; Leontievsky, A.A. Distribution of Glyphosate and Methylphosphonate Catabolism Systems in Soil Bacteria Ochrobactrum anthropi and Achromobacter sp. Appl. Microbiol. Biotechnol. 2012, 93, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Sosa, O.A.; Repeta, D.J.; DeLong, E.F.; Ashkezari, M.D.; Karl, D.M. Phosphate-Limited Ocean Regions Select for Bacterial Populations Enriched in the Carbon–Phosphorus Lyase Pathway for Phosphonate Degradation. Environ. Microbiol. 2019, 21, 2402–2414. [Google Scholar] [CrossRef]
- Karl, D.M.; Beversdorf, L.; Björkman, K.M.; Church, M.J.; Martinez, A.; Delong, E.F. Aerobic Production of Methane in the Sea. Nat. Geosci. 2008, 1, 473–478. [Google Scholar] [CrossRef]
- Harrison, K.J.; de Crécy-Lagard, V.; Zallot, R. Gene Graphics: A Genomic Neighborhood Data Visualization Web Application. Bioinformatics 2018, 34, 1406–1408. [Google Scholar] [CrossRef]
- Gebhard, S.; Cook, G.M. Differential Regulation of High-Affinity Phosphate Transport Systems of Mycobacterium smegmatis: Identification of PhnF, a Repressor of the phnDCE Operon. J. Bacteriol. 2008, 190, 1335–1343. [Google Scholar] [CrossRef]
- Kamat, S.S.; Williams, H.J.; Raushel, F.M. Intermediates in the Transformation of Phosphonates to Phosphate by Bacteria. Nature 2011, 480, 570–573. [Google Scholar] [CrossRef]
- Yang, K.; Ren, Z.; Raushel, F.M.; Zhang, J. Structures of the Carbon-Phosphorus Lyase Complex Reveal the Binding Mode of the NBD-like PhnK. Structure 2016, 24, 37–42. [Google Scholar] [CrossRef]
- Jochimsen, B.; Lolle, S.; McSorley, F.R.; Nabi, M.; Stougaard, J.; Zechel, D.L.; Hove-Jensen, B. Five Phosphonate Operon Gene Products as Components of a Multi-Subunit Complex of the Carbon-Phosphorus Lyase Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Podzelinska, K.; He, S.-M.; Wathier, M.; Yakunin, A.; Proudfoot, M.; Hove-Jensen, B.; Zechel, D.L.; Jia, Z. Structure of PhnP, a Phosphodiesterase of the Carbon-Phosphorus Lyase Pathway for Phosphonate Degradation. J. Biol. Chem. 2009, 284, 17216–17226. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Rosenkrantz, T.J.; Andreas, H.A.; Wanner, B.L. Escherichia poli phnN, Encoding Ribose 1,5-Bisphosphokinase Activity (Phosphoribosyl Diphosphate Forming): Dual Role in Phosphonate Degradation and NAD Biosynthesis Pathways. J. Bacteriol. 2003, 185, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Errey, J.C.; Blanchard, J.S. Functional Annotation and Kinetic Characterization of PhnO from Salmonella enterica. Biochemistry 2006, 45, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Hove-Jensen, B.; McSorley, F.R.; Zechel, D.L. Catabolism and Detoxification of 1-Aminoalkylphosphonic Acids: N-Acetylation by the phnO Gene Product. PLoS ONE 2012, 7, e46416. [Google Scholar] [CrossRef] [PubMed]
- Beversdorf, L.J.; White, A.E.; Björkman, K.M.; Letelier, R.M.; Karl, D.M. Phosphonate Metabolism by Trichodesmium IMS101 and the Production of Greenhouse Gases. Limnol. Oceanogr. 2010, 55, 1768–1778. [Google Scholar] [CrossRef]
- Sosa, O.A.; Burrell, T.J.; Wilson, S.T.; Foreman, R.K.; Karl, D.M.; Repeta, D.J. Phosphonate Cycling Supports Methane and Ethylene Supersaturation in the Phosphate-Depleted Western North Atlantic Ocean. Limnol. Oceanogr. 2020, 65, 2443–2459. [Google Scholar] [CrossRef]
- Teikari, J.E.; Fewer, D.P.; Shrestha, R.; Hou, S.; Leikoski, N.; Mäkelä, M.; Simojoki, A.; Hess, W.R.; Sivonen, K. Strains of the Toxic and Bloom-Forming Nodularia spumigena (Cyanobacteria) Can Degrade Methylphosphonate and Release Methane. ISME J. 2018, 12, 1619–1630. [Google Scholar] [CrossRef]
- Duhamel, S.; Diaz, J.M.; Adams, J.C.; Djaoudi, K.; Steck, V.; Waggoner, E.M. Phosphorus as an Integral Component of Global Marine Biogeochemistry. Nat. Geosci. 2021, 14, 359–368. [Google Scholar] [CrossRef]
- Repeta, D.J.; Ferrón, S.; Sosa, O.A.; Johnson, C.G.; Repeta, L.D.; Acker, M.; DeLong, E.F.; Karl, D.M. Marine Methane Paradox Explained by Bacterial Degradation of Dissolved Organic Matter. Nat. Geosci. 2016, 9, 884–887. [Google Scholar] [CrossRef]
- Zhou, Z.; Tran, P.Q.; Kieft, K.; Anantharaman, K. Genome Diversification in Globally Distributed Novel Marine Proteobacteria Is Linked to Environmental Adaptation. ISME J. 2020, 14, 2060–2077. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Chappell, P.D.; Haley, S.T.; Moffett, J.W.; Orchard, E.D.; Waterbury, J.B.; Webb, E.A. Phosphonate Utilization by the Globally Important Marine Diazotroph Trichodesmium. Nature 2006, 439, 68–71. [Google Scholar] [CrossRef]
- Walker, A. Adding Genomic “foliage” to the Tree of Life. Nat. Rev. Microbiol. 2014, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Schowanek, D.; Verstraete, W. Phosphonate Utilization by Bacterial Cultures and Enrichments from Environmental Samples. Appl. Environ. Microbiol. 1990, 56, 895–903. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for Culture of ‘Unculturable’ Bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007, 5, 0398–0431. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Babin, M.; Boss, E.; Cochrane, G.; de Vargas, C.; Follows, M.; Gorsky, G.; et al. Tara Oceans: Towards Global Ocean Ecosystems Biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Karl, D.M. Microbially Mediated Transformations of Phosphorus in the Sea: New Views of an Old Cycle. Ann. Rev. Mar. Sci. 2014, 6, 279–337. [Google Scholar] [CrossRef] [PubMed]
- Karl, D.M.; Björkman, K.M. Dynamics of Dissolved Organic Phosphorus. In Biogeochemistry of Marine Dissolved Organic Matter (Second Edition); Hansell, D.A., Carlson, C.A., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 233–334. [Google Scholar]
- Chin, J.P.; Quinn, J.P.; McGrath, J.W. Phosphate Insensitive Aminophosphonate Mineralisation within Oceanic Nutrient Cycles. ISME J. 2018, 12, 973–980. [Google Scholar] [CrossRef]
- Clark, L.L.; Ingall, E.D.; Benner, R. Marine Organic Phosphorus Cycling; Novel Insights from Nuclear Magnetic Resonance. Am. J. Sci. 1999, 299, 724–737. [Google Scholar] [CrossRef]
- Benitez-Nelson, C.R.; O’Neill, L.; Kolowith, L.C.; Pellechia, P.; Thunell, R. Phosphonates and Particulate Organic Phosphorus Cycling in an Anoxic Marine Basin. Limnol. Oceanogr. 2004, 49, 1593–1604. [Google Scholar] [CrossRef]
- Van Mooy, B.A.S.; Krupke, A.; Dyhrman, S.T.; Fredricks, H.F.; Frischkorn, K.R.; Ossolinski, J.E.; Repeta, D.J.; Rouco, M.; Seewald, J.D.; Sylva, S.P. Major Role of Planktonic Phosphate Reduction in the Marine Phosphorus Redox Cycle. Science 2015, 348, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Vila-Costa, M.; Sharma, S.; Moran, M.A.; Casamayor, E.O. Diel Gene Expression Profiles of a Phosphorus Limited Mountain Lake Using Metatranscriptomics. Environ. Microbiol. 2013, 15, 1190–1203. [Google Scholar] [CrossRef]
- Yao, M.; Henny, C.; Maresca, J.A. Freshwater Bacteria Release Methane as a By-Product of Phosphorus Acquisition. Appl. Environ. Microbiol. 2016, 82, 6994–7003. [Google Scholar] [CrossRef]
- Wang, Q.; Dore, J.E.; McDermott, T.R. Methylphosphonate Metabolism by Pseudomonas sp. Populations Contributes to the Methane Oversaturation Paradox in an Oxic Freshwater Lake. Environ. Microbiol. 2017, 19, 2366–2378. [Google Scholar] [CrossRef] [PubMed]
- Frouin, E.; Lecoeuvre, A.; Armougom, F.; Schrenk, M.O.; Erauso, G. Comparative Metagenomics Highlight a Widespread Pathway Involved in Catabolism of Phosphonates in Marine and Terrestrial Serpentinizing Ecosystems. mSystems 2022, 7, e00328-22. [Google Scholar] [CrossRef]
- Gomez-Garcia, M.R.; Davison, M.; Blain-Hartnung, M.; Grossman, A.R.; Bhaya, D. Alternative Pathways for Phosphonate Metabolism in Thermophilic Cyanobacteria from Microbial Mats. ISME J. 2011, 5, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Siles, J.A.; Starke, R.; Martinovic, T.; Parente Fernandes, M.L.; Orgiazzi, A.; Bastida, F. Distribution of Phosphorus Cycling Genes across Land Uses and Microbial Taxonomic Groups Based on Metagenome and Genome Mining. Soil. Biol. Biochem. 2022, 174, 108826. [Google Scholar] [CrossRef]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized Metabolic Functions of Keystone Taxa Sustain Soil Microbiome Stability. Microbiome 2021, 9, 35. [Google Scholar] [CrossRef]
- Oliverio, A.M.; Bissett, A.; McGuire, K.; Saltonstall, K.; Turner, B.L.; Fierer, N. The Role of Phosphorus Limitation in Shaping Soil Bacterial Communities and Their Metabolic Capabilities. mBio 2020, 11, e01718-20. [Google Scholar] [CrossRef]
- Kehler, A.; Haygarth, P.; Tamburini, F.; Blackwell, M. Cycling of Reduced Phosphorus Compounds in Soil and Potential Impacts of Climate Change. Eur. J. Soil. Sci. 2021, 72, 2517–2537. [Google Scholar] [CrossRef]
- Murphy, A.R.J.; Scanlan, D.J.; Chen, Y.; Bending, G.D.; Hammond, J.P.; Wellington, E.M.H.; Lidbury, I.D.E.A. 2-Aminoethylphosphonate Utilization in Pseudomonas putida BIRD-1 Is Controlled by Multiple Master Regulators. Environ. Microbiol. 2022, 24, 1902–1917. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, S.N.; Graham, R.L.J.; McMullan, G.; Ternan, N.G. A Role for Carbon Catabolite Repression in the Metabolism of Phosphonoacetate by Agromyces fucosus Vs2. FEMS Microbiol. Lett. 2006, 261, 133–140. [Google Scholar]
- Gardner, S.G.; McCleary, W.R. Control of the phoBR Regulon in Escherichia coli. EcoSal Plus 2019, 8, ESP-0006–2019. [Google Scholar] [CrossRef]
- Wackett, L.P.; Wanner, B.L.; Venditti, C.P.; Walsh, C.T. Involvement of the Phosphate Regulon and the psiD Locus in Carbon-Phosphorus Lyase Activity of Escherichia coli K-12. J. Bacteriol. 1987, 169, 1753–1756. [Google Scholar] [CrossRef]
- Lee, K.S.; Metcalf, W.W.; Wanner, B.L. Evidence for Two Phosphonate Degradative Pathways in Enterobacter aerogenes. J. Bacteriol. 1992, 174, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- McMullan, G.; Quinn, J.P. In Vitro Characterization of a Phosphate Starvation-Independent Carbon-Phosphorus Bond Cleavage Activity in Pseudomonas fluorescens. J. Bacteriol. 1994, 176, 320–324. [Google Scholar] [CrossRef]
- Lidbury, I.D.E.A.; Murphy, A.R.J.; Scanlan, D.J.; Bending, G.D.; Jones, A.M.E.; Moore, J.D.; Goodall, A.; Hammond, J.P.; Wellington, E.M.H. Comparative Genomic, Proteomic and Exoproteomic Analyses of Three Pseudomonas Strains Reveals Novel Insights into the Phosphorus Scavenging Capabilities of Soil Bacteria. Environ. Microbiol. 2016, 18, 3535–3549. [Google Scholar] [CrossRef]
- Quinn, J.P.; Kulakova, A.N.; Cooley, N.A.; McGrath, J.W. New Ways to Break an Old Bond: The Bacterial Carbon-Phosphorus Hydrolases and Their Role in Biogeochemical Phosphorus Cycling. Environ. Microbiol. 2007, 9, 2392–2400. [Google Scholar] [CrossRef]
- Ternan, N.G.; McMullan, G. The Utilization of 4-Aminobutylphosphonate as Sole Nitrogen Source by a Strain of Kluyveromyces fragilis. FEMS Microbiol. Lett. 2000, 184, 237–240. [Google Scholar] [CrossRef]
- Ternan, N.G.; Quinn, J.P. Phosphate Starvation-Independent 2-Aminoethylphosphonic Acid Biodegradation in a Newly Isolated Strain of Pseudomonas putida, NG2. Syst. Appl. Microbiol. 1998, 21, 346–352. [Google Scholar] [CrossRef]
- McGrath, J.W.; Ternan, N.G.; Quinn, J.P. Utilization of Organophosphonates by Environmental Microorganisms. Lett. Appl. Microbiol. 1997, 24, 69–73. [Google Scholar] [CrossRef]
- Kulakova, A.N.; Kulakov, L.A.; McGrath, J.W.; Quinn, J.P. The Construction of a Whole-Cell Biosensor for Phosphonoacetate, Based on the LysR-like Transcriptional Regulator PhnR from Pseudomonas fluorescens 23F. Microb. Biotechnol. 2009, 2, 234–240. [Google Scholar] [CrossRef]
- Maddocks, S.E.; Oyston, P.C.F. Structure and Function of the LysR-Type Transcriptional Regulator (LTTR) Family Proteins. Microbiology 2008, 154, 3609–3623. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, T.; Moldrup, P.; Ferré, T.P.A.; Olsen, P.; Rosenbom, A.E.; de Jonge, L.W. Leaching of Glyphosate and Aminomethylphosphonic Acid from an Agricultural Field over a Twelve-Year Period. Vadose Zone J. 2014, 13, 608–620. [Google Scholar] [CrossRef]
- Neft, A.; Claus, M.; Schmidt, S.; Minke, R.; Steinmetz, H. Looking Closer on Phosphorus Fractions in WWTP Effluents: Implications on the Optimisation of Phosphorus Removal. In Proceedings of the 1st IWA Austrian Young Water Professionals Conference, Vienna, Austria, 9–11 June 2010. [Google Scholar]
- Gledhill, W.E.; Feijtel, T.C.J. Environmental Properties and Safety Assessment of Organic Phosphonates Used for Detergent and Water Treatment Applications. In Detergents; de Oude, N.T., Ed.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 261–285. [Google Scholar]
- Wang, S.; Zhang, B.; Shan, C.; Yan, X.; Chen, H.; Pan, B. Occurrence and Transformation of Phosphonates in Textile Dyeing Wastewater along Full-Scale Combined Treatment Processes. Water Res. 2020, 184, 116173. [Google Scholar] [CrossRef] [PubMed]
- Studnik, H.; Liebsch, S.; Forlani, G.; Wieczorek, D.; Kafarski, P.; Lipok, J. Amino Polyphosphonates—Chemical Features and Practical Uses, Environmental Durability and Biodegradation. New Biotechnol. 2015, 32, 1–6. [Google Scholar] [CrossRef]
- Chillè, D.; Aiello, D.; Grasso, G.I.; Giuffrè, O.; Napoli, A.; Sgarlata, C.; Foti, C. Complexation of As(III) by Phosphonate Ligands in Aqueous Fluids: Thermodynamic Behavior, Chemical Binding Forms and Sequestering Abilities. J. Environ. Sci. 2020, 94, 100–110. [Google Scholar] [CrossRef]
- Castro, J.V.; Peralba, M.C.R.; Ayub, M.A.Z. Biodegradation of the Herbicide Glyphosate by Filamentous Fungi in Platform Shaker and Batch Bioreactor. J. Environ. Sci. Health B 2007, 42, 883–886. [Google Scholar] [CrossRef]
- Lipok, J.; Wieczorek, D.; Jewgiński, M.; Kafarski, P. Prospects of in Vivo 31P NMR Method in Glyphosate Degradation Studies in Whole Cell System. Enzyme Microb. Technol. 2009, 44, 11–16. [Google Scholar] [CrossRef]
- Riedel, R.; Krahl, K.; Buder, K.; Böllmann, J.; Braun, B.; Martienssen, M. Novel Standard Biodegradation Test for Synthetic Phosphonates. J. Microbiol. Methods 2023, 212, 106793. [Google Scholar] [CrossRef] [PubMed]
- Piel, S.; Grandcoin, A.; Baurès, E. L’acide Aminométhylphosphonique (AMPA) Dans Les Eaux Naturelles et Les Filières de Traitement: Origines, Comportement et Devenir. Tech. Sci. Méthodes 2019, 114, 45–56. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, W.-J.; Huang, Y.; Li, J.; Zhong, J.; Zhang, W.; Zou, Y.; Mishra, S.; Bhatt, P.; Chen, S. Insights into the Microbial Degradation and Resistance Mechanisms of Glyphosate. Environ. Res. 2022, 215, 114153. [Google Scholar] [CrossRef] [PubMed]
- Bøhn, T.; Cuhra, M.; Traavik, T.; Sanden, M.; Fagan, J.; Primicerio, R. Compositional Differences in Soybeans on the Market: Glyphosate Accumulates in Roundup Ready GM Soybeans. Food Chem. 2014, 153, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, C.M. Trends in Glyphosate Herbicide Use in the United States and Globally. Environ. Sci. Eur. 2016, 28, 3. [Google Scholar] [CrossRef] [PubMed]
- Drzyzga, D.; Lipok, J. Glyphosate Dose Modulates the Uptake of Inorganic Phosphate by Freshwater Cyanobacteria. J. Appl. Phycol. 2018, 30, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Villamar-Ayala, C.A.; Carrera-Cevallos, J.V.; Vasquez-Medrano, R.; Espinoza-Montero, P.J. Fate, Eco-Toxicological Characteristics, and Treatment Processes Applied to Water Polluted with Glyphosate: A Critical Review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 1476–1514. [Google Scholar] [CrossRef]
- Mohanty, S.S.; Das, A.P. A Systematic Study on the Microbial Degradation of Glyphosate: A Review. Geomicrobiol. J. 2022, 39, 316–327. [Google Scholar] [CrossRef]
- Peñaloza-Vazquez, A.; Mena, G.L.; Herrera-Estrella, L.; Bailey, A.M. Cloning and Sequencing of the Genes Involved in Glyphosate Utilization by Pseudomonas pseudomallei. Appl. Environ. Microbiol. 1995, 61, 538–543. [Google Scholar] [CrossRef]
- Wehrmann, A.; van Vliet, A.; Opsomer, C.; Botterman, J.; Schulz, A. The Similarities of Bar and Pat Gene Products Make Them Equally Applicable for Plant Engineers. Nat. Biotechnol. 1996, 14, 1274–1278. [Google Scholar] [CrossRef]
- Castle, L.A.; Siehl, D.L.; Gorton, R.; Patten, P.A.; Chen, Y.H.; Bertain, S.; Cho, H.-J.; Duck, N.; Wong, J.; Liu, D.; et al. Discovery and Directed Evolution of a Glyphosate Tolerance Gene. Science 2004, 304, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Borggaard, O.K.; Gimsing, A.L. Fate of Glyphosate in Soil and the Possibility of Leaching to Ground and Surface Waters: A Review. Pest. Manag. Sci. 2008, 64, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Obojska, A.; Lejczak, B.; Kubrak, M. Degradation of Phosphonates by Streptomycete Isolates. Appl. Microbiol. Biotechnol. 1999, 51, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Pipke, R.; Amrhein, N. Degradation of the Phosphonate Herbicide Glyphosate by Arthrobacter atrocyaneus ATCC 13752. Appl. Environ. Microbiol. 1988, 54, 1293–1296. [Google Scholar] [CrossRef]
- McAuliffe, K.S.; Hallas, L.E.; Kulpa, C.F. Glyphosate Degradation by Agrobacterium radiobacter Isolated from Activated Sludge. J. Ind. Microbiol. 1990, 6, 219–221. [Google Scholar] [CrossRef]
- Singh, B.; Singh, K. Microbial Degradation of Herbicides. Crit. Rev. Microbiol. 2016, 42, 245–261. [Google Scholar] [CrossRef]
- Wijekoon, N.; Yapa, N. Assessment of Plant Growth Promoting Rhizobacteria (PGPR) on Potential Biodegradation of Glyphosate in Contaminated Soil and Aquifers. Groundw. Sustain. Dev. 2018, 7, 465–469. [Google Scholar] [CrossRef]
- Correa, L.O.; Bezerra, A.F.M.; Honorato, L.R.S.; Cortez, A.C.A.; Souza, J.V.B.; Souza, E.S. Amazonian Soil Fungi Are Efficient Degraders of Glyphosate Herbicide; Novel Isolates of Penicillium, Aspergillus, and Trichoderma. Braz. J. Biol. 2023, 83, e242830. [Google Scholar] [CrossRef]
- Bujacz, B.; Wieczorek, P.; Krzysko-Lupicka, T.; Golab, Z.; Lejczak, B.; Kavfarski, P. Organophosphonate Utilization by the Wild-Type Strain of Penicillium notatum. Appl. Environ. Microbiol. 1995, 61, 2905–2910. [Google Scholar] [CrossRef]
- Fu, G.; Chen, Y.; Li, R.; Yuan, X.; Liu, C.; Li, B.; Wan, Y. Pathway and Rate-Limiting Step of Glyphosate Degradation by Aspergillus oryzae A-F02. Prep. Biochem. Biotechnol. 2017, 47, 782–788. [Google Scholar] [CrossRef]
- González-Torralva, F.; Rojano-Delgado, A.M.; Luque de Castro, M.D.; Mülleder, N.; De Prado, R. Two Non-Target Mechanisms Are Involved in Glyphosate-Resistant Horseweed (Conyza canadensis L. Cronq.) Biotypes. J. Plant Physiol. 2012, 169, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Boutet, I.; Tanguy, A.; Moraga, D. Molecular Identification and Expression of Two Non-P450 Enzymes, Monoamine Oxidase A and Flavin-Containing Monooxygenase 2, Involved in Phase I of Xenobiotic Biotransformation in the Pacific Oyster, Crassostrea gigas. Biochim. Biophys. Acta 2004, 1679, 29–36. [Google Scholar] [CrossRef]
- Hernández-Alomia, F.; Ballesteros, I.; Castillejo, P. Bioremediation Potential of Glyphosate-Degrading Microorganisms in Eutrophicated Ecuadorian Water Bodies. Saudi J. Biol. Sci. 2022, 29, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Obojska, A.; Ternan, N.G.; Lejczak, B.; Kafarski, P.; McMullan, G. Organophosphonate Utilization by the Thermophile Geobacillus caldoxylosilyticus T20. Appl. Environ. Microbiol. 2002, 68, 2081–2084. [Google Scholar] [CrossRef]
- Grandcoin, A.; Piel, S.; Baurès, E. AminoMethylPhosphonic Acid (AMPA) in Natural Waters: Its Sources, Behavior and Environmental Fate. Water Res. 2017, 117, 187–197. [Google Scholar] [CrossRef]
- Guilherme, S.; Santos, M.A.; Gaivão, I.; Pacheco, M. DNA and Chromosomal Damage Induced in Fish (Anguilla anguilla L.) by Aminomethylphosphonic Acid (AMPA)—The Major Environmental Breakdown Product of Glyphosate. J. Environ. Sci. Int. 2014, 21, 8730–8739. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.A.; Cook, A.M.; Leisinger, T. Microbial Metabolism of Sulfur and Phosphorus-Containing Xenobiotics. FEMS Microbiol. Rev. 1994, 15, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Kishore, G.M.; Jacob, G.S. Degradation of Glyphosate by Pseudomonas sp. PG2982 via a Sarcosine Intermediate. J. Biol. Chem. 1987, 262, 12164–12168. [Google Scholar] [CrossRef] [PubMed]
- Pipke, R.; Amrhein, N.; Jacob, G.S.; Schaefer, J.; Kishore, G.M. Metabolism of Glyphosate in an Arthrobacter sp. GLP-1. Eur. J. Biochem. 1987, 165, 267–273. [Google Scholar] [CrossRef]
- Sviridov, A.V.; Shushkova, T.V.; Ermakova, I.T.; Ivanova, E.V.; Epiktetov, D.O.; Leontievsky, A.A. Microbial Degradation of Glyphosate Herbicides. Appl. Biochem. Microbiol. 2015, 51, 188–195. [Google Scholar] [CrossRef]
- Pedotti, M.; Rosini, E.; Molla, G.; Moschetti, T.; Savino, C.; Vallone, B.; Pollegioni, L. Glyphosate Resistance by Engineering the Flavoenzyme Glycine Oxidase. J. Biol. Chem. 2009, 284, 36415–36423. [Google Scholar] [CrossRef] [PubMed]
- Huntscha, S.; Stravs, M.A.; Bühlmann, A.; Ahrens, C.H.; Frey, J.E.; Pomati, F.; Hollender, J.; Buerge, I.J.; Balmer, M.E.; Poiger, T. Seasonal Dynamics of Glyphosate and AMPA in Lake Greifensee: Rapid Microbial Degradation in the Epilimnion During Summer. Environ. Sci. Technol. 2018, 52, 4641–4649. [Google Scholar] [CrossRef] [PubMed]
- Peck, S.C.; van der Donk, W.A. Phosphonate Biosynthesis and Catabolism: A Treasure Trove of Unusual Enzymology. Curr. Opin. Chem. Biol. 2013, 17, 580–588. [Google Scholar] [CrossRef]
- Polidore, A.L.A.; Furiassi, L.; Hergenrother, P.J.; Metcalf, W.W. A Phosphonate Natural Product Made by Pantoea ananatis Is Necessary and Sufficient for the Hallmark Lesions of Onion Center Rot. mBio 2021, 12, e03402-20. [Google Scholar] [CrossRef]
- Jake, W.; Jerry, C.; Toshiki, N.; Happy, K.; Yeying, Z.; Kayrouz, C.M.; Pham, T.M.; Hannah, R.; Kou-San, J. Discovery of Antimicrobial Phosphonopeptide Natural Products from Bacillus velezensis by Genome Mining. Appl. Environ. Microbiol. 2023, 89, e00338-23. [Google Scholar]
- Acker, M.; Hogle, S.L.; Berube, P.M.; Hackl, T.; Coe, A.; Stepanauskas, R.; Chisholm, S.W.; Repeta, D.J. Phosphonate Production by Marine Microbes: Exploring New Sources and Potential Function. Proc. Natl. Acad. Sci. USA 2022, 119, e2113386119. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Abundance (Percentage of Expected Genomes) | |||
|---|---|---|---|---|
| Martinez et al. [8] | Villarreal-Chiu et al. [3] | Murphy et al. [39] | Lockwood et al. [36] | |
| phnX | 1% | 1.8% | <0.1% | ~2% |
| phnA | 9% | 11.2% | <10% | ~10% |
| palA | nd | 0.1% | nd | <5% |
| phnZ | 7% | 9.4% | <1% | ~20% |
| phnJ | 8% | 5.1% | <1% | >10% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruffolo, F.; Dinhof, T.; Murray, L.; Zangelmi, E.; Chin, J.P.; Pallitsch, K.; Peracchi, A. The Microbial Degradation of Natural and Anthropogenic Phosphonates. Molecules 2023, 28, 6863. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196863
Ruffolo F, Dinhof T, Murray L, Zangelmi E, Chin JP, Pallitsch K, Peracchi A. The Microbial Degradation of Natural and Anthropogenic Phosphonates. Molecules. 2023; 28(19):6863. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196863
Chicago/Turabian StyleRuffolo, Francesca, Tamara Dinhof, Leanne Murray, Erika Zangelmi, Jason P. Chin, Katharina Pallitsch, and Alessio Peracchi. 2023. "The Microbial Degradation of Natural and Anthropogenic Phosphonates" Molecules 28, no. 19: 6863. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196863