

Density Functional Calculation and Evaluation of the Spectroscopic Properties and Luminescent Material Application Potential of the N-Heterocyclic Platinum(II) Tetracarbene Complexes

Abstract
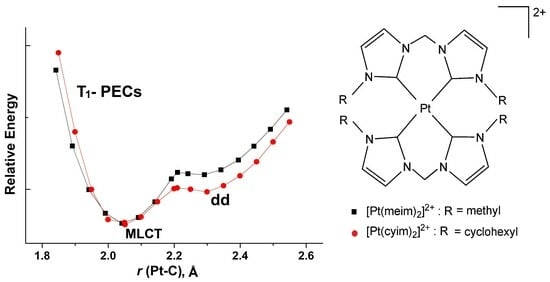
:
1. Introduction
2. Results and Discussion
3. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hahn, F.E.; Jahnke, M.C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Blacque, O.; Venkatesan, K. Highly Efficient Deep-Blue Emitters Based on cis and trans N-Heterocyclic Carbene PtII Acetylide Complexes: Synthesis, Photophysical Properties, and Mechanistic Studies. Chem. Eur. J. 2013, 19, 15689–15701. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.K.; Schwarz, J.; Frey, G.D.; Herdtweck, E.; Herrmann, W.A. Chiral, Bridged Bis(imidazolin-2-ylidene) Complexes of Palladium. J. Organomet. Chem. 2007, 692, 4560–4568. [Google Scholar] [CrossRef]
- Huh, J.-S.; Lee, D.Y.; Park, K.H.; Kwon, S.-K.; Kim, Y.-H.; Kim, J.-J. Control of the horizontal dipole ratio and emission color of deep blue tetradentate Pt(II) complexes using aliphatic spacer groups. Chem. Eng. J. 2022, 450, 137836. [Google Scholar] [CrossRef]
- McKie, R.; Murphy, J.A.; Park, S.R.; Spicer, M.D.; Zhou, S. Homoleptic Crown N-Heterocyclic Carbene Complexes. Angew. Chem. Int. Ed. 2007, 46, 6525–6528. [Google Scholar] [CrossRef] [PubMed]
- Tenne, M.; Strassner, T. Neutral platinum(II) N-heterocyclic carbene complexes with tetrazolide-tethered imidazolin-2-ylidene ligands. J. Organomet. Chem. 2016, 821, 100–105. [Google Scholar] [CrossRef]
- Albrecht, M. C4-bound Imidazolylidenes: From Curiosities to High-Impact Carbene Ligands. Chem. Commun. 2008, 3601–3610. [Google Scholar] [CrossRef]
- Hudson, Z.M.; Sun, C.; Helander, M.G.; Chang, Y.L.; Lu, Z.H.; Wang, S.N. Highly Efficient Blue Phosphorescence from Triarylboron-Functionalized Platinum(II) Complexes of N-Heterocyclic Carbenes. J. Am. Soc. Chem. 2012, 134, 13930–13933. [Google Scholar] [CrossRef]
- Shen, Y.; Kong, X.; Yang, F.; Bian, H.-D.; Cheng, G.; Cook, T.R.; Zhang, Y. Deep Blue Phosphorescence from Platinum Complexes Featuring Cyclometalated N-Pyridyl Carbazole Ligands with Monocarborane Clusters (CB11H12−). Inorg. Chem. 2022, 61, 16707–16717. [Google Scholar] [CrossRef]
- Unger, Y.; Zeller, A.; Ahrens, S.; Strassner, T. Blue Phosphorescent Emitters: New N-Heterocyclic Platinum(II) Tetracarbene Complexes. Chem. Commun. 2008, 3263–3265. [Google Scholar] [CrossRef]
- Unger, Y.; Zeller, A.; Taige, M.A.; Strassner, T. Near-UV Phosphorescent Emitters: N-Heterocyclic Platinum(II) Tetracarbene Complexes. Dalton Trans. 2009, 38, 4786–4794. [Google Scholar] [CrossRef] [PubMed]
- Bárta, O.; Pinter, P.; Císařová, I.; Strassner, T.; Štěpnička, P. Synthesis and Characterization of Cationic Platinum(II) Complexes with Two Chelating Ligands. Eur. J. Inorg. Chem. 2020, 2020, 575–580. [Google Scholar] [CrossRef]
- Bai, F.-Q.; Zhou, X.; Xia, B.-H.; Liu, T.; Zhang, J.-P.; Zhang, H.-X. Electronic structures and optical properties of neutral substituted fluorene-based cyclometalated platinum(II)—Acetylide complexes: A DFT exploration. J. Organomet. Chem. 2009, 694, 1848–1860. [Google Scholar] [CrossRef]
- Fan, H.-W.; Bai, F.-Q.; Zhang, Z.-X.; Wang, Y.; Qu, Z.-X.; Zhong, R.-L.; Zhang, H.-X. Theoretical investigation on the effect of ancillary ligand modification for highly efficient phosphorescent platinum(II) complex design. RSC Adv. 2017, 7, 17368–17376. [Google Scholar] [CrossRef]
- Feng, T.-T.; Bai, F.-Q.; Xie, L.-M.; Tang, Y.; Zhang, H.-X. Theoretical study and design of highly efficient platinum(II) complexes bearing tetradentate ligands for OLED. RSC Adv. 2016, 6, 11648–11656. [Google Scholar] [CrossRef]
- Strassner, T.; Taige, M.A. Evaluation of Functionals O3LYP, KMLYP, and MPW1K in Comparison to B3LYP for Selected Transition-Metal Compounds. J. Chem. Theory Comput. 2005, 1, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Bolink, H.J.; Cappelli, L.; Cheylan, S.; Coronado, E.; Costa, R.D.; Lardiés, N.; Nazeeruddinb, M.K.; Ortí, E. Origin of the Large Spectral Shift in Electroluminescence in a Blue Light Emitting Cationic Iridium(III) Complex. J. Mater. Chem. 2007, 17, 5032–5041. [Google Scholar] [CrossRef]
- Jamorski, C.; Casida, M.E.; Salahub, D.R. Dynamic Polarizabilities and Excitation Spectra from A Molecular Implementation of Time-Dependent Density-Functional Response Theory: N2 as a Case Study. J. Chem. Phys. 1996, 104, 5134–5147. [Google Scholar] [CrossRef]
- Petersilka, M.; Grossmann, U.J.; Gross, E.K.U. Excitation Energies from Time-Dependent Density-Functional Theory. Phys. Rev. Lett. 1996, 76, 1212–1215. [Google Scholar] [CrossRef]
- Tong, G.S.M.; Che, C.M. Emissive or Nonemissive? A Theoretical Analysis of the Phosphorescence Efficiencies of Cyclometalated Platinum(II) Complexes. Chem.-A Eur. J. 2009, 15, 7225–7237. [Google Scholar] [CrossRef] [PubMed]
- Siddique, Z.A.; Yamamoto, Y.; Ohno, T.; Nozaki, K. Structure-Dependent Photophysical Properties of Singlet and Triplet Metal-to-Ligand Charge Transfer States in Copper(I) Bis(diimine) Compounds. Inorg. Chem. 2003, 42, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Gareth Williams, J.A. Photochemistry and Photophysics of Coordination Compounds: Platinum. Top. Curr. Chem. 2007, 281, 205–268. [Google Scholar]
- Islam, A.; Ikeda, N.; Nozaki, K.; Ohno, T. Role of Higher Excited States in Radiative and Nonradiative Processes of Coordination Compounds of Ru(II) and Rh(III) in Crystal. Chem. Phys. Lett. 1996, 263, 209–214. [Google Scholar] [CrossRef]
- Durham, B.; Casper, J.V.; Nagle, J.K.; Meyer, T.J. Photochemistry of Tris(2,2′-bipyridine)ruthenium(2+) Ion. J. Am. Chem. Soc. 1982, 104, 4803–4810. [Google Scholar] [CrossRef]
- Allen, G.H.; White, R.P.; Rillema, D.P.; Meyer, T.J. Synthetic Control of Excited-State Properties. Tris-Chelate Complexes Containing the Ligands 2,2′-bipyrazine, 2,2′-bipyridine, and 2,2′-bipyrimidine. J. Am. Chem. Soc. 1984, 106, 2613–2620. [Google Scholar] [CrossRef]
- Salassa, L.; Garino, C.; Salassa, G.; Gobetto, R.; Nervi, C. Mechanism of Ligand Photodissociation in Photoactivable [Ru(bpy)2L2]2+ Complexes: A Density Functional Theory Study. J. Am. Chem. Soc. 2008, 130, 9590–9597. [Google Scholar] [CrossRef]
- Saito, K.; Nakao, Y.; Sakaki, S. Theoretical Study of Pyrazolate-Bridged Dinuclear Platinum(II) Complexes: Interesting Potential Energy Curve of the Lowest Energy Triplet Excited State and Phosphorescence Spectra. Inorg. Chem. 2008, 47, 4329–4337. [Google Scholar] [CrossRef]
- Yang, L.; Okuda, F.; Kobayashi, K.; Nozaki, K.; Tanabe, Y.; Ishii, Y.; Haga, M. Syntheses and Phosphorescent Properties of Blue Emissive Iridium Complexes with Tridentate Pyrazolyl Ligands. Inorg. Chem. 2008, 47, 7154–7165. [Google Scholar] [CrossRef]
- Yu, J.-K.; Hu, Y.-H.; Cheng, Y.-M.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Carty, A.J.; Tung, Y.-L.; Lee, S.-W.; Chi, Y.; et al. A Remarkable Ligand Orientational Effect in Osmium-Atom-Induced Blue Phosphorescence. Chem. Eur. J. 2004, 10, 6255–6264. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Evans, N.; Klein, C.; Zakeeruddin, S.M.; Moser, J.-E.; Kalyanasundaram, K.; Bolink, H.J.; Grätzel, M.; Nazeeruddin, M.K. Controlling Phosphorescence Color and Quantum Yields in Cationic Iridium Complexes: A Combined Experimental and Theoretical Study. Inorg. Chem. 2007, 46, 5989–6001. [Google Scholar] [CrossRef] [PubMed]
- Fazzi, D.; Grancini, G.; Maiuri, M.; Brida, D.; Cerullo, G.; Lanzani, G. Ultrafast Internal Conversion in A Low Band Gap Polymer for Photovoltaics: Experimental and Theoretical Study. Phys. Chem. Chem. Phys. 2012, 14, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.; Harriman, A.; Mayeux, A. The Triplet Excited State of Ruthenium(II) Bis(2,2:6,2-terpyridine): Comparison Between Experiment and Theory. Phys. Chem. Chem. Phys. 2004, 6, 1157–1164. [Google Scholar] [CrossRef]
- Benniston, A.C.; Chapman, G.; Harriman, A.; Mehrabi, M.; Sams, C.A. Electron Delocalization in a Ruthenium(II) Bis(2,2′:6′,2″-terpyridyl) Complex. Inorg. Chem. 2004, 43, 4227–4233. [Google Scholar] [CrossRef]
- Hutchison, G.H.; Ratner, M.A.; Marks, T.J. Hopping Transport in Conductive Heterocyclic Oligomers: Reorganization Energies and Substituent Effects. J. Am. Chem. Soc. 2005, 127, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Cheng, C.P.; Lao, Z.P.M. Reorganization Energies in the Transports of Holes and Electrons in Organic Amines in Organic Electroluminescence Studied by Density Functional Theory. J. Phys. Chem. A 2003, 107, 5241–5251. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Benfield, P.; Helgaker, T.; Tozer, D.J. Excitation Energies in Density Functional Theory: An Evaluation and A Diagnostic Test. J. Chem. Phys. 2008, 128, 044118. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of The Colle-Salvetti Correlation-Energy Formula into A Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Accuracy of AHn Equilibrium Geometries by Single Determinant Molecular Orbital Theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- Li, P.; Feng, L.; Li, G.; Bai, F.-Q. Effects of Electron Donating Ability of Substituents and Molecular Conjugation on the Electronic Structures of Organic Radicals. Chem. Res. Chin. Univ. 2023, 39, 202–207. [Google Scholar] [CrossRef]
- Lam, W.H.; Lam, E.S.-H.; Yam, V.W.-W. Computational Studies on the Excited States of Luminescent Platinum (II) Alkynyl Systems of Tridentate Pincer Ligands in Radiative and Nonradiative Processes. J. Am. Soc. Chem. 2013, 135, 15135–15143. [Google Scholar] [CrossRef] [PubMed]
- Vlček, A.; Zališ, S. Modeling of Charge-Transfer Transitions and Excited States in d6 Transition Metal Complexes by DFT Techniques. Coord. Chem. Rev. 2007, 251, 258–287. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1965, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron Transfer Reactions in Chemistry. Theory and Experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Pt(meim)2]2+ | [Pt(cyim)2]2+ | [Pt(meim)(cyim)]2+ | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Exp10 | S0/1Ag | T1/3Bu | MC State/3Bg | S0/1Ag | T1/3Bu | MC State/3Bg | S0/1A′ | T1/3A″ | |
| Bond length (Å) | |||||||||
| r (Pt–C1) | 2.026 | 2.032 | 2.042 | 2.292 | 2.034 | 2.051 | 2.293 | 2.031/2.036 | 2.036/2.058 |
| r (C1–N1) | 1.345 | 1.353 | 1.378 | 1.357 | 1.355 | 1.382 | 1.357 | 1.352/1.354 | 1.389/1.370 |
| r (C1–N2) | 1.352 | 1.361 | 1.384 | 1.365 | 1.364 | 1.381 | 1.366 | 1.363/1.364 | 1.395/1.376 |
| r (N2–C4) | 1.454 | 1.458 | 1.451 | 1.458 | 1.458 | 1.450 | 1.457 | 1.455/1.458 | 1.449/1.453 |
| Bite angle (degree) | |||||||||
| a (C1–Pt–C7) | 84.2 | 83.3 | 84.5 | 81.6 | 82.9 | 84.0 | 81.0 | 83.1/83.0 | 83.8/84.0 |
| a (C1–Pt–C7a) | 95.9 | 96.7 | 95.5 | 98.4 | 97.1 | 95.8 | 99.0 | 96.5/96.6 | 96.1/96.1 |
| a (N1–C1–Pt) | 133.0 | 134.2 | 133.2 | 134.6 | 134.7 | 134.2 | 134.9 | 133.9/134.7 | 132.9/134.1 |
| a (N2–C1–Pt) | 122.2 | 121.1 | 122.7 | 120.2 | 120.0 | 121.3 | 119.2 | 121.2/120.1 | 123.1/121.2 |
| a (N1–C1–N2) | 104.8 | 104.6 | 103.8 | 104.0 | 105.0 | 104.1 | 104.4 | 104.7/105.0 | 103.5/104.4 |
| Dihedral angle (degree) | |||||||||
| d (C7–C1–Pt–C7a) | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 179.6 | 178.2 |
| d (C1–C4–Pt–C7) | 124.7 | 122.2 | 125.9 | 126.6 | 118.4 | 122.7 | 123.7 | 122.0/121.1 | 124.7/123.0 |
| [Pt(meim)2]2+ | [Pt(cyim)2]2+ | TPD [36] | ||
|---|---|---|---|---|
| S0 | LUMO | 0.2% | 0.3% | |
| HOMO | 35.3% | 33.1% | ||
| T1-MLCT | HSOMO | 0.3% | 0.1% | |
| HSOMO-1 | 20.9% | 19.8% | ||
| T1-dd | HSOMO | 34.2% | 31.3% | |
| HSOMO-1 | 10.3% | 8.7% | ||
| λ(d-d)/eV | 1.456 | 1.419 | ||
| λi(h)/eV | 0.127 | 0.140 | 0.270 | |
| λi(e)/eV | 0.326 | 0.284 | 0.690 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, B.-H.; Ma, Y.-S.; Bai, F.-Q. Density Functional Calculation and Evaluation of the Spectroscopic Properties and Luminescent Material Application Potential of the N-Heterocyclic Platinum(II) Tetracarbene Complexes. Molecules 2024, 29, 524. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29020524
Xia B-H, Ma Y-S, Bai F-Q. Density Functional Calculation and Evaluation of the Spectroscopic Properties and Luminescent Material Application Potential of the N-Heterocyclic Platinum(II) Tetracarbene Complexes. Molecules. 2024; 29(2):524. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29020524
Chicago/Turabian StyleXia, Bao-Hui, Yin-Si Ma, and Fu-Quan Bai. 2024. "Density Functional Calculation and Evaluation of the Spectroscopic Properties and Luminescent Material Application Potential of the N-Heterocyclic Platinum(II) Tetracarbene Complexes" Molecules 29, no. 2: 524. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29020524