
A Bridge too Far? Comparison of Transition Metal Complexes of Dibenzyltetraazamacrocycles with and without Ethylene Cross-Bridges: X-ray Crystal Structures, Kinetic Stability, and Electronic Properties

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Complex Synthesis
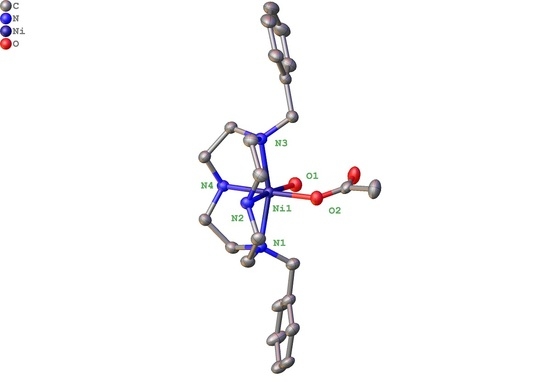
2.2. X-ray Crystal Structures
2.2.1. Complexes of 1, Dibenzylcyclen
2.2.2. Complexes of 2, Dibenzylcyclam

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Nax-M-Nax Angle | Neq-M-Neq Angle | X-M-X Angle | X Ligands | Ref |
|---|---|---|---|---|---|
| [Ni(1)(OH2)2]2+ | 158.3(2) | 92.7(4) 110.4(6) | 83.6(2) | H2O, H2O | This work |
| [Ni(1)(OAc)]+ | 160.0(5) | 102.6(4) | 62.95(17) | O—C—O (acetate) | [54] This work |
| [Ni(1)(OAc)(H2O)]+ | 160.26(7) | 97.04(7) | 88.03(6) | H2O, O (acetate) | This work |
| [Ni(Bn2Bcyclen)(OAc)(H2O)]+ | 163.52(6) | 85.59(7) | 87.61(6) | H2O, O (acetate) | [29] |
| [Ni(Bn2Bcyclen)Cl2] | 158.40(7) | 83.37(9) | 90.18(3) | Cl, Cl | [29] |
| [Cu(1)(NH3)]2+ | 150.74(7) | 145.81(8) | n/a (5-coord) | NH3 | This work |
| [(Cu(Bn2Bcyclen))2(μ-CO3)] | 155.65 | 86.50 | 66.37 | O—C—O | [64] |
| [Cu(Me2Bcyclen)(CO3)] | 154.88(7) | 85.17(7) | 66.94(6) | O—C—O (carbonate) | [28] |
| [Cu(Me2Bcyclen)(OAc)]+ | 164.04(8) | 85.00(8) | n/a (5-coord) | O (acetate) | [28] |
| [Zn(1)(OAc)]+ | 138.4(3) | 132.4(4) 126.2(12) | n/a (5-coord) | O (acetate) | This work Note: only one of the two similar independent molecules in the unit cell is presented here |
| [Zn(2)(Cl)]+ | 159.6(1) | 120.9(1) | n/a (5-coord) | Cl | [55] |
| [Zn(Bn2Bcyclen)Cl2] | 152.61 | 77.62 | 95.50 | Cl, Cl | [65] |
| [Zn(Me2Bcyclen)(OAc)(H2O)]+ | 157.59(10) | 81.80(10) | 90.83(8) | H2O, O (acetate) | [28] |
| [Co(Me2Cyclen)(CO3)]+ | 167.4(6) | 101.7(3) | 68.6(2) | O—C—O (carbonate) | [66] |
| [Co(2)(OOCCH3)]+ | 171.54(10) | 97.15(11) | 61.53(9) | O—C—O (acetate) | This work |
| [Co(Bn2Bcyclam)Cl2] | 167.7(4) | 82.5(4) | 93.27(13) | Cl, Cl | [29] |
| [Co(Me2Bcyclam)Cl2] | 172.4(2) | 81.11(13) | 97.37(4) | Cl, Cl | [67] |
| [Cu(2)]2+ | 180.00(10) | 180 | n/a (4-coord) | None | This work |
| [Cu(2)(DMF)2]2+ | 180.00 | 180 | 180.00(9) trans | DMF, DMF | [54] |
| [Cu(Bn2Bcyclam)(OAc)][OAc] | 176.11(8) | 86.05(9) | n/a (5-coord) | O (acetate) | [28] |
| [Cu(Bn2Bcyclam)(OAc)][PF6] | 176.74(8) | 85.93(8) | n/a (5-coord | O (acetate) | [28] |
| [Ni(2′)(OAc)]+ | 174.83(9) | 170.21(10) | 153.90(8) trans | O (acetate) -CH2OCH3 | This work |
| [Ni(Bn2Bcyclam)Cl2] | 170.75(15) | 84.56(16) | 92.00(5) | Cl, Cl | [29] |
| [Ni(Bn2Bcyclam)(OAc)(H2O)]+ | 163.52(6) | 85.59(7) | 87.61(6) | H2O, O (acetate) | [29] |
2.3. Kinetic Stability
2.4. UV-Visible Spectroscopy
2.5. Cyclic Voltammetry
3. Materials and Methods
3.1. General
3.2. Acid Decomplexation Studies
3.3. X-ray Crystallography Studies
3.4. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Weisman, G.R.; Rogers, M.E.; Wong, E.H.; Jasinski, J.P.; Paight, E.S. Cross-bridged cyclam-protonation and Li+ complexation in a diamond-lattice cleft. J. Am. Chem. Soc. 1990, 112, 8604–8605. [Google Scholar] [CrossRef]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, D.H. The compleat coordination chemistry. Chem. Rev. 1993, 93, 847–860. [Google Scholar] [CrossRef]
- Weisman, G.R.; Wong, E.H.; Hill, D.C.; Rogers, M.E.; Reed, D.P.; Calabrese, J.C. Synthesis and transition-metal complexes of new cross-bridged tetraamine ligands. Chem. Commun. 1996, 32, 947–948. [Google Scholar] [CrossRef]
- Wong, E.H.; Weisman, G.R.; Hill, D.C.; Reed, D.P.; Rogers, M.E.; Condon, J.S.; Fagan, M.A.; Calabrese, J.C.; Lam, K.-C.; Guzei, I.A.; et al. Synthesis and characterization of cross-bridged cyclams and pendant-armed derivatives and structural studies of their Copper(II) complexes. J. Am. Chem. Soc. 2000, 122, 10561–10572. [Google Scholar] [CrossRef]
- Sun, X.; Wuest, M.; Weisman, G.R.; Wong, E.H.; Reed, D.P.; Boswell, C.A.; Motekaitis, R.; Martell, A.E.; Welch, M.J.; Anderson, C.J. Radiolabeling and in vivo behavior of copper-64-labeled cross-bridged cyclam ligands. J. Med. Chem. 2002, 45, 469–477. [Google Scholar] [CrossRef]
- Boswell, C.A.; Sun, X.; Niu, W.; Weisman, G.R.; Wong, E.H.; Rheingold, A.L.; Anderson, C.J. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J. Med. Chem. 2004, 47, 1465–1474. [Google Scholar] [CrossRef]
- Woodin, K.S.; Heroux, K.J.; Boswell, C.A.; Wong, E.H.; Weisman, G.R.; Niu, W.J.; Tomellini, S.A.; Anderson, C.J.; Zakharov, L.N.; Rheingold, A.L. Kinetic inertness and electrochemical behavior of copper(II) tetraazamacrocyclic complexes: Possible implications for in vivo stability. Eur. J. Inorg. Chem. 2005, 2005, 4829–4833. [Google Scholar] [CrossRef]
- Sprague, J.E.; Peng, Y.; Fiamengo, A.L.; Woodin, K.S.; Southwick, E.A.; Weisman, G.R.; Wong, E.H.; Golen, J.A.; Rheingold, A.L.; Anderson, C.J. Synthesis, characterization and in vivo studies of Cu(II)-64-labeled cross-bridged tetraazamacrocycle-amide complexes as models of peptide conjugate imaging agents. J. Med. Chem. 2007, 50, 2527–2535. [Google Scholar] [CrossRef]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr. Pharm. Des. 2007, 13, 3–16. [Google Scholar] [CrossRef]
- Anderson, C.J.; Wadas, T.J.; Wong, E.H.; Weisman, G.R. Cross-bridged macrocyclic chelators for stable complexation of copper radionuclides for PET imaging. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 185–192. [Google Scholar] [PubMed]
- Odendaal, A.Y.; Fiamengo, A.L.; Ferdani, R.; Wadas, T.J.; Hill, D.C.; Peng, Y.; Heroux, K.J.; Golen, J.A.; Rheingold, A.L.; Anderson, C.J.; et al. Isomeric trimethylene and ethylene pendant-armed cross-bridged tetraazamacrocycles and in vitro/in vivo comparisions of their copper(II) complexes. Inorg. Chem. 2011, 50, 3078–3086. [Google Scholar] [CrossRef] [PubMed]
- Springborg, J.; Kofod, P.; Olsen, C.E.; Toftlund, H.; Sotofte, I. Synthesis and crystal structure of a small bicyclic tetraaza proton sponge, 1,4,7,10-tetraazabicyclo[5.5.3]pentadecane dibromide perchlorate. Acta Chem. Scand. 1995, 49, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Springborg, J.; Pretzmann, U.; Nielsen, B.; Olsen, C.E.; Sotofte, I. Synthesis of [3(5)]adamanzane, 1,5,9,13-tetraazabicyclo[7.7.3]nonadecane, by oxidative C–N cleavage of [3(6)]adamanzane, 1,5,9,13-tetraazatricyclo[7.7.3.3(5,13)]docosane and crystal structure of the tetraprotonated bromide salt of [3(5)]adamanzane. Acta Chem. Scand. 1998, 52, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Springborg, J. Adamanzanes--bi- and tricyclic tetraamines and their coordination compounds. Dalton Trans. 2003, 32, 1653–1665. [Google Scholar] [CrossRef]
- Hubin, T.J.; Alcock, N.W.; Busch, D.H. The square-pyramidal Pd(II) complex of a cross-bridged tetraazamacrocycle. Acta Crystallogr. 1999, C55, 1404–1406. [Google Scholar]
- Hubin, T.J.; Alcock, N.W.; Clase, H.J.; Busch, D.H. Potentiometric titrations and nickel(II) complexes of four topologically constrained tetraazamacrocycles. Supramol. Chem. 2001, 13, 261–276. [Google Scholar] [CrossRef]
- Collinson, S.R.; Alcock, N.W.; Hubin, T.J.; Busch, D.H. Synthesis and characterization of the novel bridged ligand 5,8-dimethyl-1,5,8,12-tetraazabicyclo 10.3.2 heptadecane and its complexes with iron(II) and manganese(II) ions. J. Coord. Chem. 2001, 52, 317–331. [Google Scholar] [CrossRef]
- Lichty, J.; Allen, S.M.; Grillo, A.I.; Archibald, S.J.; Hubin, T.J. Synthesis and characterization of the cobalt(III) complexes of two pendant-arm cross-bridged cyclams. Inorg. Chim. Acta 2004, 357, 615–618. [Google Scholar] [CrossRef]
- Khan, M.F.; Keiser, J.; Amoyaw, P.N.; Hossain, M.F.; Vargas, M.; Le, J.G.; Simpson, N.C.; Roewe, K.D.; Freeman, T.N.; Hasley, T.R.; et al. Discovery of antischistosomal drug leads based on tetraazamacrocyclic derivatives and their metal complexes. Antimicr. Agents Chemoth. 2016, 60, 5331–5336. [Google Scholar] [CrossRef] [Green Version]
- Hubin, T.J.; McCormick, J.M.; Alcock, N.W.; Busch, D.H. Synthesis and X-ray crystal structure determination of the first transition metal complexes of the tetracycles formed by tetraazamacrocycle-glyoxal condensation: PdL*Cl-2 (L = cyclam-glyoxal condensate (1), cyclen-glyoxal condensate (2)). Inorg. Chem. 1998, 37, 6549–6551. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.M.; Wilson, K.R.; Jones, D.G.; Reinheimer, E.W.; Archibald, S.J.; Prior, T.J.; Ayala, M.A.; Foster, A.L.; Hubin, T.J.; Green, K.N. Increase of direct C–C coupling reaction yield by identifying structural and electronic properties of high-spin iron tetra-azamacrocyclic complexes. Inorg. Chem. 2018, 57, 8890–8902. [Google Scholar] [CrossRef] [PubMed]
- Shircliff, A.D.; Wilson, K.R.; Cannon-Smith, D.J.; Jones, D.G.; Zhang, Z.; Chen, Z.Q.; Yin, G.C.; Prior, T.J.; Hubin, T.J. Synthesis, structural studies, and oxidation catalysis of the manganese(II), iron(II), and copper(II) complexes of a 2-pyridylmethyl pendant armed side-bridged cyclam. Inorg. Chem. Commun. 2015, 59, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Nicholson, G.; Greenman, J.; Madden, L.; McRobbie, G.; Pannecouque, C.; De Clercq, E.; Silversides, J.D.; Ullom, R.; Maples, D.L.; et al. Binding optimization through coordination chemistry: CXCR4 chemokine receptor antagonists from ultrarigid metal complexes. J. Am. Chem. Soc. 2009, 131, 3416–3417. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Huskens, D.; Daelemans, D.; Mewis, R.E.; Garcia, C.D.; Cain, A.N.; Carder Freeman, T.N.; Pannecouque, C.; De Clercq, E.; Schols, D.; et al. CXCR4 chemokine receptor antagonists: Nickel(II) complexes of configurationally restricted macrocycles. Dalton Trans. 2012, 41, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Hubin, T.J.; Amoyaw, P.N.A.; Roewe, K.D.; Simpson, N.C.; Maples, R.D.; Freeman, T.N.C.; Cain, A.N.; Le, J.G.; Archibald, S.J.; Khan, S.I.; et al. Synthesis and antimalarial activity of metal complexes of cross-bridged tetraazamacrocyclic ligands. Bioorg. Med. Chem. 2014, 22, 3239–3244. [Google Scholar] [CrossRef] [Green Version]
- Panneerselvam, J.; Jin, J.K.; Shanker, M.; Lauderdale, J.; Bates, J.; Wang, Q.; Zhao, Y.D.; Archibald, S.J.; Hubin, T.J.; Ramesh, R. IL-24 inhibits lung cancer cell migration and invasion by disrupting the SDF-1/CXCR4 signaling axis. PLoS ONE 2015, 10, e0122439. [Google Scholar] [CrossRef]
- Maples, R.D.; Cain, A.N.; Burke, B.P.; Silversides, J.D.; Mewis, R.; D’huys, T.; Schols, D.; Linder, D.P.; Archibald, S.J.; Hubin, T.J. Aspartate-based CXCR4 chemokine receptor binding of cross-bridged tetraazamacrocyclic copper(II) and zinc(II) complexes. Chem. Eur. J. 2016, 22, 12916–12930. [Google Scholar] [CrossRef] [Green Version]
- Cain, A.N.; Freeman, T.N.; Roewe, K.D.; Cockriel, D.L.; Hasley, T.R.; Maples, R.D.; Allbritton, E.M.A.; D’Huys, T.; van Loy, T.; Burke, B.P.; et al. Acetate as a model for aspartate-based CXCR4 chemokine receptor binding of cobalt and nickel complexes of cross-bridged tetraazamacrocycles. Dalton Trans. 2019, 48, 2785–2801. [Google Scholar] [CrossRef]
- Burke, B.P.; Miranda, C.S.; Lee, R.E.; Renard, I.; Nigam, S.; Clemente, G.S.; D’Huys, T.; Ruest, T.; Domarkas, J.; Thompson, J.A.; et al. Cu PET imaging of the CXCR4 chemokine receptor using a cross-bridged cyclam bis-tetraazamacrocyclic antagonist. J. Nucl. Med. 2020, 61, 123–128. [Google Scholar] [CrossRef]
- Bernier, N.; Tripier, R.; Patinec, V.; Le Baccon, M.; Handel, H. Proton-sponge behavior of new pendant armed cross-bridged bis-cyclens: Synthesis, NMR, X-ray, and potentiometric investigations. C. R. Chim. 2007, 10, 832–838. [Google Scholar] [CrossRef]
- Bernier, N.; Costa, J.; Delgado, R.; Felix, V.; Royal, G.; Tripier, R. trans-Methylpyridine cyclen versus cross-bridged trans-methylpyridine cyclen. Synthesis, acid-base and metal complexation studies (metal = Co2+, Cu2+, and Zn2+). Dalton Trans. 2011, 40, 4514–4526. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.P.; Halime, Z.; Marion, R.; Camus, N.; Delgado, R.; Platas-Iglesias, C.; Tripier, R. Monopicolinate cross-bridged cyclam comining very fast complexation with very high stability and inertness of its copper(II) complex. Inorg. Chem. 2014, 53, 5269–5279. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Regueiro-Figueroa, M.; Esteban-Gomez, D.; Tripier, R.; Tircso, G.; Kalman, F.K.; Benyei, A.C.; Toth, I.; de Blas, A.; Rodriguez-Blas, T.; et al. Complexation of Ln3+ ions with cyclam dipicolinates: A small bridge that makes huge differences in structure, equilibrium, and kinetic properties. Inorg. Chem. 2016, 55, 2227–2239. [Google Scholar] [CrossRef] [PubMed]
- AlHaddad, N.; Lelong, E.; Suh, J.-M.; Cordier, M.; Lim, M.H.; Royal, G.; Platas-Iglesias, C.; Bernard, H.; Tripier, R. Copper(II) and zinc(II) complexation with N-ethylene hydroxycyclams and consequences on the macrocyclic backbone configuration. Dalton Trans. 2022, 51, 8640–8656. [Google Scholar] [CrossRef] [PubMed]
- Uzal-Varela, R.; Patinec, V.; Tripier, R.; Valencia, L.; Maneiro, M.; Canle, M.; Platas-Iglesias, C.; Esteban-Gomez, D.; Iglesias, E. On the dissociation pathways of copper complexes relevant as PET imaging agents. J. Inorg. Biochem. 2022, 236, 111951. [Google Scholar] [CrossRef] [PubMed]
- Esteves, C.V.; Lima, L.M.; Mateus, P.; Delgado, R.; Brandao, P.; Felix, V. Cyclen derivatives with two trans-methylnitrophenolic pendant arms: A structural study of their copper(II) and zinc(II) complexes. Dalton Trans. 2013, 42, 6149–6160. [Google Scholar] [CrossRef]
- Esteves, C.; Madureira, J.; Lima, L.M.P.; Mateus, P.; Bento, I.; Delgado, R. Copper(II) and gallium(III) complexes of trans-bis(2-hydroxybenzyl) cyclen derivatives: Absence of a cross-bridge proves surprisingly more favorable. Inorg. Chem. 2014, 53, 4371–4386. [Google Scholar] [CrossRef]
- Brewer, S.M.; Schwartz, T.M.; Mekhail, M.A.; Turan, L.S.; Prior, T.J.; Hubin, T.J.; Janesko, B.G.; Green, K.N. Mechanistic insights into iron-catalized C-H bond activation and C-C coupling. Organometallics 2021, 40, 2467–2477. [Google Scholar] [CrossRef]
- Hubin, T.J.; McCormick, J.M.; Collinson, S.R.; Buchalova, M.; Perkins, C.M.; Alcock, N.W.; Kahol, P.K.; Raghunathan, A.; Busch, D.H. New iron(II) and manganese(II) complexes of two ultra-rigid, cross-bridged tetraazamacrocycles for catalysis and biomimicry. J. Am. Chem. Soc. 2000, 122, 2512–2522. [Google Scholar] [CrossRef]
- Yin, G.C.; McCormick, J.M.; Buchalova, M.; Danby, A.M.; Rodgers, K.; Day, V.W.; Smith, K.; Perkins, C.M.; Kitko, D.; Carter, J.D.; et al. Synthesis, characterization, and solution properties of a novel cross-bridged cyclam manganese(IV) complex having two terminal hydroxo ligands. Inorg. Chem. 2006, 45, 8052–8061. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; England, J.; Que, L. Iron-catalyzed olefin epoxidation and cis-dihydroxylation by tetraalkylcyclam complexes: The importance of cis-labile sites. ACS Catal. 2011, 1, 1035–1042. [Google Scholar] [CrossRef]
- Yin, G.; Danby, A.M.; Kitko, D.; Carter, J.D.; Scheper, W.M.; Busch, D.H. Olefin epoxidation by alkyl hydroperoxide with a novel cross-bridged cyclam manganese complex: Demonstration of oxygenation by two distinct reactive intermediates. Inorg. Chem. 2007, 46, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Valks, G.C.; McRobbie, G.; Lewis, E.A.; Hubin, T.J.; Hunter, T.M.; Sadler, P.J.; Pannecouque, C.; De Clercq, E.; Archibald, S.J. Configurationally restricted bismacrocyclic CXCR4 receptor antagonists. J. Med. Chem. 2006, 49, 6162–6165. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Greenman, J.; Archibald, S.J. Small molecule CXCR4 chemokine receptor antagonists: Developing drug candidates. Curr. Med. Chem. 2007, 14, 2257–2277. [Google Scholar] [CrossRef] [PubMed]
- Archibald, S.J.; Daelemans, D.; Hubin, T.J.; Huskens, D.; Schols, D.; Van Laethem, K.; De Clercq, E.; Pannecouque, C. CXCR4 chemokine receptor antagonists from ultra-rigid metal complexes profoundly inhibit HIV-1 replication, and also AMD3100-resistant strains. Antivir. Res. 2009, 82, A45. [Google Scholar] [CrossRef]
- Hung, Y.; Martin, L.Y.; Jackels, S.C.; Tait, A.M.; Busch, D.H. Ring size effects among metal complexes with macrocyclic ligands: Synthesis, stereochemistry, spectrochemistry, and electrochemistry of cobalt(III) complexes with unsubstituted, saturated tetraaza macrocycles. J. Am. Chem. Soc. 1977, 99, 4029–4039. [Google Scholar] [CrossRef]
- Martin, L.Y.; Sperati, C.R.; Busch, D.H. The spectrochemical properties of tetragonal complexes of high spin nickel(II) containing macrocyclic ligands. J. Am. Chem. Soc. 1977, 99, 2968–2981. [Google Scholar] [CrossRef]
- DeRosa, F.; Bu, X.; Ford, P.C. Synthesis and Photophysical Properties of New Chromium(III) Complexes of N-Derivatized 1,4,8,11-Tetraazacyclotetradecane Ligands cis-[Cr(1,8-R2cyclam)Cl2]Cl, where R is a pendant chromophore. exclusive formation of the cis isomer. Inorg. Chem. 2003, 42, 4171–4178. [Google Scholar] [CrossRef]
- Gasnier, A.; Royal, G.; Terech, P. Metallo-supramolecular gels based on a multitopic cyclam bis-terpyridine platform. Langmuir 2009, 25, 8751–8762. [Google Scholar] [CrossRef]
- Dong, Y.; Farquhar, S.; Gloe, K.; Lindoy, L.F.; Rumbel, B.R.; Turner, P.; Wichmann, K. Metal ion recognition. Interaction of a series of successively N-benzylated derivatives of 1,4,8,11-tetraazacyclotetradecane (cyclam) with selected transition and post-transition metal ions. Dalton Trans. 2003, 8, 1558–1566. [Google Scholar] [CrossRef]
- Dong, Y.; Lawrance, G.A.; Lindoy, L.F.; Turner, P. Macrocyclic ligand design. Interaction of a series of successively N-benzylated derivatives of 1,4,8,11-tetraazacyclotetradecane (cyclam) with copper(ii) and nickel(ii). Dalton Trans. 2003, 8, 1567–1576. [Google Scholar] [CrossRef]
- Dong, Y.; Lindoy, L.F.; Turner, P. Mononuclear and trinuclear palladium (II) complexes of single-and three-ring benzyl-or xylyl-substituted cyclam derivatives. Aust. J. Chem. 2005, 58, 339–344. [Google Scholar] [CrossRef]
- Hubin, T.J.; Walker, A.N.; Davilla, D.J.; Carder Freeman, T.N.; Epley, B.M.; Hasley, T.R.; Amoyaw, P.N.A.; Jain, S.; Archibald, S.J.; Prior, T.J.; et al. Tetraazamacrocyclic derivatives and their metal complexes as antileishmanial leads. Polyhedron 2019, 163, 42–53. [Google Scholar] [CrossRef]
- Alves, L.G.; Souto, M.; Madeira, F.; Adao, P.; Munha, R.F.; Martins, A.M. Syntheses and solid state structures of cyclam-based copper and zinc compounds. J. Organomet. Chem. 2014, 760, 130–137. [Google Scholar] [CrossRef]
- Patinec, V.; Yaouanc, J.J.; Handel, H.; Clement, J.C.; Des Abbayes, H. N1,N7-expeditious dialkylation of cyclen (1,4,7,10-tetraazacyclododecane). An astonishing reactivity of cyclen tricarbonyl molybdenum and chromium complexes. Inorg. Chim. Acta 1994, 220, 347–348. [Google Scholar] [CrossRef]
- Correia, B.B.; Brown, T.R.; Reibenspies, J.H.; Lee, H.-S.; Hancock, R.D. Exciplex formation and aggregation induced emission in Di-(N-benzyl)cyclen and its complexes—Selective fluorescence with lead(II), and as the cadmium(II) complex, with the chloride ion. Eur. J. Inorg. Chem. 2018, 33, 3736–3747. [Google Scholar] [CrossRef]
- Habata, Y.; Taniguchi, A.; Ikeda, M.; Hiraoka, T.; Matsuyama, N.; Otsuka, S.; Kuwahara, S. Argentivorous molecules bearing two aromatic side-arms: Ag+-Pi and CH-Pi interactions in the solid state and in solution. Inorg. Chem. 2013, 52, 2542–2549. [Google Scholar] [CrossRef]
- Ikeda, M.; Sah, A.K.; Iwase, M.; Murashige, R.; Ishi-i, J.-I.; Hasegawa, M.; Kachi-Terajima, C.; Park, K.-M.; Kuwahara, S.; Habata, Y. C-H…Cl-hydrogen bonds in solution and in the solid-state: HgCl2, complexes with cyclen-based cryptands. Dalton Trans. 2017, 46, 3800–3804. [Google Scholar] [CrossRef]
- Bencze, E.S.; Zonta, C.; Mancin, F.; Prins, L.J.; Scrimin, P. Distance between metal centres affects catalytic efficiancy of dinuclear Co(III) complexes in the hydrolysis of a phosphate diester [Co(Bn2Cyclen)](NO3)3. Eur. J. Org. Chem. 2018, 2018, 5375–5381. [Google Scholar] [CrossRef]
- Jeong, G.R.; Kang, S.-G.; Jeong, J.H. Synthesis of C-meso and/or C-racemic isomers of tetraaza macrocyclic copper (II) and nickel (II) complexes bearing one or two N-CH2OCH3 pendant arms. Inorg. Chim. Acta 2011, 379, 64–69. [Google Scholar] [CrossRef]
- Kang, S.-G.; Kweon, J.K.; Lee, Y.H.; Kim, J.-S.; Lee, U. Preparation of the new macropolycycle containing n-ch2-n linkages and its reaction with methanol in the presence of Cu2+ ion: Formation of a copper(II) complex bearing two N-methoxymethyl groups. Bull. Korean Chem. Soc. 2007, 28, 489–492. [Google Scholar]
- Roy, S.; Banergee, A.; Lima, S.; Horn, A.; Sampaio, R.M.S.N.; Ribeiro, N.; Correia, I.; Avecilla, F.; Fernanda, M.; Carvalho, N.N.; et al. Unusual chemistry of Cu(ii) salan complexes: Synthesis, characterization and superoxide dismutase activity. New. J. Chem. 2020, 44, 11457–11470. [Google Scholar] [CrossRef]
- Rheingold, A.L.; Wong, E.S. CCDC 1970812: Experimental Crystal Structure Determination. 2019. Available online: https://www.ccdc.cam.ac.uk/structures/search?sid=ConQuest&coden=001078&year=2019&pid=ccdc:1970812&aulast=Rheingold (accessed on 5 December 2022).
- Rheingold, A.L.; Wong, E.S. CCDC 1973827: Experimental Crystal Structure Determination. 2019. Available online: https://www.ccdc.cam.ac.uk/structures/search?sid=ConQuest&coden=001078&year=2019&pid=ccdc:1973827&aulast=Rheingold (accessed on 5 December 2022).
- Giusti, J.; Chimichi, S.; Ciampolini, M. Configurational isomerism of coordinated tetraazamacrocycles. crystal and molecular structure of carbonato (1,7-dimethyl-1,4,7,10-tetraazacyclododecane)cobalt(III) perchlorate. Inorg. Chim. Acta 1984, 88, 51–54. [Google Scholar] [CrossRef]
- Hubin, T.J.; Alcock, N.W.; Clase, H.J.; Seib, L.L.; Busch, D.H. Synthesis, characterization, and X-ray crystal structures of cobalt(II) and cobalt(III) complexes of four topologically constrained tetraazamacrocycles. Inorg. Chim. Acta 2002, 337, 91–102. [Google Scholar] [CrossRef]
- Hubin, T.J. Synthesis and coordination chemistry of topologically constrained azamacrocycles. Coord. Chem. Rev. 2003, 241, 27–46. [Google Scholar] [CrossRef]
- Matz, D.L.; Jones, D.G.; Roewe, K.D.; Gorbet, M.J.; Zhang, Z.; Chen, Z.Q.; Prior, T.J.; Archibald, S.J.; Yin, G.C.; Hubin, T.J. Synthesis, structural studies, kinetic stability, and oxidation catalysis of the late first row transition metal complexes of 4,10-dimethyl-1,4,7,10-tetraazabicyclo[6.5.2]pentadecane. Dalton Trans. 2015, 44, 12210–12224. [Google Scholar] [CrossRef]
- Hubin, T.J.; Alcock, N.W.; Morton, M.D.; Busch, D.H. Synthesis, structure, and stability in acid of copper(II) and zinc(II) complexes of cross-bridged tetraazamacrocycles. Inorg. Chim. Acta 2003, 348, 33–40. [Google Scholar] [CrossRef]
- Jones, D.G.; Wilson, K.R.; Cannon-Smith, D.J.; Shircliff, A.D.; Zhang, Z.; Chen, Z.Q.; Prior, T.J.; Yin, G.C.; Hubin, T.J. Synthesis, structural studies, and oxidation catalysis of the late-first-row-transition-metal complexes of a 2-pyridylmethyl pendant-armed ethylene cross-bridged cyclam. Inorg. Chem. 2015, 54, 2221–2234. [Google Scholar] [CrossRef]
- Barefield, E.K.; Wagner, F.; Herlinger, A.W.; Dahl, A.R. (1,4,8,11-Tetraazacyclotetradecane)Nickel(II) perchlorate and 1,4,8,11-tetraazacyclotetradecane. Inorg. Synth. 1976, 16, 220–225. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Weisman, G.R.; Ho, S.C.H.; Johnson, V. Tetracyclic tetraamines by glyoxal-macrocyclic tetraamine condensation. Tetrahedron Lett. 1980, 21, 335–338. [Google Scholar] [CrossRef]
- Royal, G.; Dahaoui-Gindrey, V.; Dahaoui, S.; Tabard, A.; Guilard, R.; Pullumbi, P.; Lecomte, C. New synthesis of trans-disubstituted cyclam macrocycles—Elucidation of the disubstitution mechanism on the basis of X-ray data and molecular modeling. Eur. J. Org. Chem. 1998, 1998, 1971–1975. [Google Scholar] [CrossRef]


















| Expected Complex | Color | Yield (g) | Yield (%) | m/z | m/z |
|---|---|---|---|---|---|
| [Co(1)(OAc)]PF6 | pink-purple | 0.506 | 71% | 499 Co(1)(OAc)+ | 439 Co(1)+ |
| [Ni(1)(OAc)]PF6 | pale sky blue | 0.324 | 46% | 498 Ni(1)(OAc)+ | 219 Ni(1)2+ |
| [Cu(1)](PF6)2 | bright blue | 0.291 | 37% | 524 Cu(1)(OAc)(H2O)+ | 222 Cu(1)2+ |
| [Zn(1)(OAc)]PF6 | light tan | 0.400 | 56% | 505 Zn(1)(OAc)+ | 464 Zn(1)(H2O)+ |
| [Co(2)(OAc)]PF6 | pale pink | 0.680 | 54% | 470 Co(2)(OAc)+ | 410 Co(2)+ |
| [Ni(2)(OAc)]PF6 | pale sky blue | 0.927 | 75% | 469 Ni(2)(OAc)+ | 205 Ni(2)2+ |
| [Cu(2)](PF6)2 | brick red | 1.055 | 72% | 560 Cu(2)(PF6)+ | 208 Cu(2)2+ |
| [Zn(2)(OAc)]PF6 | off-white | 0.945 | 76% | 475 Zn(2)(OAc)+ | 436 Zn(2)(H2O)+ |
| M-N Bond Length/Å | ||||
|---|---|---|---|---|
| Compound Identifier or CCDC Number | N-benzyl | N-CH2CH2-N Bridge | N-R | NH |
| [Ni(1)(OH2)2]Cl2 | 2.157(5) | 2.070(11) 2.050(6) | ||
| [Ni(1)(μ-OOCCH3)]PF6 | 2.162(12) 2.154(13) | 2.003(8) 2.060(8) | ||
| [Ni(1)(OOCCH3)(OH2)] (OOCCH3)∙H2O | 2.1443(17) 2.1786(17) | 2.0832(18) 2.0489(17) | ||
| 1566343 | 2.227(3) | 2.122(3) | ||
| 891843 | 2.206(3) 2.184(3) | 2.100(3) 2.089(3) | ||
| 1975750 | 2.096(5) | 2.088(5) | ||
| 1101686 | 2.107 2.111 | 2.187 2.160 | ||
| 1101688 | 2.0921 2.1035 | 2.1509 2.1581 | ||
| 891844 | 2.135(3) 2.128(3) | 2.177(3) 2.188(3) R = Me | ||
| 1891730 | 2.068(2), 2.083(2) 2.067(2), 2.079(2) | 2.096(2), 2.082(2) 2.086(2), 2.094(2) R = CH2COO | ||
| 1891731 | 2.0696(17) 2.0709(17) | 2.0841(17) 2.0970(16) R = CH2C(O)NH-Ar | ||
| Ligand | 30 °C, 1 M HCl | 50 °C, 5 M HCl | 70 °C, 5 M HCl | 90 °C, 5 M HCl | References |
|---|---|---|---|---|---|
 H2Bcyclen | <1 min | [69] | |||
 Me2Bcyclen | 36 min | <1 min | [70] | ||
 Bn2Bcyclen | 4.2 h | <1 min | [this work] [28] | ||
 Bn2cyclen = 1 | 8.47 h | <1 min | [this work] | ||
 H2Bcyclam | 11.8 min | [8] | |||
 Me2Bcyclam | 7.3 day | 79 min | [70,71] | ||
 Bn2Bcyclam | 2.38 h | 24 min | [this work] [28] | ||
 Bn2cyclam = 2 | 33 min | [this work] |
| Complex | Metal Ion | λmax in nm (ε in M−1 cm−1) [sh Indicates a Shoulder on Another Peak] | Ref | |||
|---|---|---|---|---|---|---|
| [Co(Bn2Bcyclen)(OAc)](PF6)2 | Co3+ | 380 (235) | 523 (356) | ----- | ----- | [29] |
| [Co(1)(OAc)]PF6 | Co2+ | 372sh (50) | 549 (58) | ----- | ----- | This work |
| [Co(Bn2Bcyclam)(OAc)]PF6 | Co2+ | 464sh (17) | 510 (20) | 547sh (15) | ----- | [29] |
| [Co(2)(OAc)]PF6 | Co2+ | ---- | 513 (32) | 552sh (23) | 685 (6) | This work |
| [Ni(Bn2Bcyclen)(OAc)]PF6 | Ni2+ | 334 (37) | 559 (10) | 845sh (28) | 951 (36) | [29] |
| [Ni(1)(OAc)]PF6 | Ni2+ | 364 (42) | 587 (19) | 820sh (21) | 985 (48) | This work |
| [Ni(Bn2Bcyclam)(OAc)]PF6 | Ni2+ | 354 (15) | 570 (7) | 829sh (5) | 979 (12) | [25,29] |
| [Ni(2)(OAc)]PF6 | Ni2+ | 364 (22) | 579 (20) | 814sh (20) | 980 (18) | This work |
| Cu(Bn2Bcyclen)(OAc)]PF6 | Cu2+ | 306 (6490) | 728 (140) | ----- | ----- | [28] |
| [Cu(1)(OAc)](PF6) | Cu2+ | 301 (7020) | 607 (465) | ----- | ----- | This work |
| [Cu(Bn2Bcyclam)(OAc)]PF6 | Cu2+ | 306 (6930) | 708 (150) | ----- | ----- | [28] |
| Cu(2)](PF6)2 | Cu2+ | 282 (8374) | 528 (194) | ----- | ----- | This work |
| Complex | E1/2 (V) Co3+/Co2+ | (Ea-Ec) mV | E1/2 (V) Co2+/Co+ | (Ea-Ec) mV | Ref | |
|---|---|---|---|---|---|---|
| Co(Bn2Bcyclen)(C2H3O2)2+ | +0.014 | 109 | −0.640 | 178 | [29] | |
| Co(1)(C2H3O2)+ | +0.705 (ox only) | ----- | +0.043 (red only) | ----- | This work | |
| Eox (V) unassigned | Co3+/Co2+ | (Ea-Ec) mV | E1/2 (V) #2 | (Ea-Ec) mV | ||
| Co(Bn2Bcyclam)(C2H3O2)+ | +1.226 | +0.638 +0.392 | 75 167 | ----- | ----- | [29] |
| Co(2)(C2H3O2)+ | +0.754 | +0.322 | 156 | −0.301 | 266 | This work |
| Eox (V) Ni2+/Ni3+ | E1/2 (V) Ni2+/Ni3+ | (Ea-Ec) mV | Ered (V) Ni2+/Ni+ | |||
| Ni(Bn2Bcyclen)(C2H3O2)+ | +1.170 | +1.117 | 106 | ----- | [29] | |
| Ni(1)(C2H3O2)+ | +1.230 | −1.220 | This work | |||
| Ni(Bn2Bcyclam)(C2H3O2)+ | +1.255 | ----- | ----- | ----- | [29] | |
| Ni(2)(C2H3O2)+ | +1.290 | ----- | 90 | −1.320 | This work | |
| Eox (Cu2+/3+) [V] | Ered (Cu2+/+) [V] | Eox (Cu+/2+) [V] | ||||
| Cu(Bn2Bcyclen)(C2H3O2)+ | +1.465 | −0.637 | ----- | [28] | ||
| Cu(1)(CH3CN)2+ | +1.280 | −0.470 | −0.240 | This work | ||
| Cu(Bn2Bcyclam)(OAc)+ | +1.516 | −0.641 | −0.156 | [28] | ||
| Cu(2)(CH3CN)2+ | ----- | −0.484 | −0.208 | This work |
| (M/L) Complex Formulation for Elemental Analysis | Calc C | Calc H | Calc N | Found C | Found H | Found N |
|---|---|---|---|---|---|---|
| (Co/1) [Co(C22H32N4)(C2H3O2)]PF6 | 45.50 | 5.89 | 8.84 | 45.12 | 5.52 | 8.65 |
| (Ni/1) [Ni(C22H32N4)(C2H3O2)]PF6 · 1.0 H2O | 45.52 | 5.89 | 8.85 | 45.52 | 5.89 | 8.85 |
| (Cu/1) [Cu(C22H32N4)(C2H3O2)]PF6 · 0.8 NH4PF6 | 38.41 | 5.13 | 8.96 | 38.60 | 5.01 | 9.18 |
| (Zn/1) [Zn(C22H32N4)(C2H3O2)]PF6 · 0.5 H2O | 45.69 | 5.75 | 8.88 | 45.71 | 5.39 | 8.97 |
| (Co/2) [Co(C24H36N4)(C2H3O2)]PF6 · 1.0 H2O | 47.21 | 6.25 | 8.47 | 47.45 | 6.07 | 8.53 |
| (Ni/2) [Ni(C24H36N4)(C2H3O2)]PF6 · 1.0 H2O | 47.22 | 6.25 | 8.45 | 47.54 | 6.25 | 8.29 |
| (Cu/2) [Cu(C24H36N4)](PF6)2 · 1.0 H2O | 38.33 | 5.09 | 7.45 | 38.69 | 4.74 | 7.38 |
| (Zn/2) [Zn(C24H36N4)(C2H3O2)]PF6 · 0.1 H2O | 47.91 | 6.06 | 8.60 | 47.62 | 5.82 | 8.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, A.N.; Ayala, M.A.; Mondal, S.; Bergagnini, M.C.; Bui, P.J.D.; Chidester, S.N.; Doeden, C.I.; Esjornson, L.; Sweany, B.R.; Garcia, L.; et al. A Bridge too Far? Comparison of Transition Metal Complexes of Dibenzyltetraazamacrocycles with and without Ethylene Cross-Bridges: X-ray Crystal Structures, Kinetic Stability, and Electronic Properties. Molecules 2023, 28, 895. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28020895
Walker AN, Ayala MA, Mondal S, Bergagnini MC, Bui PJD, Chidester SN, Doeden CI, Esjornson L, Sweany BR, Garcia L, et al. A Bridge too Far? Comparison of Transition Metal Complexes of Dibenzyltetraazamacrocycles with and without Ethylene Cross-Bridges: X-ray Crystal Structures, Kinetic Stability, and Electronic Properties. Molecules. 2023; 28(2):895. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28020895
Chicago/Turabian StyleWalker, Ashlie N., Megan A. Ayala, Somrita Mondal, Mackenzie C. Bergagnini, Phuong John D. Bui, Stephanie N. Chidester, Chad I. Doeden, Louise Esjornson, Brian R. Sweany, Leslie Garcia, and et al. 2023. "A Bridge too Far? Comparison of Transition Metal Complexes of Dibenzyltetraazamacrocycles with and without Ethylene Cross-Bridges: X-ray Crystal Structures, Kinetic Stability, and Electronic Properties" Molecules 28, no. 2: 895. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28020895