
Virtual Screening Strategy to Identify Retinoic Acid-Related Orphan Receptor γt Modulators
 , , , , , and
, , , , , and
Abstract
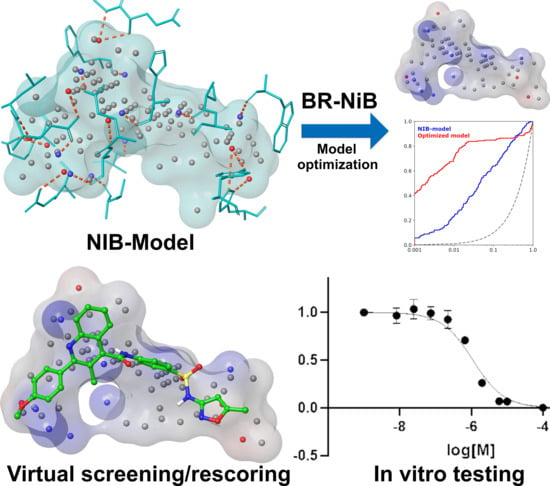
:
1. Introduction
2. Results and Discussion
Shape-Focused Pha Modeling and Docking Rescoring
3. Pharmacophore Filtering Focuses Compound Selection to Inverse Agonists
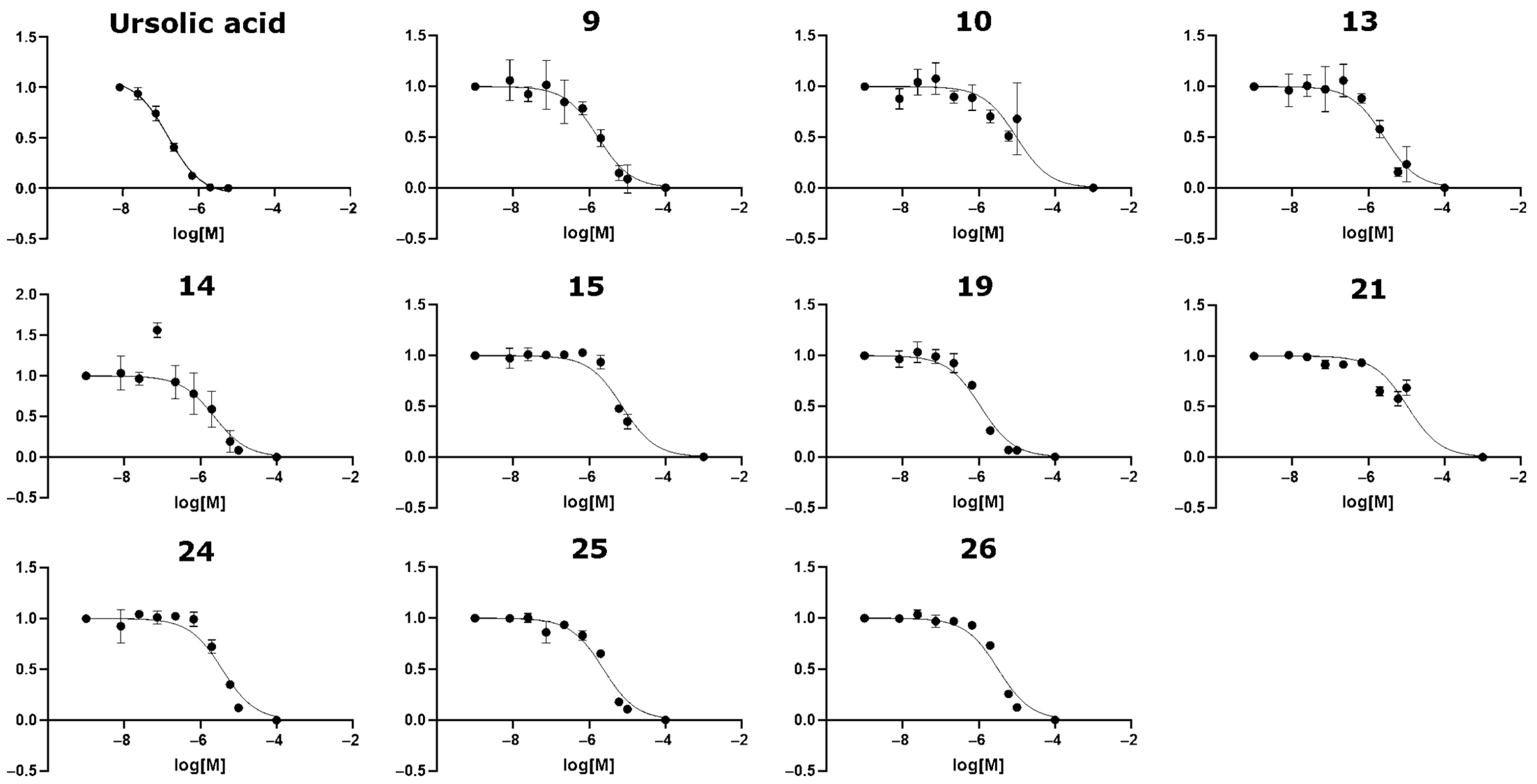
4. Selecting Compounds for Experimental Testing
5. Novel Virtual Screening Strategy Yielded an Effective Hit Rate of 29%
6. Possible Binding Modes of the Most Active Inverse Agonists 9 and 19
7. Materials and Methods
7.1. Protein Structures and Preparation
7.2. Ligand Structures and Preparation
7.3. Nib Models
7.4. Molecular Docking and Rescoring
7.5. Optimization of Nib Models
7.6. Pharmacophore Point Filtering
7.7. Compound Selection
7.8. Experimental Testing
7.9. Molecular Similarity Analysis
7.10. Physicochemical and Admet Property Calculations
7.11. Figure Preparation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ABS | Allosteric binding site |
| ADME | Absorption, distribution, metabolism, and excretion |
| BEDROC | Boltzmann-enhanced discrimination of receiver operating characteristic |
| RMSD | Root-mean-square deviation |
| R-NiB | Negative image-based rescoring |
| BR-NiB | Brute force negative image-based |
| LBR-NiB | Ligand-enhanced brute force negative image-based |
| LBD | Ligand binding domain |
| NIB | Negative image-based |
| PDB | Protein Data Bank |
| RORγt | Retinoic acid-related orphan receptor γt |
| VS | Virtual screening |
| PHA | Pharmacophore |
| ESP | Electrostatic potential |
| MM/GBSA | Molecular mechanics/generalized Born surface area |
References
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, virtual high throughput screening and in silico fragment-based drug design. J. Cell. Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Niinivehmas, S.; Ahinko, M.; Lätti, S.; Pentikäinen, O.T.; Postila, P.A. Improving docking performance using negative image-based rescoring. Front. Pharmacol. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Niinivehmas, S.P.; Salokas, K.; Lätti, S.; Raunio, H.; Pentikäinen, O.T. Ultrafast protein structure-based virtual screening with Panther. J. Comput. Aided. Mol. Des. 2015, 29, 989–1006. [Google Scholar] [CrossRef]
- Vainio, M.J.; Puranen, J.S.; Johnson, M.S. ShaEP: Molecular overlay based on shape and electrostatic potential. J. Chem. Inf. Model. 2009, 49, 492–502. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Lätti, S.; Pentikäinen, O.T.; Postila, P.A. Getting Docking into Shape Using Negative Image-Based Rescoring. J. Chem. Inf. Model. 2019, 59, 3584–3599. [Google Scholar] [CrossRef]
- Jokinen, E.M.; Gopinath, K.; Kurkinen, S.T.; Pentikainen, O.T. Detection of binding sites on SARS-CoV-2 Spike protein receptor-binding domain by molecular dynamics simulations in mixed solvents. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2021, 18, 1281–1289. [Google Scholar] [CrossRef]
- Gopinath, K.; Jokinen, E.M.; Kurkinen, S.T.; Pentikäinen, O.T. Screening of Natural Products Targeting SARS-CoV-2–ACE2 Receptor Interface–A MixMD Based HTVS Pipeline. Front. Chem. 2020, 8, 1084. [Google Scholar] [CrossRef]
- Virtanen, S.I.; Pentikäinen, O.T. Efficient virtual screening using multiple protein conformations described as negative images of the ligand-binding site. J. Chem. Inf. Model. 2010, 50, 1005–1011. [Google Scholar] [CrossRef]
- Niinivehmas, S.P.S.P.; Virtanen, S.I.S.I.; Lehtonen, J.V.J.V.; Postila, P.A.P.A.; Pentikäinen, O.T.O.T. Comparison of virtual high-throughput screening methods for the identification of phosphodiesterase-5 inhibitors. J. Chem. Inf. Model. 2011, 51, 1353–1363. [Google Scholar] [CrossRef]
- Jokinen, E.M.; Postila, P.A.; Ahinko, M.; Niinivehmas, S.; Pentikäinen, O.T. Fragment- and negative image-based screening of phosphodiesterase 10A inhibitors. Chem. Biol. Drug Des. 2019, 94, 1799–1812. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Lehtonen, J.V.; Pentikäinen, O.T.; Postila, P.A. Optimization of Cavity-Based Negative Images to Boost Docking Enrichment in Virtual Screening. J. Chem. Inf. Model. 2022, 62, 1100–1112. [Google Scholar] [CrossRef]
- López-López, E.; Fernández-de Gortari, E.; Medina-Franco, J.L. Yes SIR! On the structure–inactivity relationships in drug discovery. Drug Discov. Today 2022, 27, 2353–2362. [Google Scholar] [CrossRef]
- Lätti, S.; Niinivehmas, S.; Pentikäinen, O.T. Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. J. Cheminform. 2016, 8, 45. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Lehtonen, J.V.; Pentikäinen, O.T.; Postila, P.A. Ligand-Enhanced Negative Images Optimized for Docking Rescoring. Int. J. Mol. Sci. 2022, 23, 7871. [Google Scholar] [CrossRef]
- Huang, M.; Bolin, S.; Miller, H.; Ng, H.L. Rorγ structural plasticity and druggability. Int. J. Mol. Sci. 2020, 21, 5329. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, X.Y.; Wu, D.H.; Xu, Y. ROR nuclear receptors: Structures, related diseases, and drug discovery. Acta Pharmacol. Sin. 2015, 36, 71–87. [Google Scholar] [CrossRef]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009, 7, nrs-07003. [Google Scholar] [CrossRef]
- Mickael, M.E.; Bhaumik, S.; Basu, R. Retinoid-Related Orphan Receptor RORγt in CD4+ T-Cell–Mediated Intestinal Homeostasis and Inflammation. Am. J. Pathol. 2020, 190, 1984–1999. [Google Scholar] [CrossRef]
- Weawer, C.T.; Elson, C.O.; Fouser, L.A.; Kolls, J.K. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu. Rev. Pathol. 2013, 8, 477–512. [Google Scholar]
- Igaki, K.; Nakamura, Y.; Komoike, Y.; Uga, K.; Shibata, A.; Ishimura, Y.; Yamasaki, M.; Tsukimi, Y.; Tsuchimori, N. Pharmacological Evaluation of TAK-828F, a Novel Orally Available RORγt Inverse Agonist, on Murine Colitis Model. Inflammation 2019, 42, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Soroosh, P.; De Leon-Tabaldo, A.; Luna-Roman, R.; Sablad, M.; Rozenkrants, N.; Yu, J.; Castro, G.; Banie, H.; Fung-Leung, W.P.; et al. Pharmacologic modulation of RORγt translates to efficacy in preclinical and translational models of psoriasis and inflammatory arthritis. Sci. Rep. 2016, 6, 37977. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.R.; Lyda, B.; Kamenecka, T.M.; Griffin, P.R. Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor γ is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol. 2014, 66, 579–588. [Google Scholar] [CrossRef]
- Fukase, Y.; Sato, A.; Tomata, Y.; Ochida, A.; Kono, M.; Yonemori, K.; Koga, K.; Okui, T.; Yamasaki, M.; Fujitani, Y.; et al. Identification of novel quinazolinedione derivatives as RORγt inverse agonist. Bioorganic Med. Chem. 2018, 26, 721–736. [Google Scholar] [CrossRef]
- Kumar, N.; Lyda, B.; Chang, M.R.; Lauer, J.L.; Solt, L.A.; Burris, T.P.; Kamenecka, T.M.; Griffin, P.R. Identification of SR2211: A potent synthetic RORγ-selective modulator. ACS Chem. Biol. 2012, 7, 672–677. [Google Scholar] [CrossRef]
- Sun, N.; Xie, Q.; Dang, Y.; Wang, Y. Agonist Lock Touched and Untouched Retinoic Acid Receptor-Related Orphan Receptor-γt (RORγt) Inverse Agonists: Classification Based on the Molecular Mechanisms of Action. J. Med. Chem. 2021, 64, 10519–10536. [Google Scholar] [CrossRef]
- Jetten, A.M.; Cook, D.N. (Inverse) Agonists of Retinoic Acid–Related Orphan Receptor γ: Regulation of Immune Responses, Inflammation, and Autoimmune Disease. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 371–390. [Google Scholar] [CrossRef]
- Sun, N.; Guo, H.; Wang, Y. Retinoic acid receptor-related orphan receptor gamma-t (RORγt) inhibitors in clinical development for the treatment of autoimmune diseases: A patent review (2016-present). Expert Opin. Ther. Pat. 2019, 29, 663–674. [Google Scholar] [CrossRef]
- Sun, N.; Yuan, C.; Ma, X.; Wang, Y.; Gu, X.; Fu, W. Molecular mechanism of action of RORγt agonists and inverse agonists: Insights from molecular dynamics simulation. Molecules 2018, 23, 3181. [Google Scholar] [CrossRef]
- Scheepstra, M.; Leysen, S.; Van Almen, G.C.; Miller, J.R.; Piesvaux, J.; Kutilek, V.; Van Eenennaam, H.; Zhang, H.; Barr, K.; Nagpal, S.; et al. Identification of an allosteric binding site for RORγt inhibition. Nat. Commun. 2015, 6, 8833. [Google Scholar] [CrossRef]
- Kallen, J.; Izaac, A.; Be, C.; Arista, L.; Orain, D.; Kaupmann, K.; Guntermann, C.; Hoegenauer, K.; Hintermann, S. Structural States of RORγt: X-ray Elucidation of Molecular Mechanisms and Binding Interactions for Natural and Synthetic Compounds. ChemMedChem 2017, 12, 1014–1021. [Google Scholar] [CrossRef]
- Lugar, C.W.; Clarke, C.A.; Morphy, R.; Rudyk, H.; Sapmaz, S.; Stites, R.E.; Vaught, G.M.; Furness, K.; Broughton, H.B.; Durst, G.L.; et al. Defining Target Engagement Required for Efficacy in Vivo at the Retinoic Acid Receptor-Related Orphan Receptor C2 (RORγt). J. Med. Chem. 2021, 64, 5470–5484. [Google Scholar] [CrossRef]
- Tan, J.; Liu, H.; Huang, M.; Li, N.; Tang, S.; Meng, J.; Tang, S.; Zhou, H.; Kijlstra, A.; Yang, P.; et al. Small molecules targeting RORγt inhibit autoimmune disease by suppressing Th17 cell differentiation. Cell Death Dis. 2020, 11, 697. [Google Scholar] [CrossRef]
- Song, Y.; Xue, X.; Wu, X.; Wang, R.; Xing, Y.; Yan, W.; Zhou, Y.; Qian, C.N.; Zhang, Y.; Xu, Y. Identification of N -phenyl-2-(N -phenylphenylsulfonamido)acetamides as new RORγ inverse agonists: Virtual screening, structure-based optimization, and biological evaluation. Eur. J. Med. Chem. 2016, 116, 13–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, X.; Jin, X.; Song, Y.; Li, J.; Luo, X.; Song, M.; Yan, W.; Song, H.; Xu, Y. Discovery of 2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide derivatives as new RORγ inhibitors using virtual screening, synthesis and biological evaluation. Eur. J. Med. Chem. 2014, 78, 431–441. [Google Scholar] [CrossRef]
- Rauhamäki, S.; Postila, P.A.; Lätti, S.; Niinivehmas, S.; Multamäki, E.; Liedl, K.R.; Pentikäinen, O.T. Discovery of Retinoic Acid-Related Orphan Receptor γt Inverse Agonists via Docking and Negative Image-Based Screening. ACS Omega 2018, 3, 6259–6266. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, H.; Liu, Z.; Zheng, L.; Yu, Z.; Cao, S.; Fang, W.; Wu, L.; Li, W.; Liu, G.; et al. wSDTNBI: A novel network-based inference method for virtual screening. Chem. Sci. 2022, 13, 1060–1079. [Google Scholar] [CrossRef]
- Damm-Ganamet, K.L.; Arora, N.; Becart, S.; Edwards, J.P.; Lebsack, A.D.; McAllister, H.M.; Nelen, M.I.; Rao, N.L.; Westover, L.; Wiener, J.J.M.; et al. Accelerating Lead Identification by High Throughput Virtual Screening: Prospective Case Studies from the Pharmaceutical Industry. J. Chem. Inf. Model. 2019, 59, 2046–2062. [Google Scholar] [CrossRef]
- Karaś, K.; Sałkowska, A.; Walczak-Drzewiecka, A.; Ryba, K.; Dastych, J.; Bachorz, R.A.; Ratajewski, M. The cardenolides strophanthidin, digoxigenin and dihydroouabain act as activators of the human RORγ/RORγT receptors. Toxicol. Lett. 2018, 295, 314–324. [Google Scholar] [CrossRef]
- Li, Z.; Liu, T.; He, X.; Bai, C. The evolution paths of some reprehensive scaffolds of RORγt modulators, a perspective from medicinal chemistry. Eur. J. Med. Chem. 2022, 228, 113962. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Anderson, M.; Collin, D.; Muegge, I.; Wan, J.; Brennan, D.; Kugler, S.; Terenzio, D.; Kennedy, C.; Lin, S.; et al. Structural studies unravel the active conformation of apo RORγt nuclear receptor and a common inverse agonism of two diverse classes of RORγt inhibitors. J. Biol. Chem. 2017, 292, 11618–11630. [Google Scholar] [CrossRef] [PubMed]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Lätti, S.T.; Niinivehmas, S.; Pentikäinen, O.T. Sdfconf: A Novel, Flexible, and Robust Molecular Data Management Tool. J. Chem. Inf. Model. 2022, 62, 9–15. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Lehtonen, J.V.; Still, D.J.; Rantanen, V.V.; Ekholm, J.; Bjorkland, D.; Iftikhar, Z.; Huhtala, M.; Repo, S.; Jussila, A.; Jaakkola, J.; et al. BODIL: A molecular modeling environment for structure-function analysis and drug design. J. Comput. Aided. Mol. Des. 2004, 18, 401–419. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Ahinko, M.; Kurkinen, S.T.; Niinivehmas, S.P.; Pentikäinen, O.T.; Postila, P.A. A Practical Perspective: The Effect of Ligand Conformers on the Negative Image-Based Screening. Int. J. Mol. Sci. 2019, 20, 2779. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of ant colony optimization to structure-based drug design. In Lecture Notes in Computer Science Vol. 4150: Ant Colony Optimization and Swarm Intelligence-ANTS2006 Proceedings, Brussels, Belgium, 4–7 September 2006; Springer: Berlin/Heidelberg, Germany, 2006; pp. 247–258. [Google Scholar]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Compound Name or SPECS ID | IC50 (µM) 1 | MM/GBSA Energy (kcal/mol) | Max Tanimoto Similarity 3 |
|---|---|---|---|---|
| Control | Ursolic acid | 0.2 | - | - |
| 1 | AE-641/30156032 | Nb | −102.79 | - |
| 2 | AF-399/15599217 | NS | −98.34 | - |
| 3 | AF-399/41318865 | Nb | −115.72 | - |
| 4 | AF-399/41895766 | Nb | −102.31 | - |
| 5 | AF-399/42017292 | Nb | −103.83 | - |
| 6 | AF-399/42017398 | Nb | −98.77 | - |
| 7 | AF-399/42017933 | Nb | −107.31 | - |
| 8 | AG-205/33688028 | Nb | −100.63 | - |
| 9 | AG-205/36940103 | 1.7 | −105.53 | 0.149 |
| 10 | AG-219/11789371 | 9.7 2 | −106.91 | 0.093 |
| 11 | AG-219/12748006 | Nb | −99.60 | - |
| 12 | AG-690/11570086 | Nb | −114.9 | - |
| 13 | AG-690/40752975 | 2.6 | −98.67 | 0.127 |
| 14 | AG-690/40753951 | 2.3 | −99.59 | 0.121 |
| 15 | AH-487/40936254 | 7.3 | −110.82 | 0.140 |
| 16 | AH-487/41654264 | Nb | −112.1 | - |
| 17 | AK-918/11909161 | NS | −102.78 | - |
| 18 | AK-968/12162001 | Nb | −93.12 | - |
| 19 | AK-968/15253414 | 1.1 | −97.45 | 0.171 |
| 20 | AK-968/15359742 | Nb | −98.30 | - |
| 21 | AK-968/15607331 | 10.7 2 | −117.16 | 0.131 |
| 22 | AK-968/15608930 | Nb | −99.75 | - |
| 23 | AK-968/41022069 | Nb | −104.65 | - |
| 24 | AN-648/15596220 | 3.8 | −110.34 | 0.462 |
| 25 | AN-989/41838307 | 2.4 | −108.44 | 0.109 |
| 26 | AN-989/41838397 | 3.2 | −113.62 | 0.114 |
| 27 | AO-081/15386326 | Nb | −107.94 | - |
| 28 | AO-081/15386957 | Nb | −103.42 | - |
| Compound | MW 1 | QPlogKhsa 2 | % Human Oral Absorption 3 | HBD 4 | HBA 5 | QPPCaco 6 | AlogP 7 | RO5 8 | #rtvFG 9 |
|---|---|---|---|---|---|---|---|---|---|
| 9 | 509.994 | 0.297 | 100 | 1 | 10.65 | 2766.242 | 4.894 | 1 | 0 |
| 13 | 547.647 | 1.63 | 93.086 | 1 | 6.5 | 991.865 | 4.795 | 2 | 1 |
| 14 | 517.621 | 1.597 | 90.076 | 1 | 5.75 | 819.461 | 4.812 | 2 | 1 |
| 15 | 614.842 | 0.686 | 40.411 | 1 | 10 | 4.28 | 2.823 | 1 | 0 |
| 19 | 528.581 | 0.432 | 78.699 | 2 | 10.25 | 220.29 | 4.802 | 1 | 0 |
| 24 | 587.773 | 0.277 | 46.497 | 1 | 11.7 | 17.451 | 1.776 | 1 | 1 |
| 25 | 538.62 | 1.131 | 87.639 | 3 | 7 | 859.654 | 5.444 | 2 | 1 |
| 26 | 520.63 | 1.116 | 86.241 | 3 | 7 | 804.649 | 5.239 | 2 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jokinen, E.M.; Niemeläinen, M.; Kurkinen, S.T.; Lehtonen, J.V.; Lätti, S.; Postila, P.A.; Pentikäinen, O.T.; Niinivehmas, S.P. Virtual Screening Strategy to Identify Retinoic Acid-Related Orphan Receptor γt Modulators. Molecules 2023, 28, 3420. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28083420
Jokinen EM, Niemeläinen M, Kurkinen ST, Lehtonen JV, Lätti S, Postila PA, Pentikäinen OT, Niinivehmas SP. Virtual Screening Strategy to Identify Retinoic Acid-Related Orphan Receptor γt Modulators. Molecules. 2023; 28(8):3420. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28083420
Chicago/Turabian StyleJokinen, Elmeri M., Miika Niemeläinen, Sami T. Kurkinen, Jukka V. Lehtonen, Sakari Lätti, Pekka A. Postila, Olli T. Pentikäinen, and Sanna P. Niinivehmas. 2023. "Virtual Screening Strategy to Identify Retinoic Acid-Related Orphan Receptor γt Modulators" Molecules 28, no. 8: 3420. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28083420