
Computational Analysis of Histamine Protonation Effects on H1R Binding

1
Division of Bioinformatics, Institute of Biochemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 91054 Erlangen, Germany
2
Erlangen National High Performance Computing Center (NHR@FAU), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 91058 Erlangen, Germany
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(9), 3774; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093774
Submission received: 29 March 2023
/
Revised: 21 April 2023
/
Accepted: 24 April 2023
/
Published: 27 April 2023
(This article belongs to the Special Issue Computational Approaches: Drug Discovery and Design in Medicinal Chemistry and Bioinformatics II)
Abstract
:Despite numerous studies investigating histamine and its receptors, the impact of histamine protonation states on binding to the histamine H-receptor (HR) has remained elusive. Therefore, we assessed the influence of different histamine tautomers (-tautomer, -tautomer) and charge states (mono- vs. dicationic) on the interaction with the ternary histamine-HR-Gq complex. In atomistic molecular dynamics simulations, the -tautomer formed stable interactions with the receptor, while the -tautomer induced a rotation of the histamine ring by 180° and formed only weaker hydrogen bonding interactions. This suggests that the -tautomer is more relevant for stabilization of the active ternary histamine-HR-Gq complex. In addition to the two monocationic tautomers, the binding of dicationic histamine was investigated, whose interaction with the HR had been observed in a previous experimental study. Our simulations showed that the dication is less compatible with the ternary histamine-HR-Gq complex and rather induces an inactive conformation in the absence of the Gq protein. Our data thus indicate that the charge state of histamine critically affects its interactions with the HR. Ultimately these findings might have implications for the future development of new ligands that stabilize distinct HR activation states.
1. Introduction
The histamine H receptor (HR) is a G protein-coupled receptor (GPCR) that is expressed in many different cell types, including neurons, immune cells, vascular endothelial cells, and smooth muscle cells of the airway or intestinal epithelium [1]. The HR plays an important role in type I hypersensitivity reactions in which histamine is released from mast cells, binds to the receptor, and leads to its activation [2]. Because of its special role in hypersensitivity reactions, the histamine H receptor is one of the most important targets in the treatment of allergic reactions as well as sleep disorders and vomiting [1,3].
HR signal transduction is mediated mainly by the Gq family. After activation and subsequent HR-Gq dissociation, the G subunit can stimulate the phospholipase C, which cleaves the membrane-bound lipid phosphatidylinositol-4,5-bisphosphate (PIP2) into the secondary messengers inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). While IP3, upon binding to the IP3 receptor, causes the release of secondary messengers such as calcium ions (), DAG, as well as the released , ensure the stimulation of protein kinase C (PKC) [4,5]. The latter causes the activation of the NF-B pathway for the immune response signal and thus enables the cell to respond according to the signaled information [6,7,8].
In 2021, the structure of a ternary histamine-HR-Gq complex was published [9], which represented the first experimental structure of histamine in complex with a histamine receptor. This structure allowed the proposal of a “squash to activate and expand to deactivate” mechanism [9]. In this process, the agonist histamine activates the receptor by forming hydrogen bonds with residues in the transmembrane helices H3 and H6 to ”squash” the ligand-binding pocket on the extracellular side while creating a lever-like tension that opens the cavity for the G protein on the intracellular side. In contrast, bulky antagonists expand the orthosteric binding pocket, keeping the transmembrane helices in place and the G-protein binding pocket closed [9]. Despite this important mechanistic information gained from the histamine-HR-Gq ternary complex, one structural detail still remained unclear: since hydrogen atoms are not resolved due to the limited resolution (3.3 Å) of this cryo-EM structure, the protonation state of the bound histamine could not be determined.
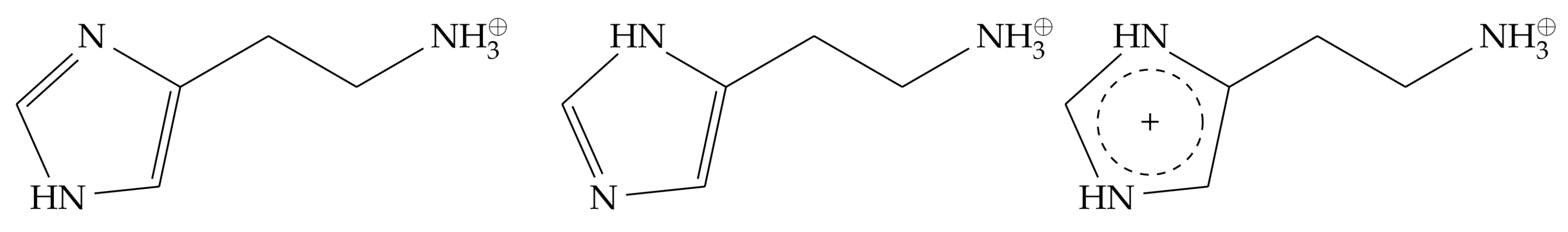
In principle, histamine can exist in two different charge states, either a monocationic or a dicationic form (Figure 1). The monocationic form contains a charged ammonium group in the aliphatic sidechain, while the imidazole ring is singly protonated and consequently uncharged. Depending on the position of the proton, two tautomeric forms exist, referred to as -histamine and -histamine (Figure 1). The -tautomer (pKa 6.16) was reported to be a slightly weaker base than the -tautomer (pKa 6.79) [10]. Whereas most previous modeling studies assumed a monocationic histamine as a ligand [11,12,13], an NMR study by Ratnala et al. [14] has shown that the H1R can bind two different charge states of histamine, suggesting that dicationic histamine might also represent a physiologically relevant form. Based on the structure of the histamine-HR-Gq ternary complex, we, therefore, investigated which histamine charge state best stabilizes the active conformation of the receptor. For that purpose, we performed molecular dynamics simulations to investigate the HR binding properties of different histamine protonation states (i.e., monocationic - and -tautomers as well as the dicationic form).
2. Results
2.1. Initial Structural Analysis of the Histamine-HR-Gq Complex
The cryo-EM structure of the ternary histamine-HR-Gq complex (PDB: 7DFL) provided the first experimental information about the interactions of histamine in the orthosteric binding pocket. However, due to the limited resolution of 3.3 Å, protons are not visible in the structure. Therefore, we first performed an analysis of the static structure to characterize the polar interactions formed between histamine and the H1R (Figure 2). D1073.32 acts as an anchor residue, establishing a salt bridge to the charged ammonium group of the histamine. The side chains of residues T112, N198, and Y431, which are in the vicinity of the imidazole nitrogens, can function both as donors and as acceptors of hydrogen bonds. This suggests that each tautomer (i.e., - or -protonated) or even a diprotonated imidazole ring could be accommodated in the binding pocket (Figure 2b).
Since the protonation state of histamine cannot be unambiguously determined on the basis of the static structure, we performed comparative molecular dynamics (MD) simulations of the two monocationic tautomers and the dicationic form of histamine. These simulations are described in Section 2.2 and Section 2.3. In the respective analyses, amino acids are labeled with superscripts according to the Ballesteros–Weinstein (BW) nomenclature [15], which is explained in more detail at the end of the methods section.
2.2. Monocationic Histamine Tautomers
The first set of simulations aimed to study the interaction of histamine tautomers with the active HR. To ensure maintenance of the active state, the 5-helix of the Gq-protein was included in the simulations in addition to the HR. Depending on the histamine (HSM) tautomer investigated, these simulations are hereafter referred to as HR-HSM--5 or HR-HSM--5. In the case of -protonation, the initial interaction of the histamine imidazole ring with Y4316.51 formed in the crystal structure did not remain stable (Figure 3).
In the first nanoseconds of the simulation, the hydrogen bond to the tyrosine hydroxy group was lost, leading to a rotation of histamine in the binding pocket that affects both the imidazole ring and the aliphatic sidechain. This motion resulted in a rotation of the imidazole ring by about 180°, so that the protonated -nitrogen interacted with the residue S1113.36 (Figure 3b). In contrast, this type of rotation has no effect on the salt bridge of the histamine ammonium group to the strictly conserved D1073.32, which remained stable (Figure 4a). This conformation was adopted throughout most of the simulation, with infrequent fluctuations of the imidazole ring allowing recurrent short-lived interactions with Y4316.51 (Figure 4b).
In simulations of the -tautomer (HR-HSM--5), no 180° rotation of the ring was observed, and the histamine adopted in both simulation runs a conformation similar to the experimental binding mode from the cryoEM (Figure 2b). The interactions to N1985.461 and T1123.37 observed in the experimental structure were preserved in the case of the protonated nitrogen, with the interaction with N1985.461 preferentially formed in about 90% of all structures. This interaction pattern already emerged at the beginning of the simulation after about 50 ns. The interaction with Y4316.51, which was suggested from the crystal structure, was also observed (Figure 2b), but only in the form of transient interactions (Figure 4b) and only when no simultaneous interaction with T1123.37 was formed (Figure 5b).
A comparison of the key hydrogen bonds (Figure 6) showed that these were more stable for the -tautomer than for the -tautomer. The distances between the hydrogen of the protonated -nitrogen and the oxygen of N1985.461 (Figure 6b) were stable in both runs, whereas larger fluctuations occured for the interaction of the -tautomer with S1113.36 (Figure 6a).
It should be noted that the salt bridge of the ammonium group in the aliphatic histamine sidechain to the strongly conserved D1073.32 is preserved in all tautomer simulations with Gq(5). Despite this common principle, there are essentially two findings that render the -tautomer more likely than the -tautomer for interacting with the active HR:
- (i)
- The conformation of the -histamine fits better to the binding mode from the experimental structure, and no 180° rotational fluctuations of the ring are observed during the simulations as in the -tautomer.
- (ii)
- The hydrogen bond network of the imidazole ring is more stable for the -tautomer (Figure 6).
A second set of simulations was used to investigate the extent to which the G protein stabilizes the histamine interaction. For this purpose, simulations were performed in the complete absence of Gq.
In the simulations without the Gq(5) helix, both histamine tautomers displayed a significantly less stable binding. This is particularly evident from the histamine-D1073.32 distance: independently of the tautomer, significant fluctuations of the distance of histamine with its anchor moiety were observed (Figure 7), which suggested a weaker binding compared to the ternary complex investigated above (Figure 4a).
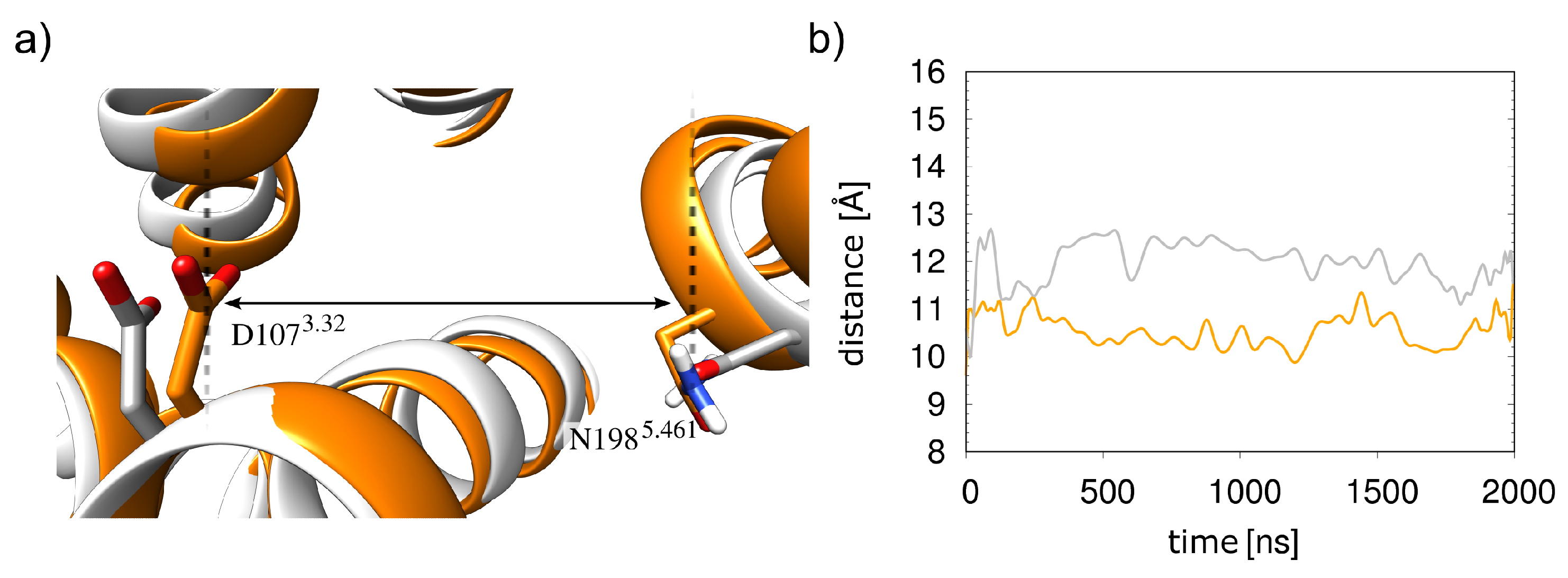
This increased mobility of the histamine results from an opening of the orthosteric pocket compared to the 5-bound receptor. The opening leads to a weaker interaction with histamine since D1073.32 and N1985.461, the major interacting residues, are approximately 1 Å farther apart (Figure 8). In summary, the presence of the Gq protein thus stabilizes the HR-histamine interactions.
2.3. Dicationic Histamine
The dicationic form of histamine (), whose interaction with the HR was demonstrated using NMR spectroscopy [14], showed significantly different behavior in the MD simulations than the monocationic form (). While in all simulations of the monocationic state, the presence of the Gq5 helix stabilized the interaction, the dicationic histamine dissociated in one of the simulations that included the 5 helix. The other simulation run of this system also showed high flexibility of the histamine.
The molecular origin for this instability can be rationalized with a close examination of the histamine dissociation from the orthosteric pocket (Figure 9). First, the charged imidazole ring shifted from its original binding position and contacted D1073.32 (Figure 9b) just as the ammonium group of the histamine side chain did. By increasing the local positive charge density near the negatively charged D1073.32, an easier release of the otherwise tightly bound ammonium group of histamine could occur. Therefore, histamine was able to interact with E18145.51, which extends into the orthosteric pocket (Figure 9c). Hydrogen bonding to the peptide group between T18245.52 and D183 helped stabilize this position (Figure 9c). Subsequently, E18145.51 shifted outwards, allowing contact formation with D186 (Figure 9d). This further exposed the ligand to the solvent, allowing complete dissociation to occur shortly thereafter (Figure 9e).
In the simulations without an 5 helix, remained bound in the orthosteric pocket in both simulation runs. In contrast to run2, which only showed increased fluctuations of in the orthosteric pocket, a drastic change in binding mode was observed in run1 (Figure 10), which was evaluated in more detail below.
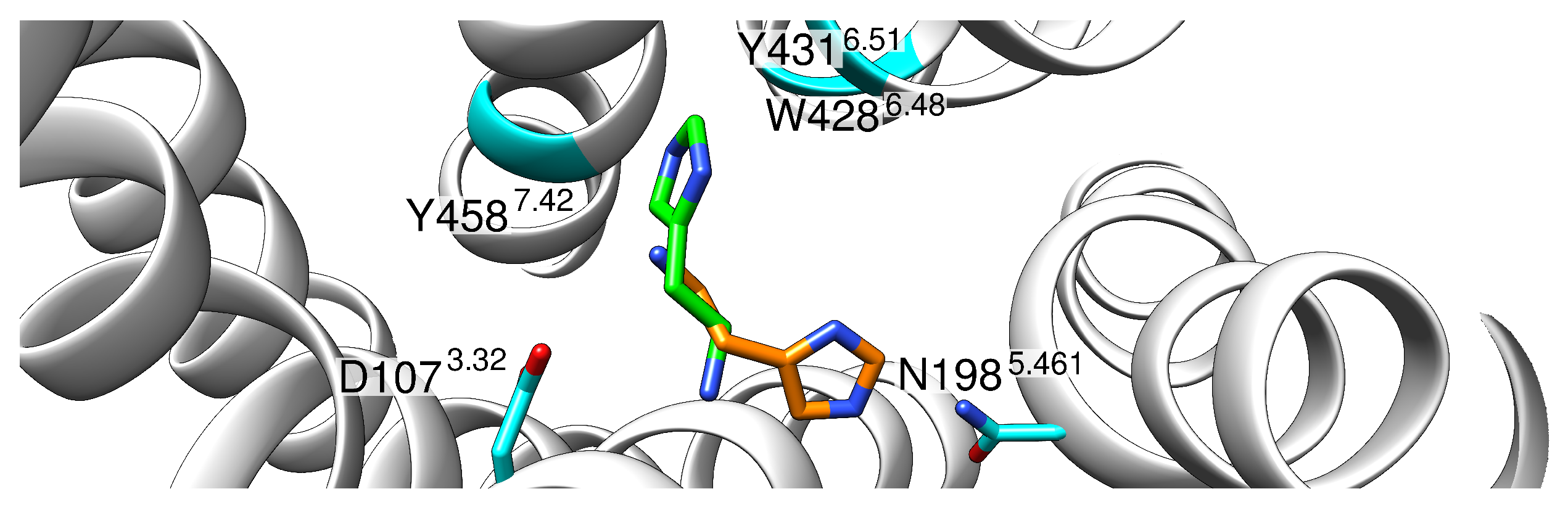
In the newly occupied position, the ammonium group was still tightly bound to D1073.32, but the imidazole ring assumed a position rotated by 120° where it was stabilized with interactions with the side chains of residues Y4587.42 and Y4316.51, as well as W4286.48.
The shift in the binding mode of histamine was accompanied by an inward movement of the intracellular side of H6, as is often observed during GPCR inactivation. To further investigate this finding, the distance between the R1253.50 and E4106.30 residues that form the ionic lock in other receptors was analyzed.
Analysis of the ionic lock distances revealed a strong decrease and formation of a stable salt bridge in run1 of the 5-free simulation. As shown in Figure 11a, there was an inward movement of H6 that favors the formation of the ionic lock.
Further, for (run2), in which no hlionic lock is observed (Figure 11b), stronger fluctuations in the H3-H6 distance are visible than for the monocationic tautomers. Therefore, despite the differences in the exact motion observed, both simulation runs indicate that is significantly less compatible with the active HR conformation than and might rather represent an interaction partner of the inactive HR. This conclusion is supported by the observation that even in the presence of the 5 helix, higher fluctuations are observed for the simulations with compared to (Figure 11c).
3. Discussion
In solution, histamine can exist in two different tautomers, a - and a -form. In contrast to previous studies on histamine protonation, which were based on quantum mechanical calculations in aqueous solution [17,18], the present study explicitly considered the influence of the receptor. Our MD simulations revealed clear differences between both tautomers with respect to the interaction patterns and the stability of the binding conformations. In the simulations, the -tautomer initially established a stable interaction pattern with the receptor after minimization and formed polar interactions with D1073.32, Y4316.51, and N1985.461 (Figure 3a). However, in the MD simulation, this mode of interaction proved unstable and resulted in a 180° rotation of the histamine ring in the binding pocket (Figure 3). This caused a loss of contacts with N1985.461, and alternative contacts were formed with S1113.36 instead. A mutation study by Xia et al., however, showed that mutation of S111 to alanine has little effect on ligand-induced HR activation, indicating that S111 does not participate in the forming of the hydrogen bond network with the ligand [9]. This finding, together with the 180° rotation of the ring, argues against the physiological relevance of the -tautomer. In contrast, the preferred conformation of the -tautomer is in good agreement with the structure from the cryo-EM and formed stable interactions to N1985.461 in the MD-simulations (Figure 5). This finding also agrees well with mutation studies describing the importance of N1985.461 for HR activation [19]. The results show the advantages of molecular dynamics compared to static structural analysis. Based on the crystal structure and static analysis, the differences in conformational stability would not have been apparent. However, one should keep in mind that the initial HR coordinates used might also affect the relative tautomer stability. Therefore, it cannot completely be ruled out that slight rearrangements of the sidechains in the orthosteric pocket of the active HR will allow for more favorable interactions of the -tautomer. This issue is beyond the scope of the present work, but might be addressed by a more complex simulation setup in the future. For example, one could constrain the -protonated histamine in its initial conformation (thereby preventing 180° rotation) and only relax the HR conformation using MD simulations. If the HR remains stable under these conditions, longer MD simulations without histamine restraints would allow verifying the existence of an alternative stable binding mode for the -tautomer. Another alternative to address this question could be docking studies for different histamine tautomers as ligands, explicitly considering the flexibility of the HR side chains in the orthosteric pocket.
Another finding of this work was the stabilizing effect of the G protein on the histamine-HR interaction. When the receptor is stabilized with the G protein, histamine occupies a stable position in the receptor (Figure 4). Without the G protein, larger fluctuations are observed (Figure 7), which can be explained by an opening of the orthosteric binding pocket in the absence of the G protein (Figure 8). This is consistent with the observation that the water-accessible region of the orthosteric binding pocket is larger in inactive GPCRs compared to the active conformation [9,20]. These findings are also in line with previous assumptions regarding the HR activation mechanism, in which the agonist favors contraction of the orthosteric pocket [9]. However, according to the results of the simulations performed here, histamine alone can only partially stabilize the closed form of the orthosteric binding pocket in the absence of the Gq protein.
An NMR study by Ratnala et al. [14] showed that HR-bound histamine can exist both in a monocationic form and in a dicationic form with an additionally charged imidazole ring. The HR binding properties of this dicationic form were therefore investigated using MD-simulations analogous to the tautomers described above. The results of these simulations indicate that the active HR prefers the monocationic form. In particular, this is evident from the higher mobility of the dicationic histamine, which also led to the dissociation of the histamine in the presence of the G protein in one of the simulation runs (Figure 9). Additionally, in both simulations without G protein, the dication induces conformational instability in the active HR (Figure 11b) and even induced a rearrangement towards an inactive conformation in one of the simulation runs (Figure 10). For better statistical significance of this inactivation motion, more and longer simulation runs would be needed, but nevertheless the already conducted simulations offer first evidence for the compatibility of the dicationic histamine with the inactive HR (Figure 11). An independent indication of such a role was already found in the study carried out by Ratnala et al., who proposed a protonation-dependent switch for the HR, analogous to rhodopsin [14]. According to the results of the current study, such a switch could be that dicationic histamine binds to the inactive HR and then promotes the activation of the HR by a transition to the monocationic form. However, further studies required to confirm this mechanism are both experimentally and theoretically challenging: experimentally, measurements would be necessary at different pH values corresponding to the different histamine protonation states. However, this is accompanied by the problem of HR stability, whose structure is also strongly influenced by pH. This makes it difficult to clearly assign observed effects to a change in the histamine protonation state. With simulations, it would first have to be validated that the dicationic histamine was indeed the preferred ligand of the inactive HR. A verification of the postulated protonation-dependent switch for the HR [14] would further require simulating a proton transfer from the dicationic histamine to the HR. This is not possible with conventional MD but will require MD simulations at constant pH to allow proton transfer between the ligand and protein.
In summary, our study was able to establish protonation-dependent differences in the receptor binding properties of histamine and indicate that the monocationic -tautomer is the preferred ligand of the active HR. These differences in the binding behavior were not apparent from the static structural models of the complexes, but could only be captured by simulating the dynamics. This emphasizes the value of MD simulations as a method for the atomistic investigation of protein-ligand complexes and thus also for the development of active substances with tailor-made binding properties.
4. Materials and Methods
The coordinates from the PDB entry 7DFL [9] were used as the starting structure for the simulations of the ternary histamine-H1R-Gq complex with its ligand histamine. Residues 224–401 forming the intracellular loop region (ICL3) between H5 and H6 were deleted from the expression construct for this cryo-EM structure [9]. Xia et al. also demonstrated that the ICL3-deleted receptor still responds to ligand binding, although with increased EC50 compared wild-type receptor [9]. According to the AlphaFold-2 model (https://alphafold.ebi.ac.uk/entry/P35367 (accessed on 2 April 2023)), the ICL3 does not contain significant elements of secondary structure and does not adopt a stable 3D fold. Therefore, microsecond MD simulations appear insufficient for an exhaustive conformational sampling of this loop. Such simulations would most probably result in heterogeneous conformations and interactions of the ICL3 that are difficult to interpret due to the lack of proper statistics. For that reason, we have replaced the missing residues 222–404 with a 9-residue GSGSGSGSG-spacer in the system preparation. According to our experience, using a spacer is superior to simulations with unlinked transmembrane helices because the spacer allows for keeping the helix ends in close spatial proximity, as observed in the experimental structures.
Of the Gq protein, only the 5 helix (residues 334–359), which represents the major HR interaction site, was retained in the complex. Six systems were generated that differ by the presence/absence of the Gq(5) helix and the histamine protonation state (Table 1). For the lipid environment, a preequilibrated DOPC membrane was used similar to prior publications [21].
All simulations were performed using Amber [22]. The force field implementation FF14SB [23] was used for proteins, while lipid14 [24] was used for DOPC molecules. The relevant histamine forms were parameterized separately following the same steps as in [11,21]. Before calculating RESP atomic charges, a structural optimization was carried out, and a frequency calculation ensured the minimum found. For this, R.E.D. with GAMESS [25] was used. Missing parameters were automatically assigned using parmchk2 from the AmberTools suite [22]. RESP charges were derived using the R.E.D. server [26]. Water was described via the TIP3P model [27]. The system was neutralized using and ions for which the Li and Merz 12-6 model was applied [28]. The system was embedded in a simulation box of rectangular shape (Figure 12). Each system was minimized and equilibrated according to a uniform protocol.
The minimization consisted of three successive steps with restraints applied to different subsets of atoms (first to all atoms except water molecules, then to C atoms only, and finally without any restraints). During minimization, 2500 steps of the steepest descent algorithm were applied, followed by 2500 steps of the conjugate gradient algorithm. A harmonic potential with a force constant of 10 kcal·mol·Å was used for the atomic restraints. Membrane equilibration was performed in 300 consecutive Gromacs [29] simulations of 100 ps each. At this stage, water molecules, diffused into the membrane, were removed while the receptor and ligand atoms were held with a force constant of 5 kcal·mol·Å. The temperature was kept constant at 310 K using a Berendsen thermostat [30]. Surface tension coupling was applied with a reference pressure of 1 bar and a reference surface tension of 1.1 nm·bar. After membrane equilibration, the systems were converted to an Amber format and underwent a final equilibration cycle analogous to the steps performed previously. The SHAKE algorithm [31] allowed a time step of 2 fs during the equilibration and production runs. Periodic boundary conditions were set for the x, y, and z directions. A summary of all simulation runs performed can be found in Table 1.
Structural analyses were performed with the tool cpptraj from Amber [22]. To allow for a comparison to other GPCRs, HR residues are labeled by superscripts according to the Ballesteros–Weinstein (BW) nomenclature [15] in addition to their HR sequence position. In the BW numbering scheme, the most conserved position of each transmembrane helix is assigned the number 50. For example, in HR, D732.50 indicates that D73 is the most conserved residue of transmembrane helix 2. The other residues in the N- and C-directions are numbered relative to this position (e.g., A722.49 is N-terminally adjacent to D732.50). Residues with three-digit superscripts (e.g., 461 for N1985.461) indicate an extension of the original BW nomenclature that was introduced for nonconserved positions by the GPCRdb [32].
Author Contributions
Conceptualization, H.S. and M.C.; methodology, M.C. and A.H.C.H.; validation, M.C.; formal analysis, M.C.; investigation, M.C.; resources, H.S.; data curation, M.C.; writing—original draft preparation, M.C.; writing—review and editing, H.S., A.H.C.H. and M.C.; visualization, M.C.; supervision, H.S.; funding acquisition, H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG), GRK1910 (project number 222125149). The authors gratefully acknowledge the Gauss Centre for Supercomputing e.V. (www.gauss-centre.eu) for funding this project by providing computing time on the GCS Supercomputer SuperMUC at Leibniz Supercomputing Centre (www.lrz.de, project pr74su).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data is available upon request.
Acknowledgments
The authors gratefully acknowledge the computing resources and support provided by the Erlangen Regional Computing Center (RRZE) and by NHR@FAU.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Sample Availability
Not applicable.
References
- Hill, S.J. Distribution, properties, and functional characteristics of three classes of histamine receptor. Pharmacol. Rev. 1990, 42, 45–83. [Google Scholar]
- Simons, F.E.R. Advances in H1-antihistamines. N. Engl. J. Med. 2004, 351, 2203–2217. [Google Scholar] [CrossRef]
- Akdis, C.A.; Simons, F.E.R. Histamine receptors are hot in immunopharmacology. Eur. J. Pharmacol. 2006, 533, 69–76. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling. Nature 1993, 361, 315–325. [Google Scholar] [CrossRef]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef]
- Smit, M.; Hoffmann, M.; Timmerman, H.; Leurs, R. Molecular properties and signalling pathways of the histamine H1 receptor. Clin. Exp. Allergy 1999, 29, 19–28. [Google Scholar] [CrossRef]
- Fernández-Nogueira, P.; Noguera-Castells, A.; Fuster, G.; Recalde-Percaz, L.; Moragas, N.; López-Plana, A.; Enreig, E.; Jauregui, P.; Carbó, N.; Almendro, V.; et al. Histamine receptor 1 inhibition enhances antitumor therapeutic responses through extracellular signal-regulated kinase (ERK) activation in breast cancer. Cancer Lett. 2018, 424, 70–83. [Google Scholar] [CrossRef]
- Francis, H.; Glaser, S.; DeMorrow, S.; Gaudio, E.; Ueno, Y.; Venter, J.; Dostal, D.; Onori, P.; Franchitto, A.; Marzioni, M.; et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am. J. Physiol. Cell Physiol. 2008, 295, C499–C513. [Google Scholar] [CrossRef]
- Xia, R.; Wang, N.; Xu, Z.; Lu, Y.; Song, J.; Zhang, A.; Guo, C.; He, Y. Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat. Commun. 2021, 12, 2086. [Google Scholar] [CrossRef]
- Ganellin, C. The tautomer ratio of histamine. J. Pharm. Pharmacol. 1973, 25, 787–792. [Google Scholar] [CrossRef]
- Söldner, C.A.; Horn, A.H.; Sticht, H. Binding of histamine to the H1 receptor—A molecular dynamics study. J. Mol. Model. 2018, 24, 346. [Google Scholar] [CrossRef]
- Durant, G.J.; Ganellin, C.R.; Parsons, M.E. Chemical differentiation of histamine H1-and H2-receptor agonists. J. Med. Chem. 1975, 18, 905–909. [Google Scholar] [CrossRef]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef]
- Ratnala, V.R.; Kiihne, S.R.; Buda, F.; Leurs, R.; de Groot, H.J.; DeGrip, W.J. Solid-state NMR evidence for a protonation switch in the binding pocket of the H1 receptor upon binding of the agonist histamine. J. Am. Chem. Soc. 2007, 129, 867–872. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Elsevier: Amsterdam, The Netherlands, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef]
- Worth, G.A.; King, P.M.; Richards, W.G. Histamine tautomerism and its mode of action. Biochim. Biophys. Acta (BBA)—Gen. Subj. 1990, 1036, 158–161. [Google Scholar] [CrossRef]
- Karpińska, G.; Dobrowolski, J.C.; Mazurek, A.P. Tautomerism of histamine revisited. J. Mol. Struct. THEOCHEM 1996, 369, 137–144. [Google Scholar] [CrossRef]
- Bruysters, M.; Pertz, H.H.; Teunissen, A.; Bakker, R.A.; Gillard, M.; Chatelain, P.; Schunack, W.; Timmerman, H.; Leurs, R. Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur. J. Pharmacol. 2004, 487, 55–63. [Google Scholar] [CrossRef]
- Warne, T.; Edwards, P.C.; Doré, A.S.; Leslie, A.G.; Tate, C.G. Molecular basis for high-affinity agonist binding in GPCRs. Science 2019, 364, 775–778. [Google Scholar] [CrossRef]
- Conrad, M.; Söldner, C.A.; Miao, Y.; Sticht, H. Agonist binding and G protein coupling in histamine H2 receptor: A molecular dynamics study. Int. J. Mol. Sci. 2020, 21, 6693. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. Tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.v.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
Figure 1.
Different histamine protonation forms investigated in this work. Left: monocationic -protonated histamine tautomer. Middle: monocationic -tautomer. Right: dicationic histamine, in which both the and the positions are protonated.
Figure 1.
Different histamine protonation forms investigated in this work. Left: monocationic -protonated histamine tautomer. Middle: monocationic -tautomer. Right: dicationic histamine, in which both the and the positions are protonated.

Figure 2.
HR-histamine interaction in the ternary histamine-HR-Gq complex. (a) Overall topology of the complex. The receptor is shown as a white ribbon, the G in orange, G in cyan, G in green, and a stabilizing antibody in red. (b) Enlargement of the histamine binding site. The histamine and interacting residues of the receptor are shown as sticks. Short distances between polar groups are highlighted with dashed lines indicating the distance in Angstrom (Å). The Ballesteros–Weinstein numbering scheme [15] is used to indicate residue position.
Figure 2.
HR-histamine interaction in the ternary histamine-HR-Gq complex. (a) Overall topology of the complex. The receptor is shown as a white ribbon, the G in orange, G in cyan, G in green, and a stabilizing antibody in red. (b) Enlargement of the histamine binding site. The histamine and interacting residues of the receptor are shown as sticks. Short distances between polar groups are highlighted with dashed lines indicating the distance in Angstrom (Å). The Ballesteros–Weinstein numbering scheme [15] is used to indicate residue position.

Figure 3.
Hydrogen bonding patterns of the ternary histamine HR-Gq(5) complex with the -histamine tautomer (HR-HSM--5). (a) Initial histamine position. (b) Predominant binding mode with a hydrogen bond between the imidazole ring and S1113.36. Blue dashed lines indicate distances below 2.5 Å. H1–H7 denote the transmembrane helices of the receptor.
Figure 3.
Hydrogen bonding patterns of the ternary histamine HR-Gq(5) complex with the -histamine tautomer (HR-HSM--5). (a) Initial histamine position. (b) Predominant binding mode with a hydrogen bond between the imidazole ring and S1113.36. Blue dashed lines indicate distances below 2.5 Å. H1–H7 denote the transmembrane helices of the receptor.

Figure 4.
Interactions of the different histamine tautomers. (a) Distance between the nitrogen of the histamine ammonium group and the carboxyl oxygens of the D1073.32. (b) Distance between the -hydrogen of the imidazole ring and the oxygen of the hydroxy group of Y4316.51 for the -tautomer shown in black and red lines for run1 and run2, respectively. Distance between the -nitrogen atom of the imidazole ring and the hydrogen of the hydroxy group of the Y4316.51 for the -tautomer shown in blue and green lines for run1 and run2, respectively. In both representations, explicit values are highlighted by dots, and running averages are shown as lines.
Figure 4.
Interactions of the different histamine tautomers. (a) Distance between the nitrogen of the histamine ammonium group and the carboxyl oxygens of the D1073.32. (b) Distance between the -hydrogen of the imidazole ring and the oxygen of the hydroxy group of Y4316.51 for the -tautomer shown in black and red lines for run1 and run2, respectively. Distance between the -nitrogen atom of the imidazole ring and the hydrogen of the hydroxy group of the Y4316.51 for the -tautomer shown in blue and green lines for run1 and run2, respectively. In both representations, explicit values are highlighted by dots, and running averages are shown as lines.

Figure 5.
Hydrogen bonding patterns of the ternary histamine-HR-Gq(5) complex with the -histamine tautomer (HR-HSM--5) (a) Hydrogen bonding interaction between N1985.461 and T1123.37. (b) Hydrogen bonding interaction between N1985.461 and Y4316.51.
Figure 5.
Hydrogen bonding patterns of the ternary histamine-HR-Gq(5) complex with the -histamine tautomer (HR-HSM--5) (a) Hydrogen bonding interaction between N1985.461 and T1123.37. (b) Hydrogen bonding interaction between N1985.461 and Y4316.51.

Figure 6.
Key hydrogen bonds for different histamine tautomers in the HR-5 complex. (a) Distance between the hydrogen of the protonated -nitrogen and the O-oxygen of S1113.36. (b) Distance between the hydrogen of the protonated nitrogen and the O-oxygen of N1985.461.
Figure 6.
Key hydrogen bonds for different histamine tautomers in the HR-5 complex. (a) Distance between the hydrogen of the protonated -nitrogen and the O-oxygen of S1113.36. (b) Distance between the hydrogen of the protonated nitrogen and the O-oxygen of N1985.461.

Figure 7.
Interaction of histamine with D1073.32 in the absence of Gq(5). Distances between the ammonium nitrogen of histamine and the carboxyl oxygens of D1073.32 for (a) the -tautomer and (b) the -tautomer.
Figure 7.
Interaction of histamine with D1073.32 in the absence of Gq(5). Distances between the ammonium nitrogen of histamine and the carboxyl oxygens of D1073.32 for (a) the -tautomer and (b) the -tautomer.

Figure 8.
Histamine-binding pocket in the presence and absence of Gq(5). (a) The structure from the simulations with and without 5-helix are shown in orange and white, respectively. (b) Distances between the D1073.32 carboxylate and the N1985.461 side-chain nitrogen as measured during the simulations, in the same colors as in (a).
Figure 8.
Histamine-binding pocket in the presence and absence of Gq(5). (a) The structure from the simulations with and without 5-helix are shown in orange and white, respectively. (b) Distances between the D1073.32 carboxylate and the N1985.461 side-chain nitrogen as measured during the simulations, in the same colors as in (a).

Figure 9.
Dissociation of the . (a) Histamine positions observed during dissociation. Histamine is shown by carbon atoms highlighted in green in sticks The starting position from the crystal structure is shown in orange, the receptor in white band representation. (b–e) show the structures from the HR--5 run 2 at 0 ns, 100 ns, 240 ns, and 346 ns. Interactions are shown by dashed lines.
Figure 9.
Dissociation of the . (a) Histamine positions observed during dissociation. Histamine is shown by carbon atoms highlighted in green in sticks The starting position from the crystal structure is shown in orange, the receptor in white band representation. (b–e) show the structures from the HR--5 run 2 at 0 ns, 100 ns, 240 ns, and 346 ns. Interactions are shown by dashed lines.

Figure 10.
Rearranged binding position of . Histamine coordinates before (orange) and after (green) rearrangement. Histamine is shown with green/orange carbon atoms, and interacting polar residues are shown in cyan. Positions of the aromatic residues Y4587.42, Y4316.51, as well as W4286.48, are marked while their sidechains are not displayed to enhance clarity.
Figure 10.
Rearranged binding position of . Histamine coordinates before (orange) and after (green) rearrangement. Histamine is shown with green/orange carbon atoms, and interacting polar residues are shown in cyan. Positions of the aromatic residues Y4587.42, Y4316.51, as well as W4286.48, are marked while their sidechains are not displayed to enhance clarity.

Figure 11.
(a) Overlay of helices H3 and H6 for the inactive (3RZE [16]; orange) and active (7DFL [9]; cyan) HR and with a representative structure from simulation run 1 of without Gq(5). Residues R125 and E410 are marked red and blue, respectively, in the band plot. Distance between R1253.50 to E4106.30 (b) without Gq(5) and (c) with Gq(5).
Figure 11.
(a) Overlay of helices H3 and H6 for the inactive (3RZE [16]; orange) and active (7DFL [9]; cyan) HR and with a representative structure from simulation run 1 of without Gq(5). Residues R125 and E410 are marked red and blue, respectively, in the band plot. Distance between R1253.50 to E4106.30 (b) without Gq(5) and (c) with Gq(5).

Figure 12.
Overview of the simulated system. The HR and the Gq 5-helix are shown as white and orange tubes, respectively. Phospholipids are shown in a space-filled presentation with carbons in orange. Only a subset of the phospholipids is shown to allow for a side-view of the embedded receptor.
Figure 12.
Overview of the simulated system. The HR and the Gq 5-helix are shown as white and orange tubes, respectively. Phospholipids are shown in a space-filled presentation with carbons in orange. Only a subset of the phospholipids is shown to allow for a side-view of the embedded receptor.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of the simulations performed for the histamine-HR-Gq system. The table describes the composition of the systems, i.e., in which protonation state the ligand histamine or the Gq-5 helix (✓) was present. The symbol (×) indicates the absence of the respective component in the arrangement.
Table 1.
Overview of the simulations performed for the histamine-HR-Gq system. The table describes the composition of the systems, i.e., in which protonation state the ligand histamine or the Gq-5 helix (✓) was present. The symbol (×) indicates the absence of the respective component in the arrangement.
| System Name | Runs × Time | Histamine | 5 Helix | # Atoms | # DOPC |
|---|---|---|---|---|---|
| HR-HSM- | 2 × 2 s | × | 125,354 | 277 | |
| HR-HSM--5 | 2 × 2 s | ✓ | 125,794 | 277 | |
| HR-HSM- | 2 × 2 s | × | 124,943 | 278 | |
| HR-HSM--5 | 2 × 2 s | ✓ | 125,392 | 278 | |
| HR- | 2 × 2 s | × | 124,942 | 278 | |
| HR--5 | 2 × 2 s | ✓ | 125,391 | 278 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Conrad, M.; Horn, A.H.C.; Sticht, H. Computational Analysis of Histamine Protonation Effects on H1R Binding. Molecules 2023, 28, 3774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093774
AMA Style
Conrad M, Horn AHC, Sticht H. Computational Analysis of Histamine Protonation Effects on H1R Binding. Molecules. 2023; 28(9):3774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093774
Chicago/Turabian StyleConrad, Marcus, Anselm H. C. Horn, and Heinrich Sticht. 2023. "Computational Analysis of Histamine Protonation Effects on H1R Binding" Molecules 28, no. 9: 3774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093774