
Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements


School of Chemistry, University College Dublin, Belfield, D04 V1W8 Dublin, Ireland
Molecules 2024, 29(6), 1409; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29061409
Submission received: 6 March 2024
/
Revised: 18 March 2024
/
Accepted: 20 March 2024
/
Published: 21 March 2024
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
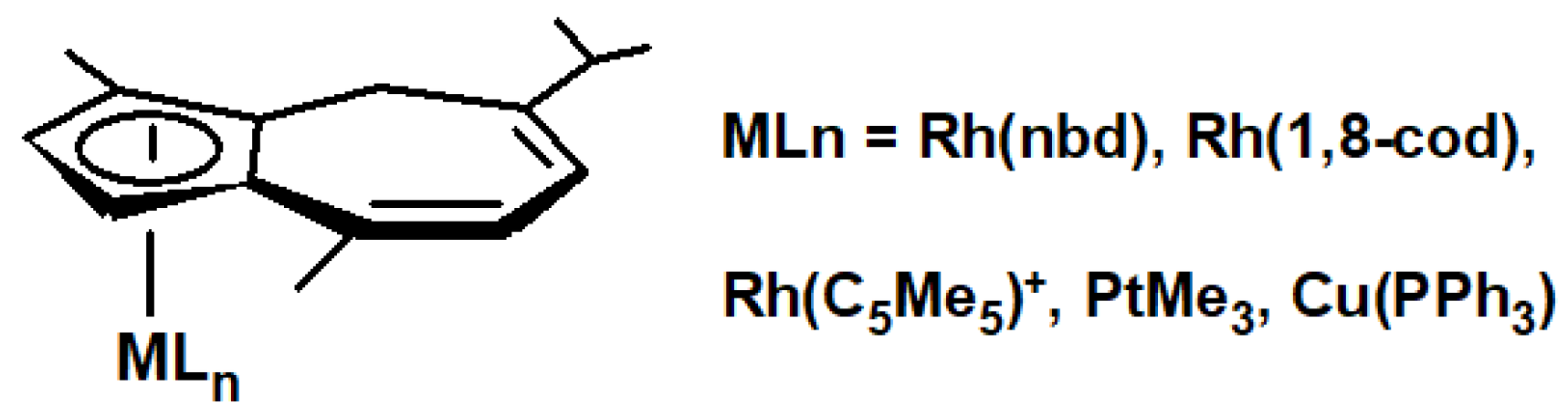{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The impact of organometallic chemistry on the terpene field only really blossomed in the 1960s and 1970s with the realisation that carbon–carbon bond formation under mild conditions could be achieved by using nickel or iron carbonyls as synthetic reagents. Concomitantly, the development of palladium derivatives capable of the controlled coupling of isoprene units attracted the attention of numerous highly talented researchers, including future Nobel laureates. We discuss briefly how early work on the syntheses of simple monoterpenes soon progressed to sesquiterpenes and diterpenes of increasing complexity, such as humulene, flexibilene, vitamin A, or pheromones of commercial value, in particular those used in perfumery (muscone, lavandulol), or grandisol and red scale pheromone as replacements for harmful pesticides. As the field progressed, there has been more emphasis on developing organometallic routes to enantiopure rather than racemic products, as well as gaining precise mechanistic data on the transformations, notably the course of metal-promoted molecular rearrangements that have long been a feature of terpene chemistry. We note the impact of the enormously enhanced analytical techniques, high-field NMR spectroscopy and X-ray crystallography, and their use to re-examine the originally proposed structures of terpenes and their organometallic derivatives. Finally, we highlight the very recent ground-breaking use of the crystalline sponge method to acquire structural data on low-melting or volatile terpenes. The literature cited herein covers the period 1959 to 2023.
1. Introduction
Terpenoids (or isoprenoids) found commonly in essential oils of plants have long fascinated chemists, not only for their outstanding structural diversity, but also for the versatility of their molecular rearrangements. The elucidation of their structures and biosynthetic pathways, and the rationalisation of their rearrangement behaviour is one of the most satisfying successes of organic chemistry and owes much to such pioneers as Otto Wallach, Leopold Ružička, Konrad Bloch and Feodor Lynen, and John Cornforth (Nobel laureates in 1910, 1939, 1964 and 1975, respectively). While the pleasantly agreeable odours of many volatile terpenes have led to their widespread use in the perfume industry, in their own natural world, they can function as protection against predators, as pheromones for sexual attraction, as trail pheromones, and in a multitude of other ways [1]. We note that, strictly speaking, terpenes are oligomers or polymers of isoprene (C5H8), and depending on the number of isoprene units are categorized as monoterpenes (C10H16), sesquiterpenes (C15H24), diterpenes (C20H32), etc. Those molecules with a different formula, such as geraniol (C10H18O) or citral (C10H16O), are known as monoterpenoids rather than monoterpenes [2].
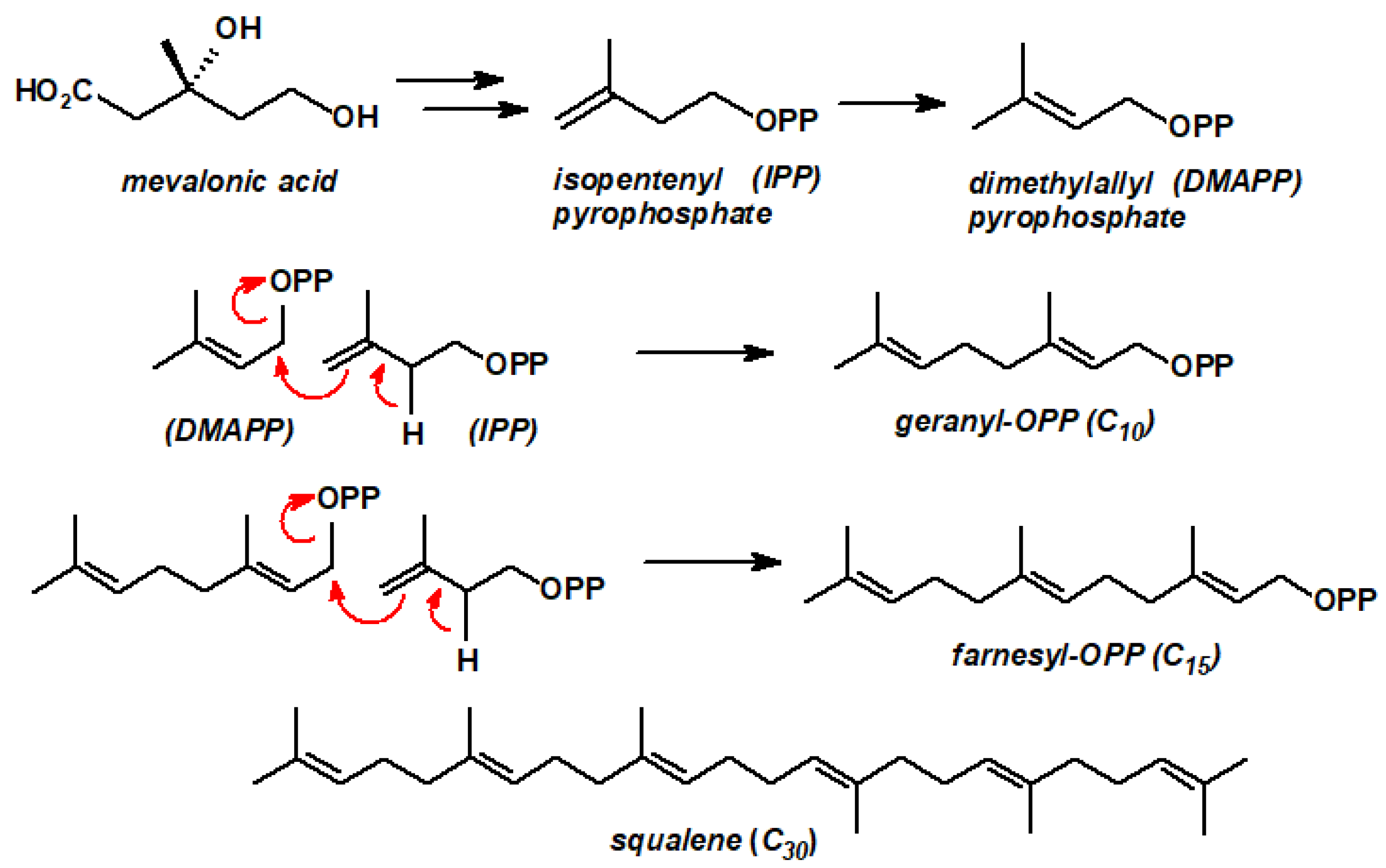
Although terpenes appear superficially to be assembled simply by combining isoprenes, in fact, the crucial intermediate in their biosynthetic pathway is mevalonic acid (derived originally from acetate) that is converted in several steps into isopentenyl pyrophosphate (IPP), which itself undergoes an enzyme-catalysed rearrangement to form its more reactive dimethylallyl pyrophosphate isomer (DMAPP). Nucleophilic attack by IPP at the C-1 site of DMAPP with the concomitant elimination of pyrophosphate, an excellent leaving group, yields geranyl-OPP, the precursor to myriad (C10) monoterpenes. As shown in Scheme 1, subsequent alkylation of geranyl-OPP by IPP leads to farnesyl-OPP, the precursor of the sesquiterpenes (C15), while the head-to-head coupling of farnesyl units produces squalene (C30), a progenitor of the steroid skeleton.
As shown in Scheme 2, the 2,3-epoxide of the triterpene squalene, when folded appropriately on an enzyme surface, undergoes a series of hydride and methyl 1,2-shifts leading to lanosterol (Scheme 2), and ultimately to human sex hormones, such as testosterone and estradiol [3].
Since these biosynthetic routes to terpenes are mediated by enzymes, they yield enantiopure products that find extensive applications in catalytic asymmetric syntheses. This aspect of their chemistry, i.e., the formation of coordination compounds possessing chiral terpenoid ligands, such as those based on the camphor or pinane framework, 1 or 2, respectively (Figure 1), has been comprehensively reviewed [4].
However, our focus here is on the organometallic chemistry of terpenoids, with the aim of providing a short introduction to the field, and an overview of its development in recent decades. We begin with the pioneering work on the syntheses of simple terpenoids in the 1960s and 1970s, and the subsequent extension to more complex molecular targets. In all cases, the organometallic steps in the synthetic sequences are those highlighted. We then discuss the reactions and molecular rearrangements of terpenoids when treated with organometallic reagents, finally moving towards the current status and future goals of the field.
2. Synthetic Overview
The use of transition metals in terpenoid synthesis is a long-established approach. Typically, in 1959, a large-scale route to citral was developed in which the α-acetoxy-alkyne, 3, was treated with silver carbonate in acetic acid to form the allene 4, which yielded citral, 5, upon saponification (Scheme 3) [5].
This area gained considerable impetus in the 1960s and 1970s when the development of new and improved transition-metal-based carbon–carbon coupling reactions, many of them catalytic in nature, was a major focus of a number of leading investigators, several of whom received Nobel Prizes for their seminal contributions. In many instances, researchers chose terpenoids as synthetic targets to illustrate the utility and versatility of their own new-found reaction sequences.
One general method takes advantage of the ability of metal carbonyls, such as Fe(CO)5 or Ni(CO)4, to discard CO ligands and undergo oxidative addition reactions with carbon–halogen linkages, while another major approach uses palladium(II) salts as precursors to Pd(0) systems, generally stabilized by phosphines; another, now very widely adopted, procedure is based on the generation of π-allyl-nickel or -palladium intermediates. It is, noteworthy, however, that many researchers now focus their synthetic efforts on the use of first-row (3d) transition elements [6] rather than heavy precious-metal catalysts, not only for their lower cost and ready availability, but also for their low toxicity. We shall describe representative examples of all the abovementioned methodologies where they have been used in synthetic routes to isoprenoids.
3. Organonickel Complexes as Coupling Reagents
3.1. Dehalogenations Using Ni(CO)4
The earliest use of Ni(CO)4 in carbon–carbon bond formation is to be found in a Belgian Patent filed in 1943 [7]; but the first journal publication appeared in 1951, when Webb and Borcherdt reported that a wide range of allylic halides could be coupled to form the corresponding diallyl (1,5-diene) products with the elimination of carbon monoxide (Scheme 4) [8]. However, they emphasized the need for great care in handling such a volatile (b.p. 43 °C) and toxic reagent.
This reaction requires the loss of CO ligands to generate vacant sites available for oxidative addition by the allyl halide, and the formation of an (η3-allyl)nickel carbonyl halide, 6, in equilibrium with the diallyl complex, 7, both of which react irreversibly with allyl bromide in a polar solvent to ultimately yield the 1,5-diene product and NiBr2 (Scheme 5).
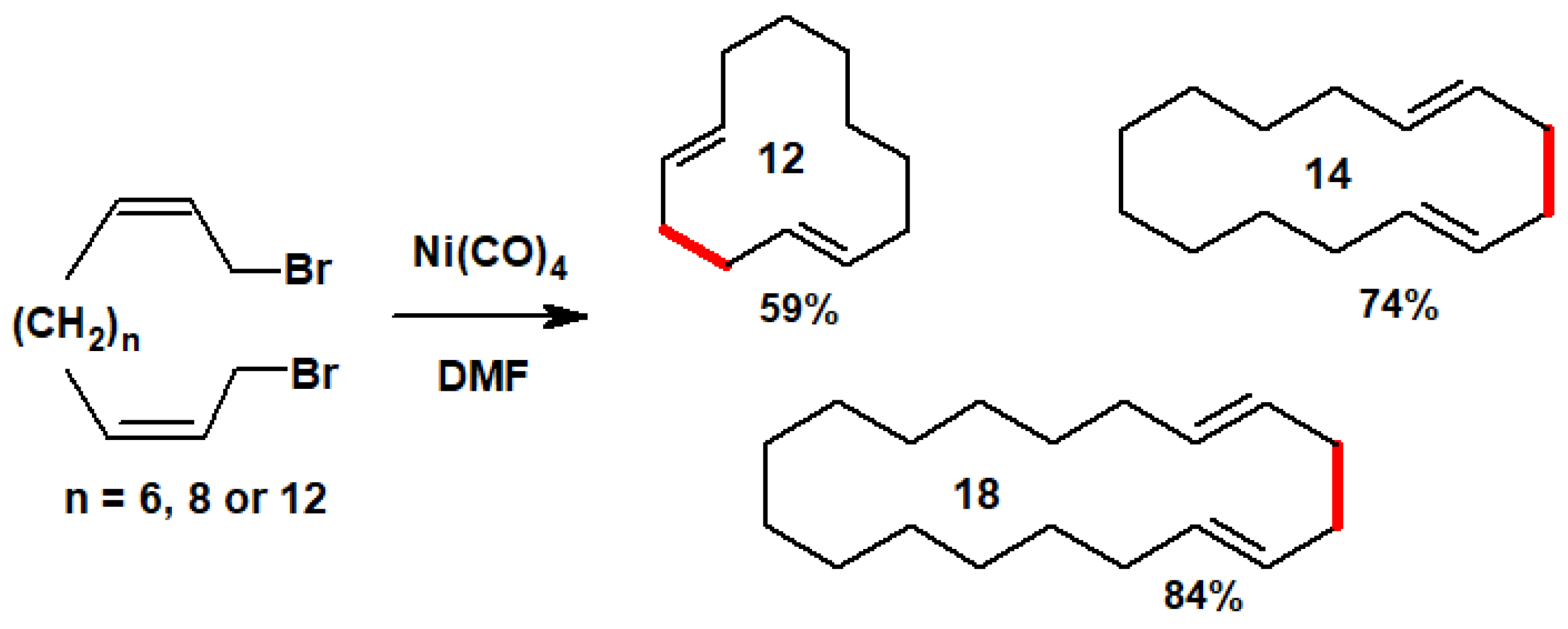
Despite the potentially hazardous nature of Ni(CO)4, this concept was brilliantly exploited [9,10] by E.J. Corey (Nobel 1990), who extended it to long-chain α,ω-dibromo-diene systems, thereby forming large-ring 1,5-dienes containing up to 18 carbons in modest-to-good yields [11], as in Scheme 6.
This approach offered a route to humulene, (2,6,6,9-tetramethyl-trans,trans,trans-cycloundeca-1,4,8-triene), a monocyclic sesquiterpene containing an 11-membered ring and found in the essential oils of Humulus lupulus (hops), from which it derives its name. To this end, treatment of the dibromide 8 with Ni(CO)4 in N-methylpyrrolidone delivered 9, possessing a cis C4-C5 double bond; subsequent photolysis with diphenyl disulphide in cyclohexane brought about the required cis-trans isomerisation to form humulene, 10, in modest yield (Scheme 7) [12]. A second report, from the group of O.P Vig in Chandighar, India, using essentially this same route, appeared in 1976 [13].
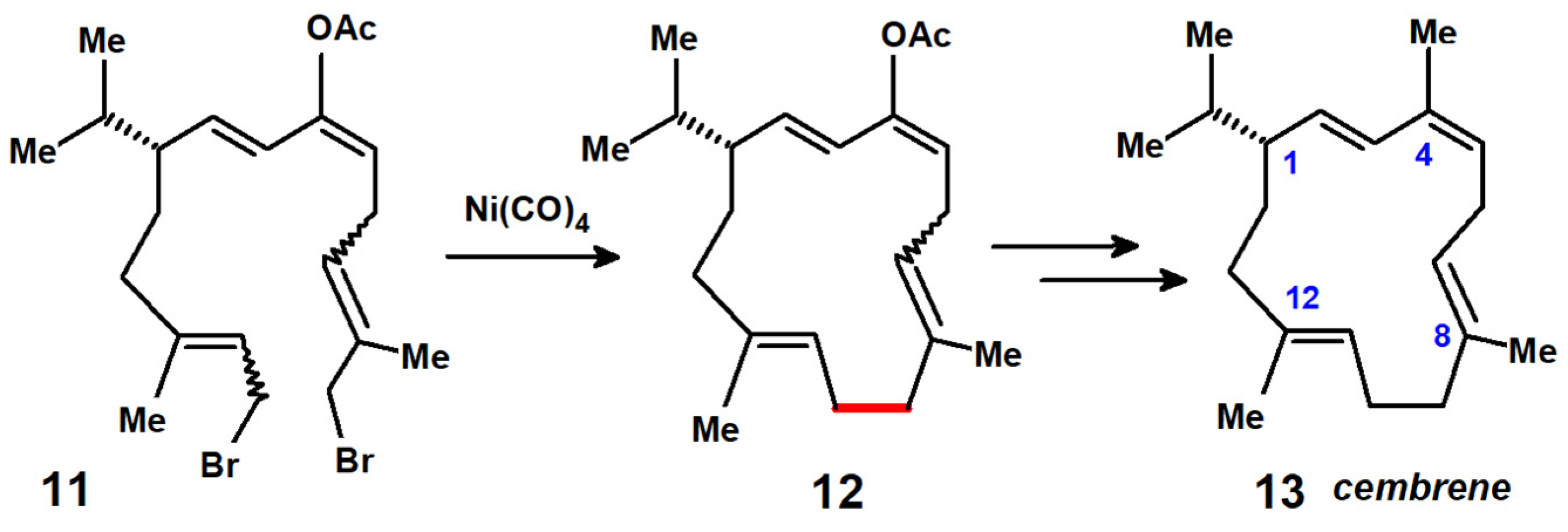
In related work, Dauben’s group used this approach to prepare cembrene (1-isopropyl-4,8,12-trimethylcyclododeca-2,4,7,11-tetraene), a monocyclic diterpene found in pine oil and which contains a 14-membered ring [14]. The key feature shown in Scheme 8 depicts the nickel tetracarbonyl-mediated ring closure of the acetate derivative 11 to form 12. To complete the synthesis, Jones oxidation of the acetate functionality to ketone, reaction with MeLi and the dehydration of the resulting alcohol delivered cembrene, 13, spectroscopically identical to the authentic material whose X-ray crystal structure appears as Figure 2 [15].
3.2. π-Allyl-Nickel Complexes
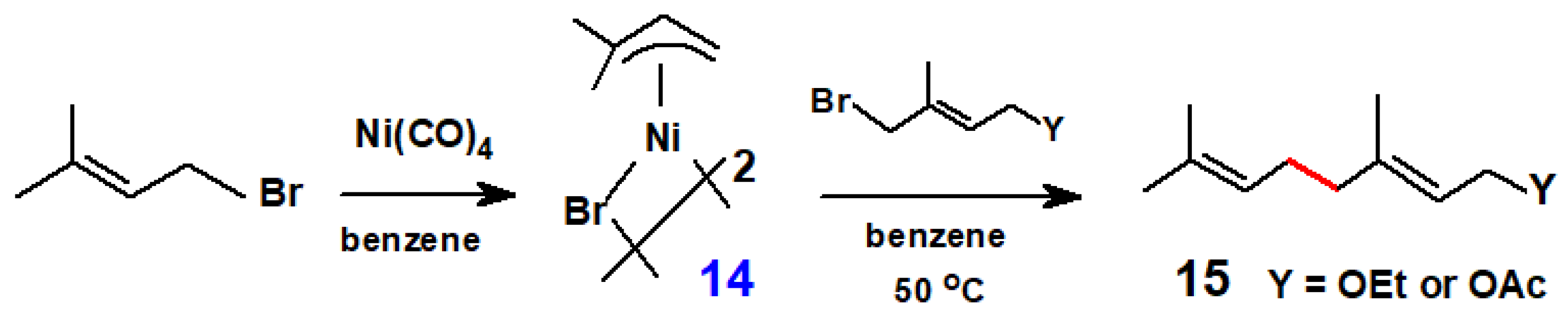
Continuing in this vein, Sato reported that the treatment of (1,1-dimethyl-π-allyl)nickel bromide dimer, 14, with 4-acetoxy-1-bromo-2-methyl-2-butene (or its 4-ethoxy analogue) gave geranyl acetate (or geranyl ethyl ether), 15, in moderate yield (Scheme 9) [16].
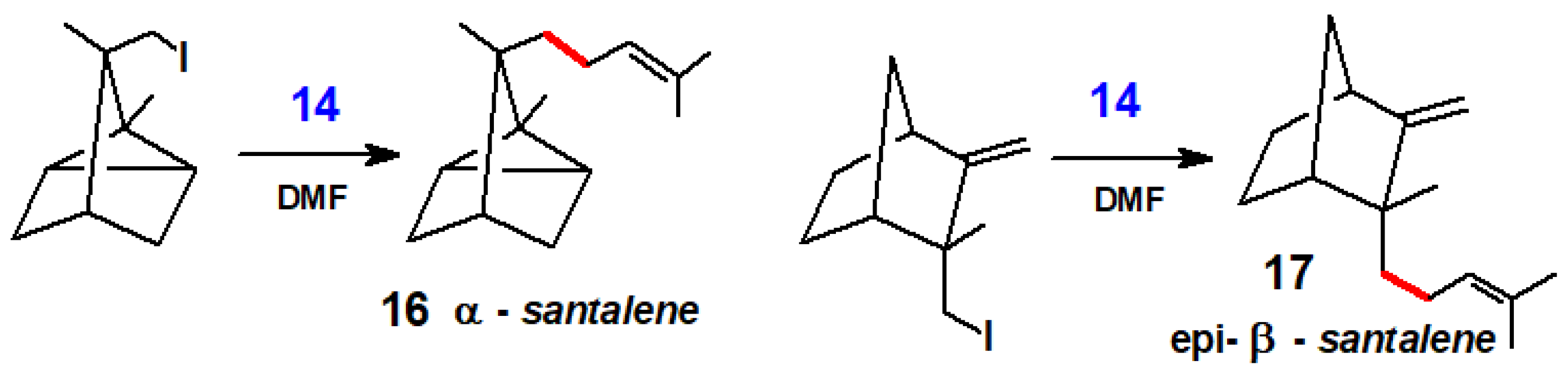
Likewise, Corey and Semmelhack used this approach to complete the syntheses of α-santalene, 16, and epi-β-santalene, 17, by prenylation of their iodomethyl side chains (Scheme 10) [10].
The direct involvement of π-allyl-nickel intermediates in such processes was unequivocally demonstrated in now classic studies by the Wilke group at the Max Planck-Institut für Kohlenforschung in Mülheim, Germany. They not only isolated and characterized many such species derived from 1,3-butadiene and a source of “naked nickel” (from a Ni(0) precursor, e.g., bis(1,5-cyclooctadiene)nickel), but also elucidated how the various coupling products are dependent on the associated ligands (phosphines, phosphites, CO) and on the ligand-to-metal ratio. This was eloquently reviewed in a virtuoso contribution by Günther Wilke himself [17].
3.3. Syntheses of Grandisol and of Muscone
For our specific purposes, we concentrate on two such π-allyl-nickel complexes: (i) the C8H12Ni system, 18, derived from the coupling of two butadiene units, and (ii) the C12H18Ni homologue, 19, arising from incorporation of three butadiene monomers. In the former case, elimination of the nickel moiety yields cis-1,2-divinylcyclobutane, 20, whereas the latter leads to 1,5,9-cyclododecatriene, 21 (Scheme 11).
Since the widespread use of pesticides has been severely criticised because of its harmful effects on human and wildlife, an alternative approach to pest control has been proposed, involving the use of insect pheromones. To this end, the formation of cis-1,2-divinylcyclobutane from 18 prompted Ed Billups from Rice University in Houston, Texas, to carry out the same reaction, but starting from isoprene, with the goal of preparing the important monoterpenoid grandisol, a key constituent of the male boll weevil pheromone. Gratifyingly, the reaction of a large excess of isoprene with (cod)2Ni and tris(2-biphenylyl) phosphite furnished the desired dimer, 22, in a 15% yield. Subsequent hydroboration with disiamylborane in THF at 0 °C followed by treatment of the organoborane with alkaline peroxide yielded (±)-grandisol, 23 (52%), as shown in Scheme 12 [18].
The synthetic availability of dodecatrienylnickel, 19, and its known ring closure to 1,5,9-cyclododecatriene, 21, offered the possibility of an incorporation of an additional small unit, such as allene, thereby extending the molecular skeleton to form a 14-membered carbon chain. As shown in Scheme 13, this was successfully achieved and led to the extended bis-allyl-nickel complex, 24 [19]. Subsequent incorporation of carbon monoxide resulted in the formation of the 15-membered ring species, 25, that also possessed a ketone functionality, and the hydrogenation of the double bonds yielded (±)-muscone, 26, that is obtainable in small quantities as its (R)-(-)-enantiomer from a glandular secretion of the male musk deer.
In an improved synthesis of muscone, also by the Baker/Cookson group [20], the incorporation of allene formed 24; subsequently, the reaction with tert-butyl isocyanide formed the imine, 27, that was then converted into the previously prepared ketone, 25, thereby raising the overall yield to 40%. Muscone is now manufactured commercially, starting from (+)-citronellal, and is widely used in perfumery.
3.4. Organocobalt Complexes as Substitutes for Ni(CO)4
Despite the evident versatility of Ni(CO)4 in synthesis, its toxicity has prompted the search for alternative methodologies. Perhaps the most useful reagent as a replacement is chlorotris(triphenylphosphine)cobalt(I), (Ph3P)3CoCl, readily preparable as an air-stable crystalline solid. Typically, its use in the coupling of allyl halides has been exemplified in a report by the Yamada group in Tokyo [21]. As illustrated in Scheme 14, the reaction of geranyl and farnesyl halides, 28 and 29, respectively, and also of their geometrical isomers, 30 and 31, with (Ph3P)3CoCl in benzene gave the corresponding geometrically pure coupling products 32–35, under mild and non-basic conditions. This method provided a convenient route to squalenes.
In similar vein, nucleophilic attack by an alkyllithium on a carbonyl ligand of Ni(CO)4 leads to alkylnickel carbonylates that react with allyl halides, such as geranyl bromide, to yield ketones. However, with the aim of avoiding the use of such a toxic material as nickel tetracarbonyl, Hegedus and Perry sought to develop a safer and more convenient reagent that could bring about the same transformations. Isoelectronic cobalt nitrosyl tricarbonyl, Co(CO)3NO, is also a volatile, toxic liquid, but it reacts with triphenylphosphine to form (Ph3P)Co(CO)2NO, 36, an orange, air-stable, non-volatile crystalline solid that is readily prepared and easily handled; gratifyingly, it also behaves as an efficient acylating agent (Scheme 15) [22].
4. Organoiron Complexes as Coupling Reagents
4.1. Oxaallyl-Fe(II) Intermediates
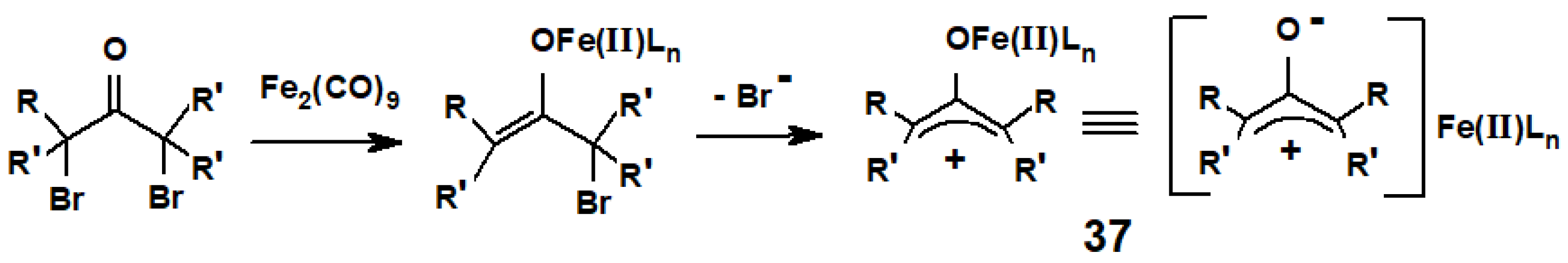
Ryōri Noyori (Nobel 2001) discovered that diiron enneacarbonyl, Fe2(CO)9—which slowly dissociates in solution to form Fe(CO)5 and the reactive coordinatively unsaturated moiety Fe(CO)4—reacts with α,α′-dibromo ketones to form oxaallyl-Fe(II) intermediates, 37, that behave as allyl cations since the negative charge is masked by complexation with the Fe(II) ion (Scheme 16) [23].
These species undergo cycloadditions of the type “3 + 2 → 5”; typically, the reaction of Fe2(CO)9 with 1,3-dibromo-1,3-diphenylpropan-2-one yields 1-phenyl-2-indanone (70%). The proposed mechanism (Scheme 17) invokes nucleophilic attack on the allyl cation, 38, by the adjacent aromatic ring to form 39, that in turn undergoes aromatisation to yield the final product [24,25].
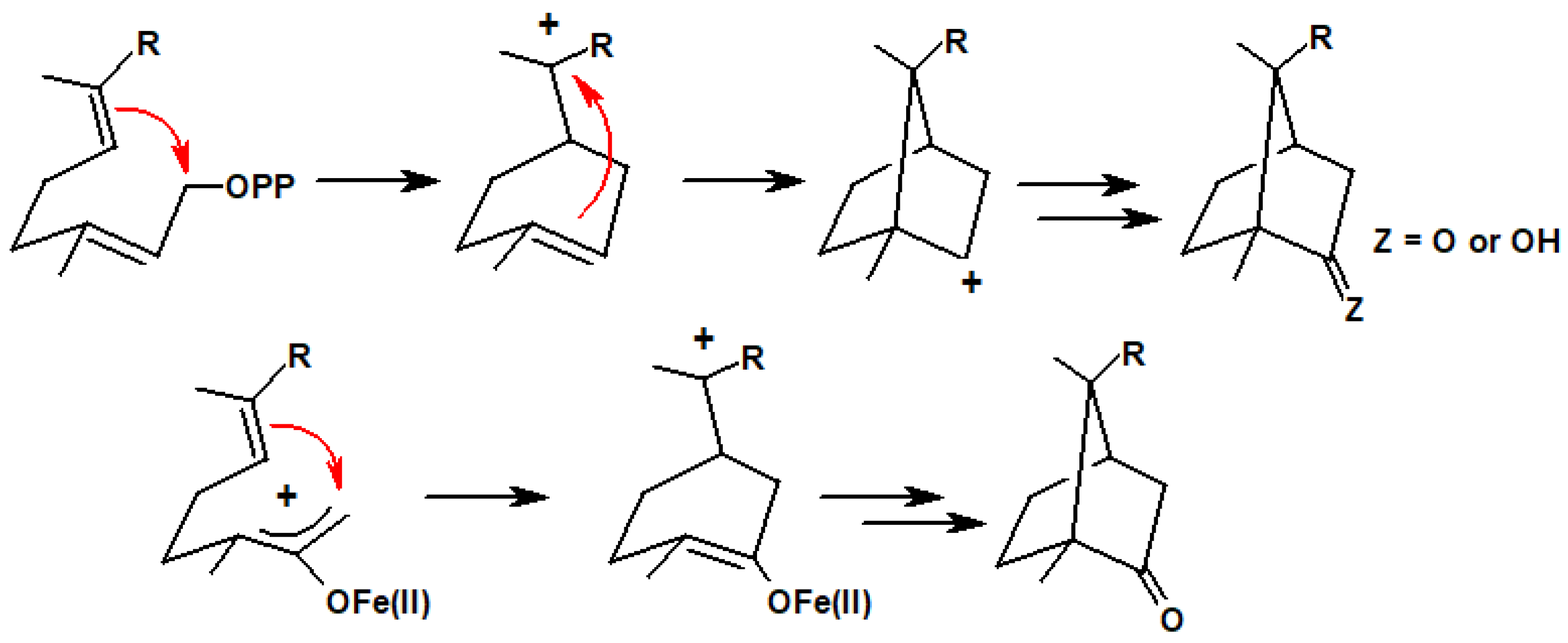
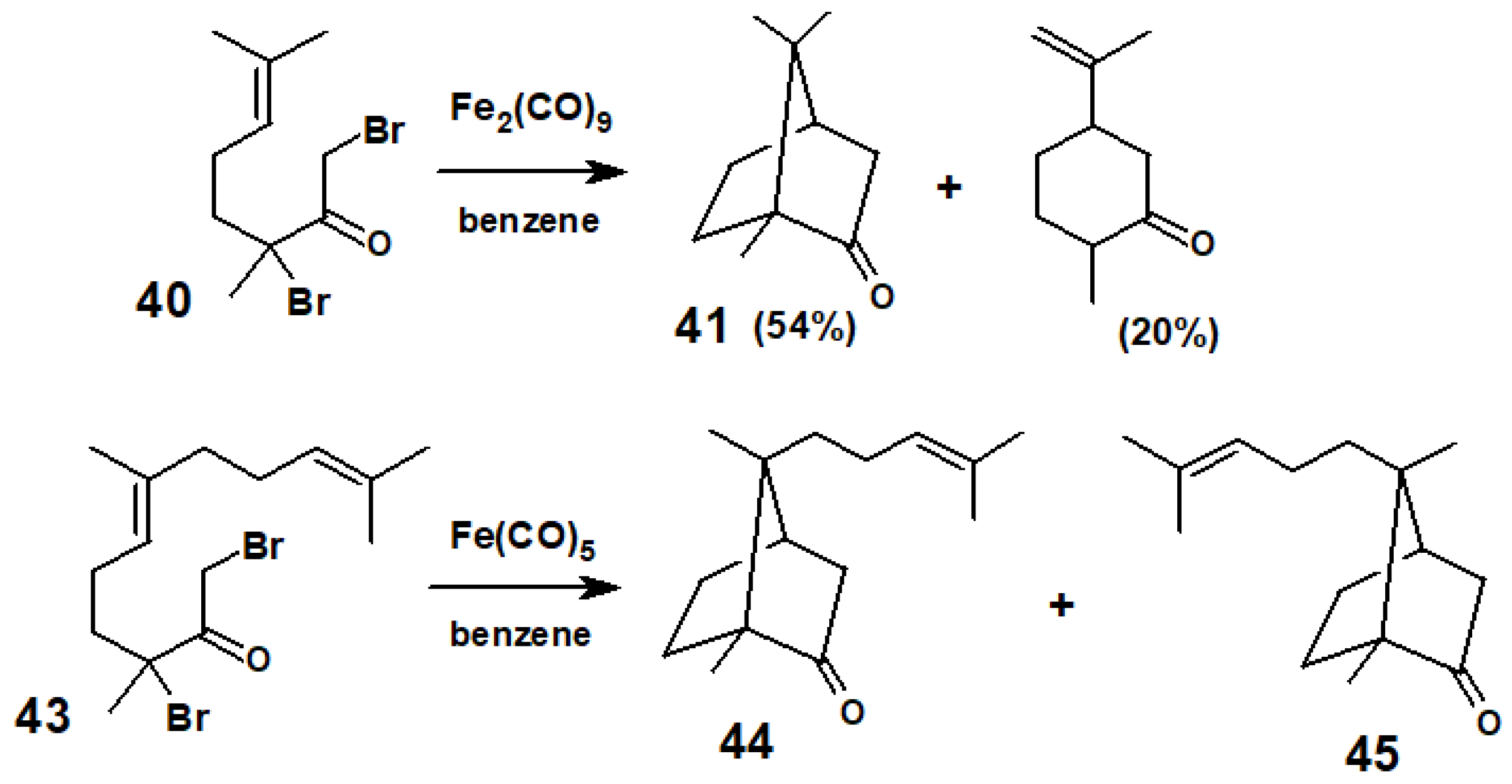
Inspired by a biogenetic hypothesis for the formation of camphor, Noyori envisaged an iron carbonyl-mediated analogous reaction sequence (Scheme 18). Gratifyingly, when the dibromo ketone 40 (obtained from geraniol) and Fe2(CO)9 were heated in benzene in a pressure vessel, the major products were (±)-camphor, 41, and (±)-dihydrocarvone, 42, as shown in Scheme 19. Similarly, treatment of the dibromo ketone 43 (prepared from farnesol) and Fe(CO)5 delivered a 2:1 mixture of (±)-campherenone, 44, and (±)-epicampherenone, 45, in a 58% yield. Further examples of other natural product syntheses using this approach are given in a review by Noyori and Hayakawa [26].
4.2. Allyl-Iron(Dicarbonyl)(η5-Cyclopentadienyl) Complexes
A very different route to terpenoids, reported by Celebuski and Rosenblum [27], is based on the unique ability of (η1-allyl)Fp complexes, 46, where Fp = (C5H5)Fe(CO)2, to flip easily and reversibly between monohapto (σ) and dihapto (π) bonding modes. Molecules of this type react with electrophiles such as the π-complexed ethyl vinyl ether, 47, to yield the coupled product 48; in this example, the σ-bonded and π-bonded Fp moieties have exchanged bonding modes (Scheme 20).
This methodology has been exploited in the syntheses of terpenoids, as exemplified in Scheme 21 and Scheme 22. (Isobutenyl)Fp reacts with dioxolenium tetrafluoroborate to form 49 that, when deprotonated, yields the geometric isomers of 50 essentially quantitatively. Subsequent treatment with prenyl iodide delivers the ethylene acetal 51; hydrolysis and reduction with sodium borohydride yields (±)-lavandulol, 52, which is found in lavender oil, and whose R-enantiomer has a lemon-like or citrus fruit odour, and is used in some perfumes.
A somewhat more challenging target was a pheromone of the California red scale insect, a major pest of citrus fruit, that requires bringing together 1-Fp-2-methyl-2,6-heptadiene, 53, and 1-iodo-3-methyl-5-acetoxy-2-pentene, 54, preparable by the ring-opening of 5,6-dihydro4-methyl-2H-pyran, to form the desired product, 55.
5. Palladium Complexes as Coupling Reagents
Palladium-mediated carbon–carbon coupling procedures are now a standard weapon in the synthetic chemist’s armoury. Many researchers made very significant contributions, but the Nobel regulations limit the number of chemistry laureates in a single year to three, and Richard Heck, Ei-ichi Negishi and Akira Suzuki all shared the award in 2010.
5.1. The Heck Reaction
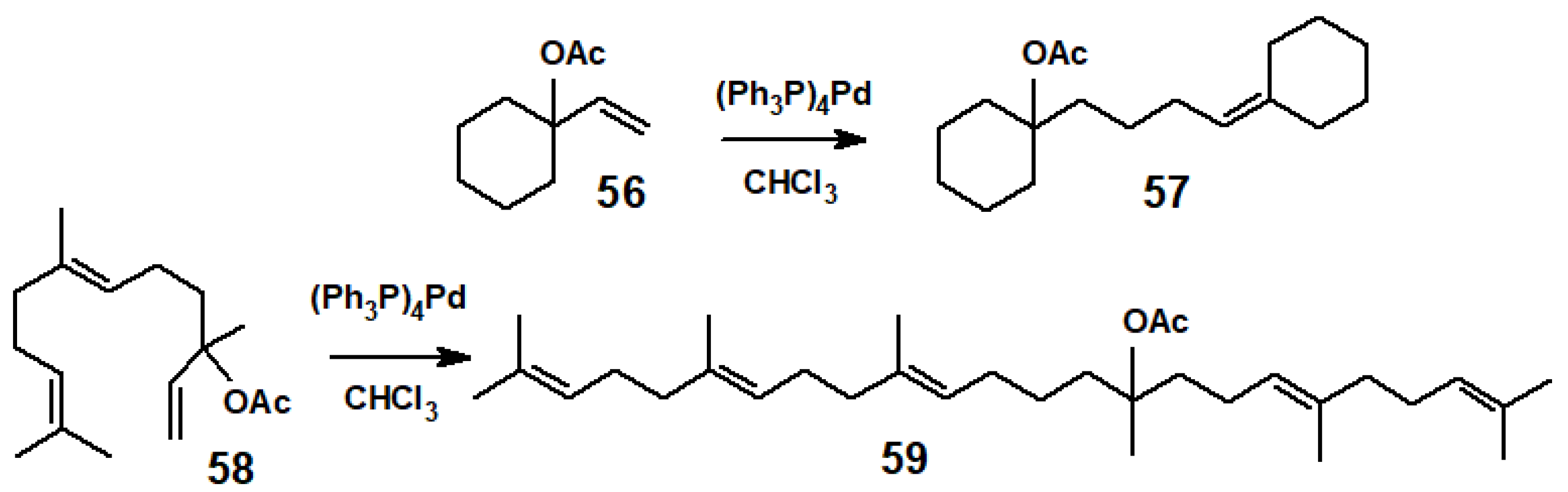
Following a report by Heck in 1968 that palladium(II) salts induced coupling between allylic halides and arylmercurials [28], in 1970, Francis McQuillin at the University of Newcastle, U.K., found that simple monoterpenes underwent dimerisation when treated with tetrakis(triphenylphosphine)palladium(0) in chloroform, and extended this to its reaction with 1-vinylcyclohexyl acetate, 56, to form 1-(4-cyclohexylidenebutyl)cyclohexyl acetate, 57 (Scheme 23). Moreover, the same reaction with nerolidyl acetate, 58, also yielded a dimer, tentatively identified as 59, the acetate derivative of squalene [29].
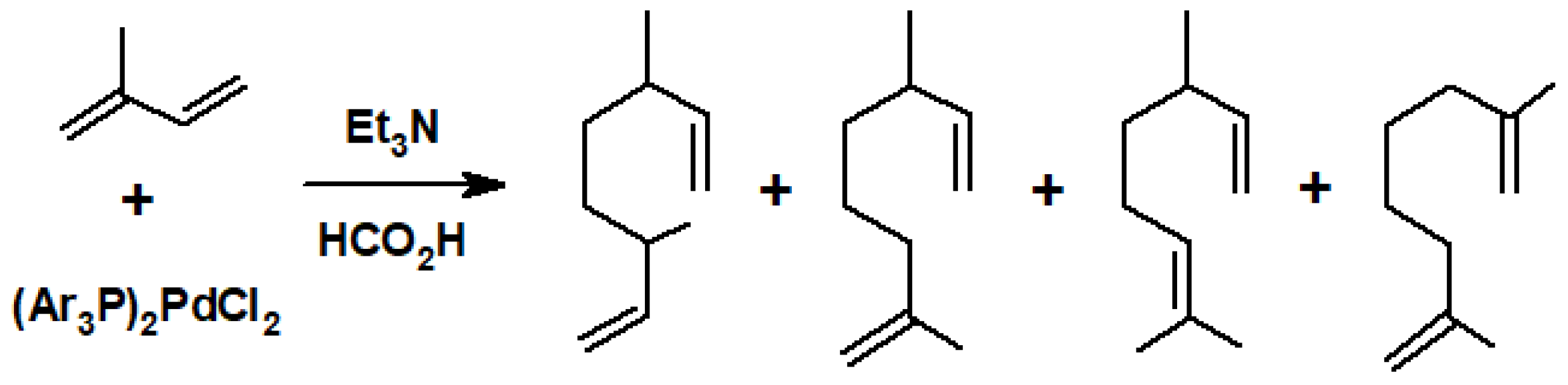
Shortly thereafter, Heck reported that isoprene was dimerised when treated with complexes of the type (Ar3P)2PdCl2 in the presence of triethylamine and formic acid to yield a number of isomeric products (Scheme 24) in various ratios depending on the identity of the phosphine, the reaction time, and the solvent [30]. In addition, it was found that (Ar3P)2Pd(OAc)2 catalysed the vinylation of 4-methyl-1,3-pentadiene via the sequence depicted in Scheme 25 [31].
5.2. Prenylations and Geranylations Using π-Allyl-Palladium Complexes
A major breakthrough was the development of the Trost–Tsuji approach [32], whereby nucleophilic attack on a π-allyl-palladium complex brings about carbon–carbon coupling under stereochemical control [33,34]. Application to the synthesis of complex natural products has been comprehensively reviewed not only by Fairlamb and colleagues [35], but also by Barry Trost himself [36,37].
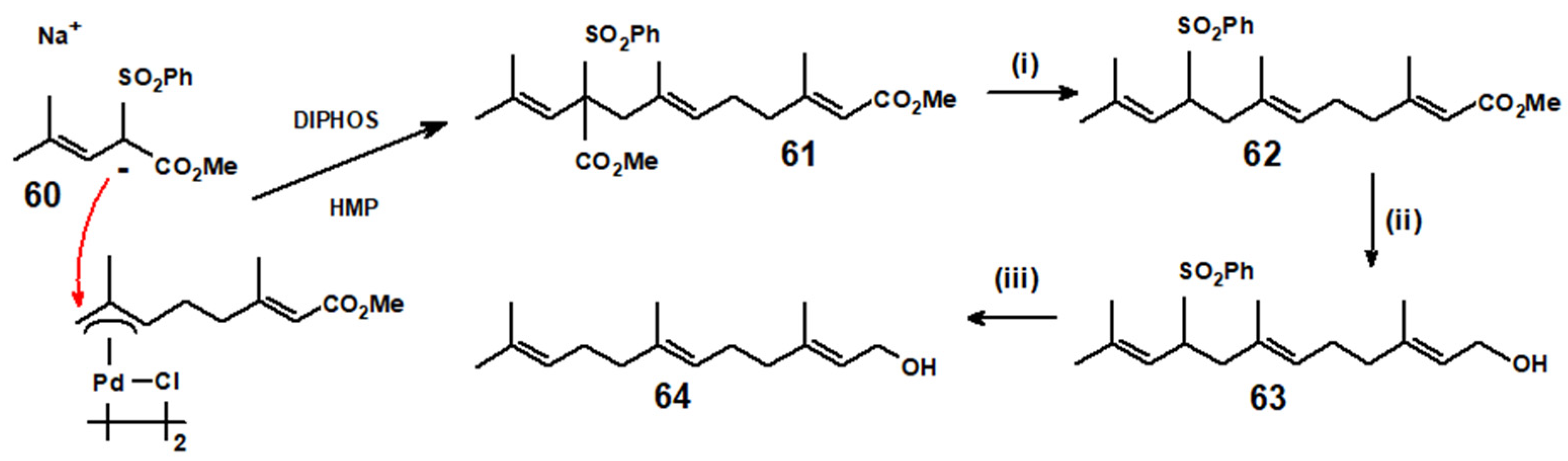
The success of this approach is based on the preference for (η3-allyl)palladium complexes to form non-conjugated double bonds, and the ability of sulphur-stabilised carbanions to attack almost exclusively at the less-substituted allyl terminus. We focus here on prenylation, as exemplified in Scheme 26, in which the monoterpenoid methyl geraniate is converted into the sesquiterpenoid (E,E)-farnesol and then into the diterpenoid (E,E,E)-geranylgeraniol. Nucleophilic attack by the stabilised anion, 60, leads to 61, that is sequentially decarbomethoxylated, to form 62, the terminal ester functionality reduced to the alcohol 63, and finally desulphonylated to yield (E,E)-farnesol, 64. As shown in Scheme 27, the analogous sequential procedure, but now starting from methyl farnesoate to form the allyl-palladium complex 65, delivered (E,E,E)-geranylgeraniol, 66 [38].
We note, however, that later work by Trost revealed that “reverse prenylation” (Scheme 28) can also be achieved depending on the choice of Pd catalyst, and on the solvent, where toluene favoured prenylation; but in dichloromethane, the branched isomer was strongly preferred. They also reported examples of direct geranylation [39].
In related work, Goliaszewski and Schwartz reported that allylic coupling arising from the reaction of bis(allylic)palladium species was facilitated by using π-acidic ligands, rather than phosphines [40]. Maleic anhydride was found to be effective in this regard, and the 1,5-dienic products formed were predominantly head-to-head adducts, as exemplified by the prenylation of pinene to form 67, as shown in Scheme 29.
5.3. Palladium-Mediated Routes to Humulene, Grandisol, Vitamin A and Mokupalide
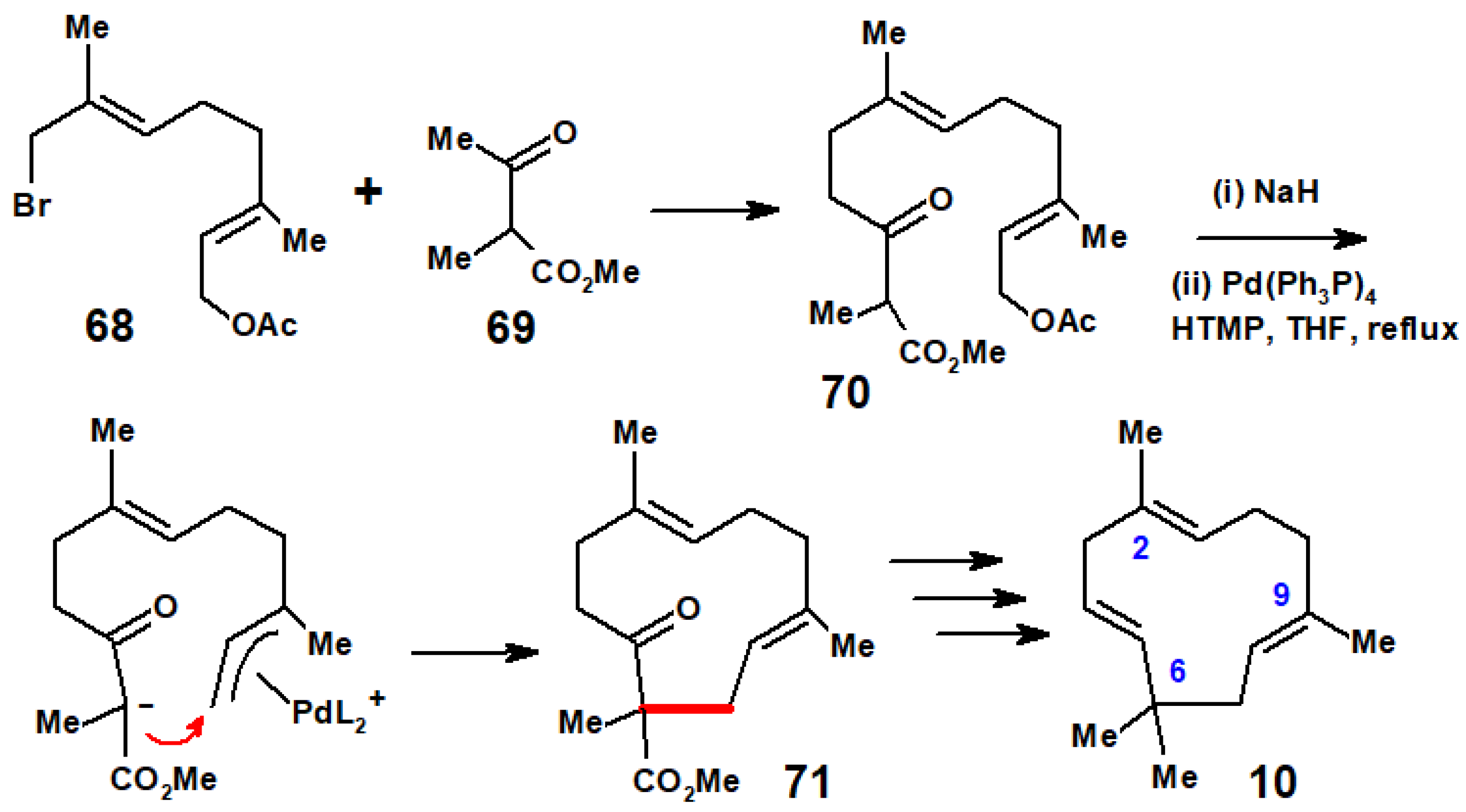
The first organometallic route to humulene, by Corey and Hamanaka, used Ni(CO)4 to couple two bromomethyl termini with the elimination of NiBr2 [12]. However, this same monocyclic sesquiterpene was also targeted by several groups using their newly developed palladium coupling techniques. Inspired by the biological formation of humulene via anti-Markovnikov cyclisation of farnesol pyrophosphate (Scheme 30), the Nozaki group at the University of Kyoto, Japan, designed a (π-allyl)palladium system poised to accept nucleophilic attack by a carbanion stabilised by a neighbouring ester functionality, as in Scheme 31. The E,E-bromide 68, derived from geranyl acetate, reacted with the dianion of methyl α-methylacetoacetate, 69, to form the keto ester 70 which, when deprotonated and slowly added to a solution containing (Ph3P)4Pd, led to the 11-membered ring system 71; further manipulation furnished humulene [41].
The approach to humulene taken by Akira Suzuki, not surprisingly, took advantage of his own methodology, whereby the terminal alkyne, 72, was converted sequentially into the disiamylborane derivative 73 prior to palladium-mediated ring closure, as in Scheme 32 [42].
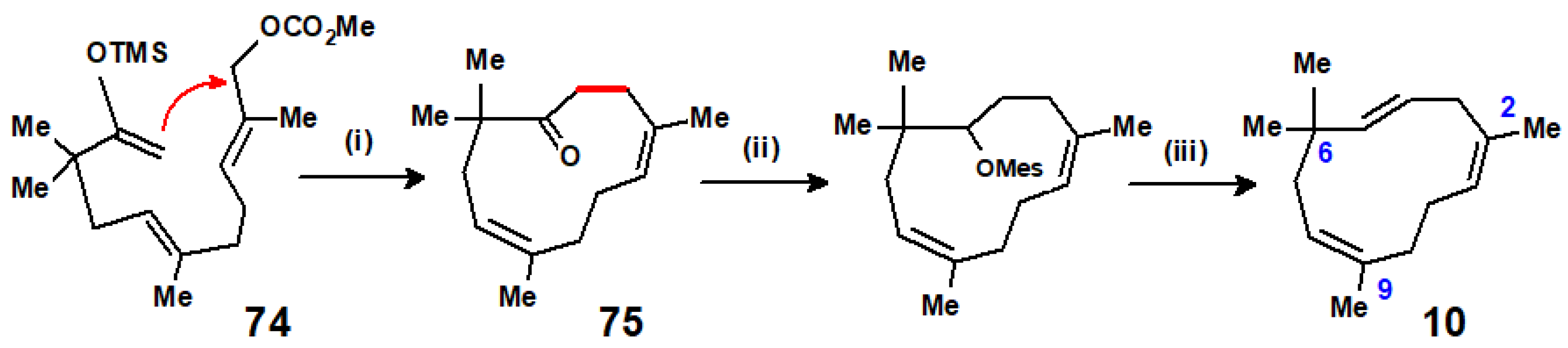
Yet, another short route to humulene was reported by Hu and Corey, whereby the allylic carbonate/silyl ether, 74, underwent palladium coupling to form the cycloundecadienone 75, as in Scheme 33. Subsequent mesylation and elimination delivered humulene in excellent yield [43].
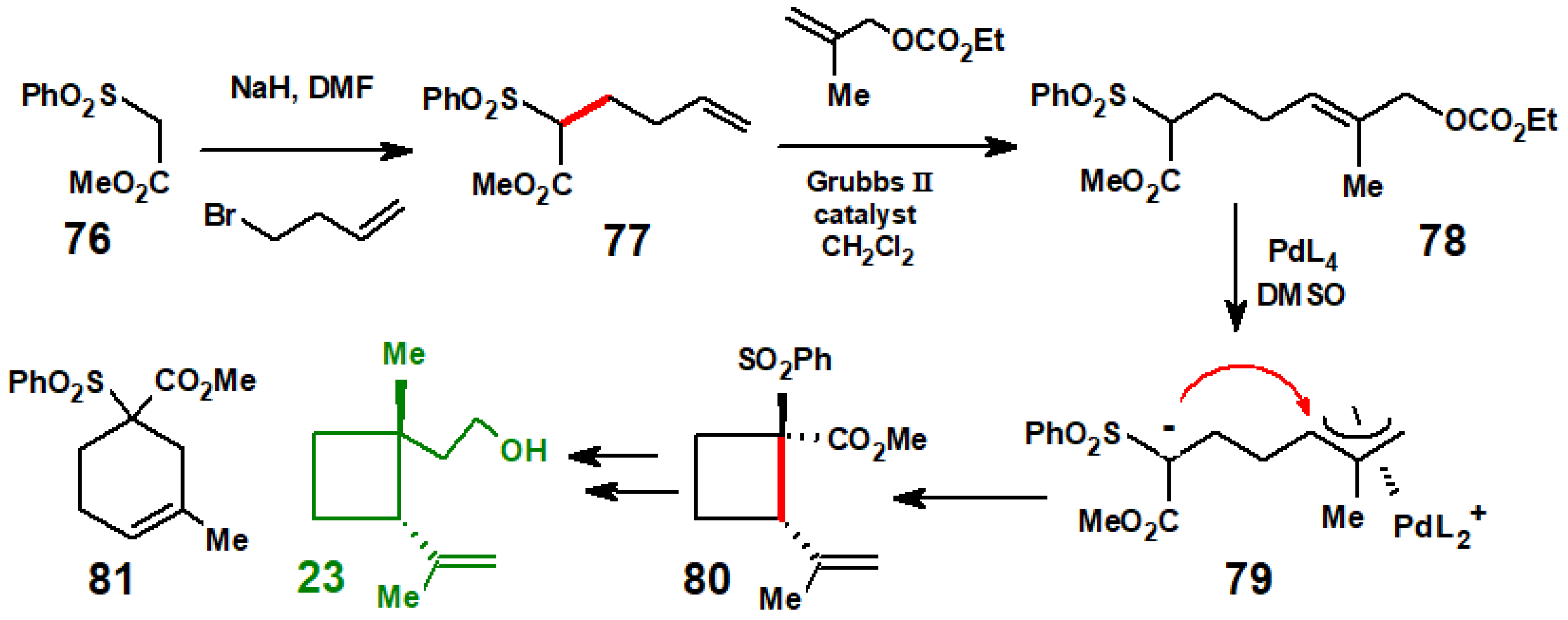
In 1973, the first organometallic route to grandisol, 23, involved the nickel(0)-mediated coupling of isoprene prior to hydroboration [18]. Almost four decades later, the group led by Y-G. Suh at Seoul National University, Korea, reported a synthesis (Scheme 34) in which the anion of methyl benzenesulphonyl acetate, 76, and 4-bromo-1-butene reacted to form 77, that in turn underwent metathesis with ethyl (2-methylallyl) carbonate to yield 78. Treatment with Pd(PPh3)4 in DMSO generated the allyl-palladium species, 79, in which the sulphur-stabilised anion preferentially attacked intramolecularly in a 4-exo-trig fashion to form the desired cyclobutane ring, 80. Further manipulation furnished grandisol [44]. The regioisomeric cyclohexene 81 was a minor side product.
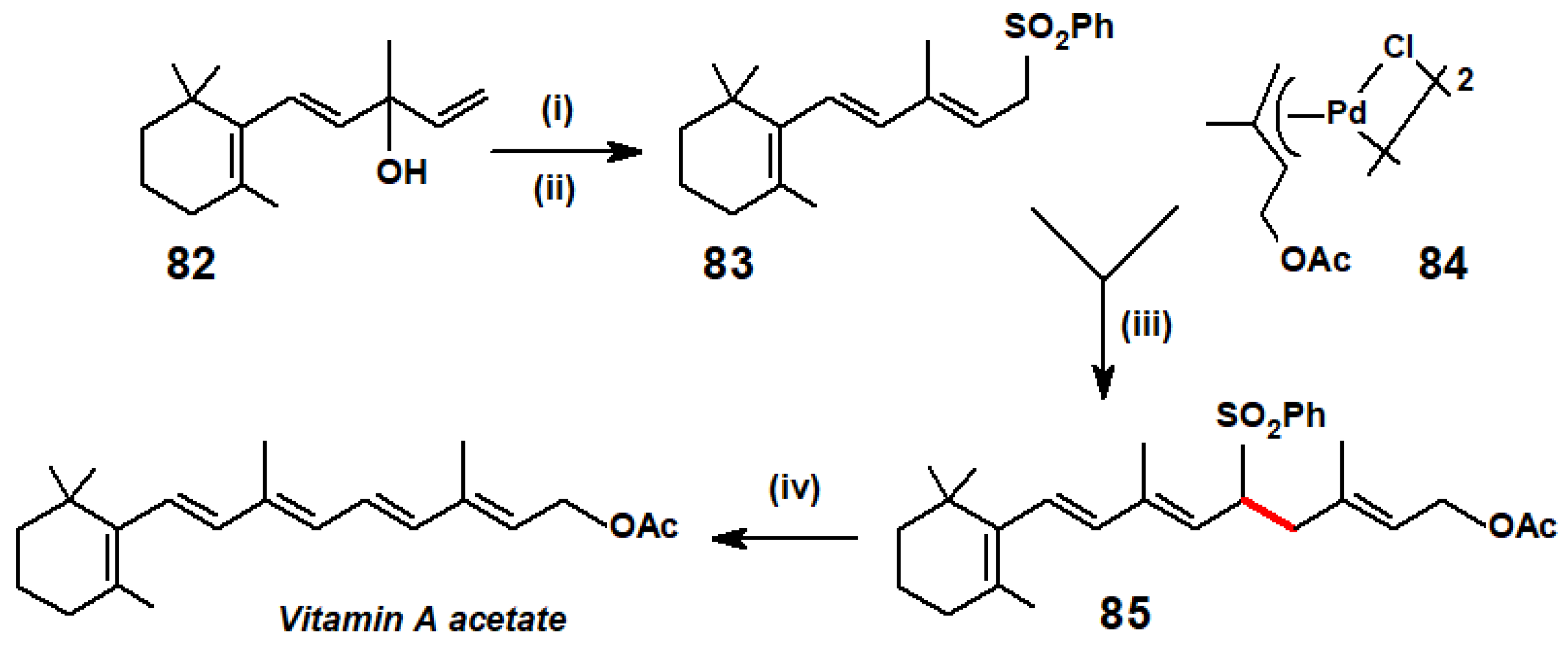
Manchand’s synthesis of vitamin A acetate in many ways resembles Trost’s procedure for the prenylation of methyl geraniate (Scheme 26). Starting from vinyl-β-ionol, 82, bromination and introduction of the phenylsulphonyl group yielded 83, the anion of which underwent nucleophilic attack on the allyl-palladium dimer, 84, derived from prenyl acetate, to generate 85. Desulphonylation completed the synthesis (Scheme 35) [45].
An elegant example of a Pd coupling process facilitating the linking of multiple prenyl units was reported by Negishi [46], and is shown in Scheme 36. Starting from the terminal alkyne 86, treatment with Me3Al—Cl2Zr(C5H5)2 and then with iodine generates the (E)-β-methyl-iodoalkene, 87. Chain extension via palladium coupling with a homopropargylic zinc chloride to form 88, followed by a second carbometallation and iodination, formed 89. Finally, reaction with n-BuLi and paraformaldehyde led to the tetraenol 90, which was subsequently converted by Sum and Weiler [47] into the C30 isoprenoid mokupalide, derived from a marine sponge.
6. Terpenoid Synthesis Using Titanium and Zirconium Reagents
6.1. Schwartz’s Hydrozirconation Reaction
In conjunction with allyl-palladium complexes, the cis addition of (C5H5)2Zr(H)Cl to terminal alkynes, thus generating vinylzirconium(IV) intermediates, reported by Schwartz [48], provides another convenient approach to carbon–carbon coupling. This concept has been exploited in the terpene field, notably in the elegant work of McMurry as exemplified in Section 6.2.
6.2. McMurry Dicarbonyl Coupling
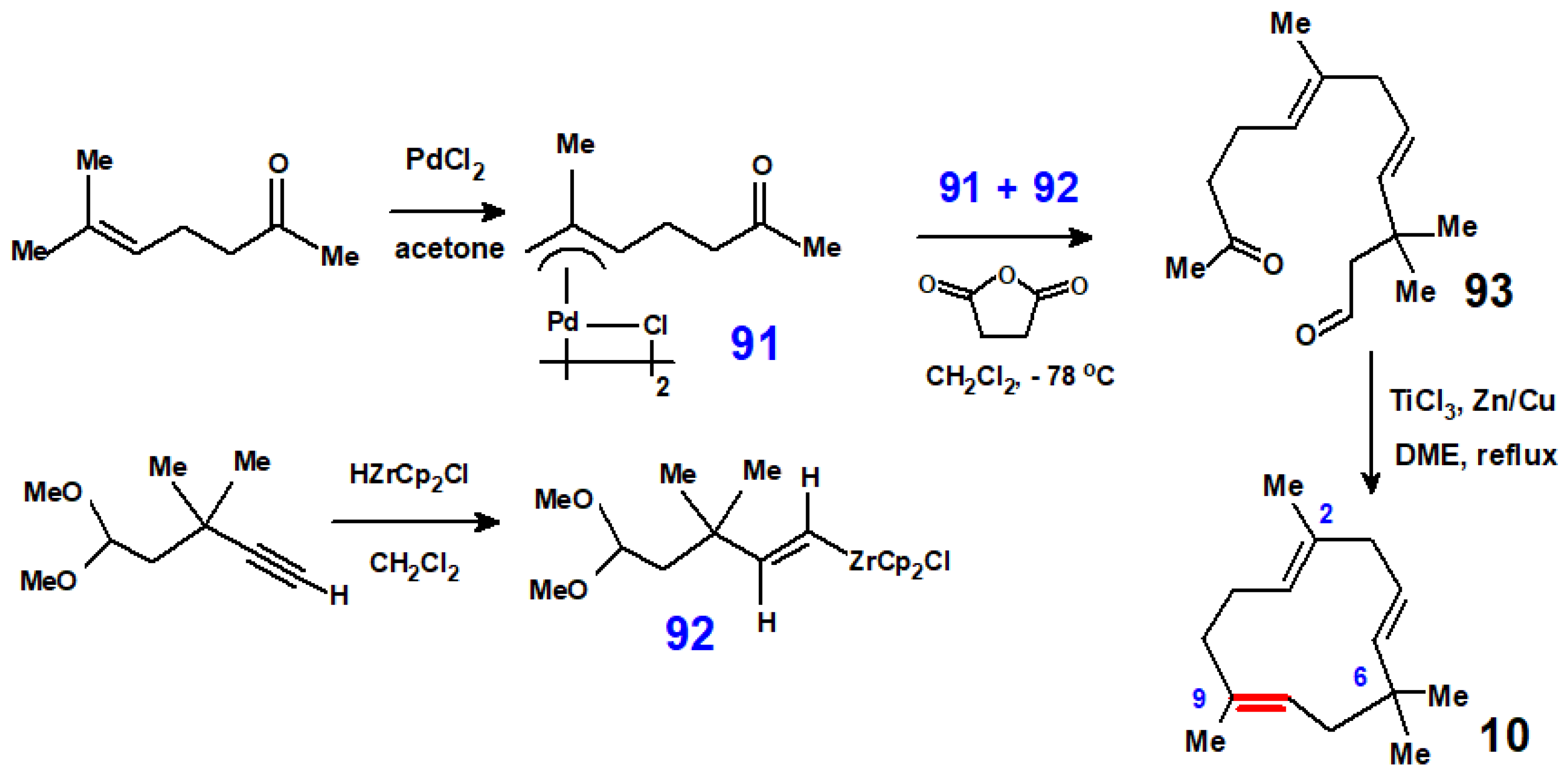
The titanium-induced dicarbonyl coupling reaction devised by John McMurry at Cornell University, Ithaca, New York, brings about deoxygenative coupling and carbon–carbon double bond formation by reaction with TiCl3/Zn-Cu [49]. This has been brilliantly applied to terpenoid syntheses, and provides probably the most efficient organometallic route to humulene (Scheme 37). The required allyl-palladium precursor, 91, was obtained by the reaction of commercially available 6-methyl-5-hepten-2-one with PdCl2 and was allowed to react with the vinyl zirconium complex, 92, prepared by hydrozirconation of 5,5-dimethoxy-3,3-dimethyl-1-pentyne. The resulting ketoaldehyde, 93, was subjected to McMurry coupling conditions to yield humulene in good yield in only four steps [50], an outstanding achievement!
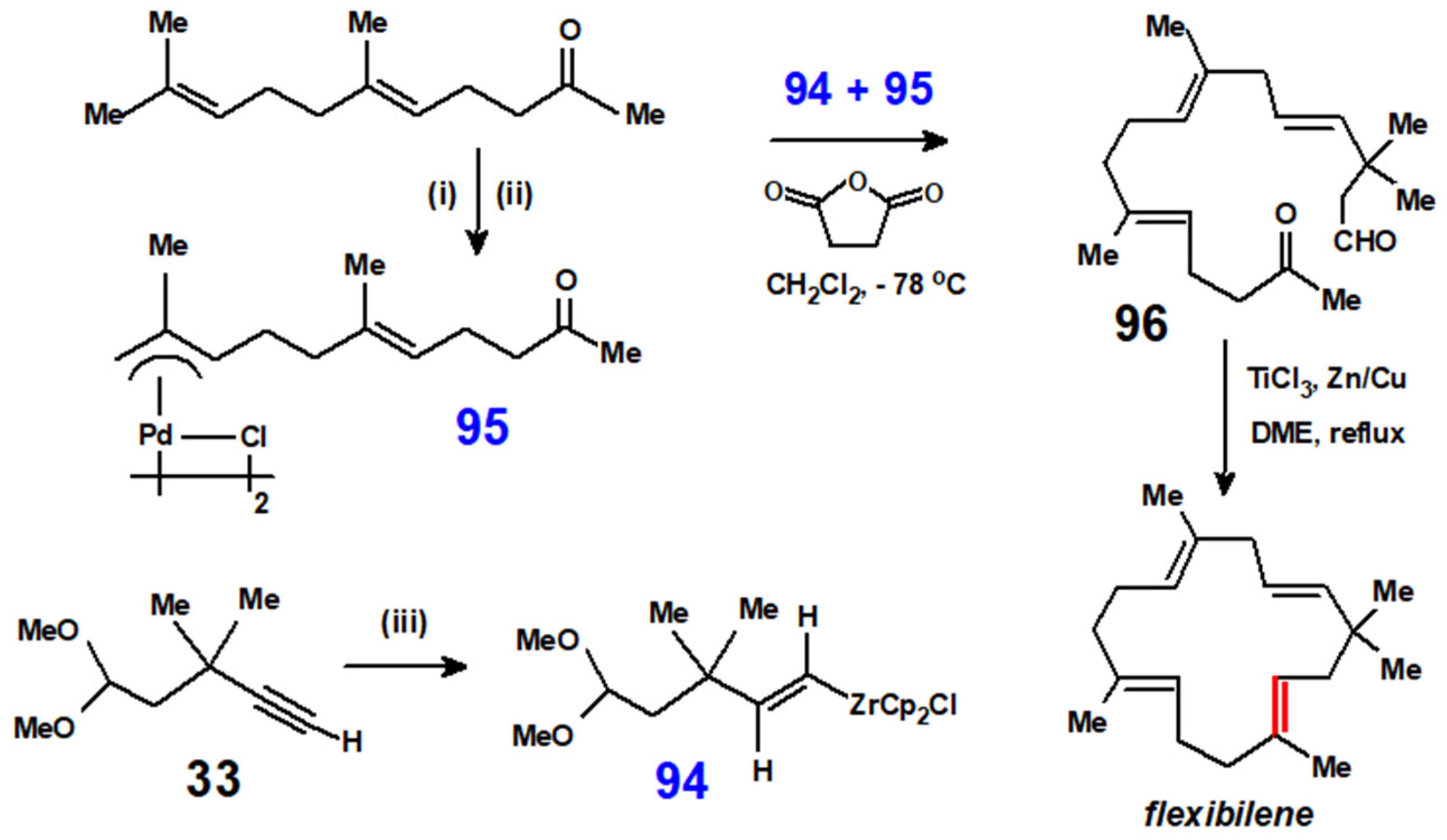
This same technique was utilised again to synthesise the first 15-membered ring diterpene to have been found in nature, flexibilene, 2,6,6,9,13-pentamethylcyclopentadeca-1,4,8,12-tetraene, that was originally isolated from the soft coral Sinularia flexibilis. Once again, hydrozirconation was used to obtain the required vinyl reagent, 94, while its allyl-palladium counterpart, 95, was prepared from geranyl acetone (Scheme 38). Under McMurry coupling conditions, the resulting keto-aldehyde, 96, cyclised in a 78% yield [51], and the final product was characterised by comparison with an authentic sample.
Another example of this efficient approach to ring-closing is given by the synthesis of (±)-δ-araneosene, 97, by Hu and Corey that closely parallels their route to humulene, as shown previously (Scheme 33) [43]. The initial stages follow the palladium-mediated formation of the 11-membered ring with the difference that one of the methyl substituents at C(6) has been replaced by an extended chain bearing an isopropyl and a methylidene group, as in 98. Ozonolysis of this latter functionality delivered the diketone 99 poised to undergo McMurry coupling and thereby yield the final product (Scheme 39).
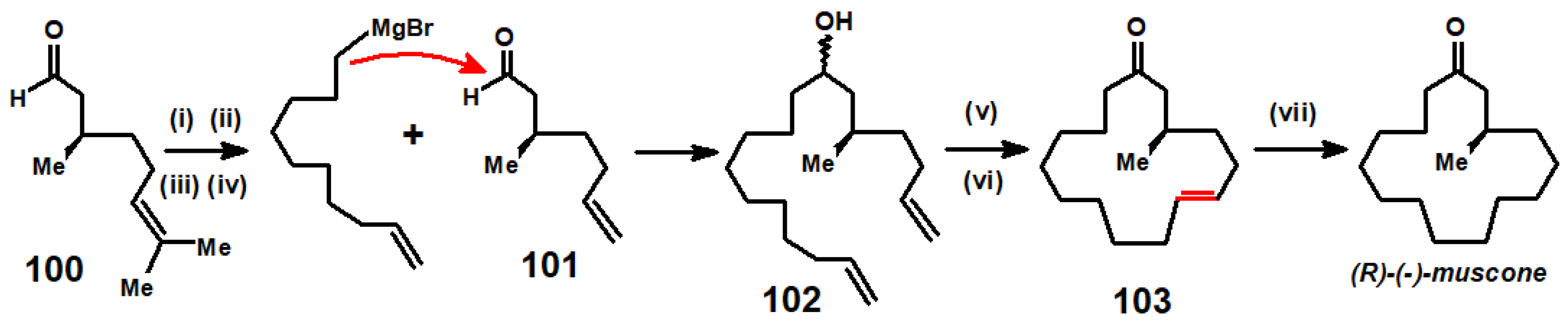
An interesting parallel may be drawn between McMurry’s approach, whereby two oxygens are abstracted from adjacent carbonyl groups, thus leading to the formation of a carbon–carbon double bond and ring-closing metathesis (RCM), in which a pair of terminal alkenes can exchange partners and eliminate ethylene. In the terpene field, this latter methodology was used to advantage in an ingenious route to (R)-(-)-muscone [52]. Commercially available (+)-citronellal, 100, was reduced to citronellol, protected as an ether with t-BuMe2SiCl, and then treated with OsO4/NaIO4 to cleave the isopropenyl linkage, followed by Wittig olefination to give 101, which was allowed to react with the Grignard derived from 10-bromodec-1-ene to form alcohol 102. After oxidation to ketone, RCM using the Grubbs II catalyst produced the 15-membered cycloalkene, 103, that yielded (R)-(-)-muscone upon hydrogenation (Scheme 40).
6.3. Ti(III) and Zr(III) as Dimerisation Catalysts
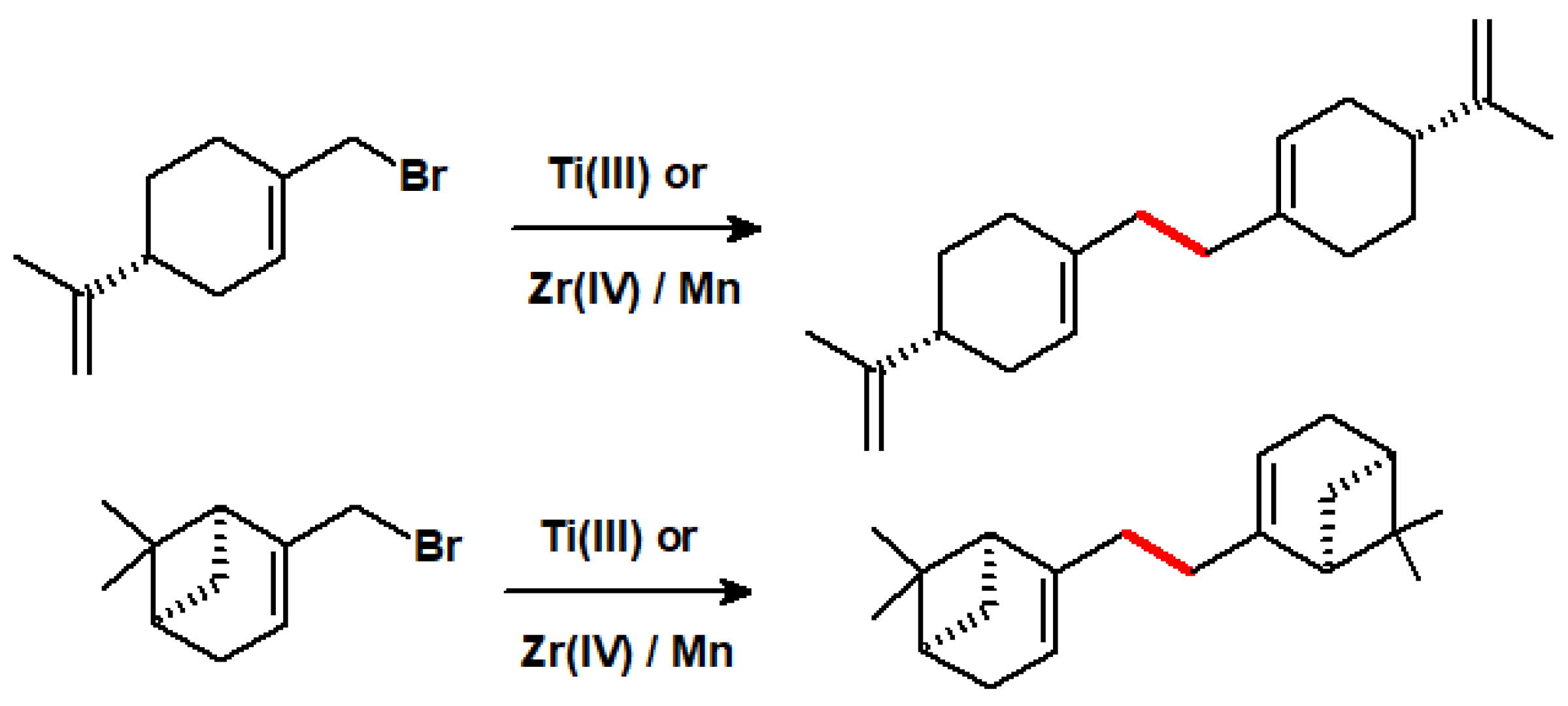
Group 4′s metal-mediated coupling of allylic halides has been described by Barrero who found that a Ti(III) catalyst, prepared from titanocene dichloride and powdered manganese, efficiently brings about a number of dimerisation processes. The analogous Zr(III) system, made from Cp2ZrCl2 and Mn, behaves similarly and, interestingly, the addition of Lewis acids, such as collidine hydrochloride, allows catalyst loading to be as low as 0.05 equivalents [53]. Typical examples are shown in Scheme 41.
7. The Golden Route to Carenes
While carene and its homologues are conventionally prepared via cyclopropanation of unsaturated ketones, in an elegant new approach, Fürstner and Hannen hypothesised that metal-catalysed rearrangement of a propargyl acetate to a metal carbene could provide a more concise synthetic protocol. As indicated in Scheme 42, starting from the alkyne-metal π-complex, 104, migration of the acetate via a cyclic intermediate, 105, to form the metal carbene complex 106 could then bring about cyclopropane formation [54]. The completion of the route to the carene skeleton would merely require a conversion of the acetate to ketone and an introduction of a double bond.
Gratifyingly, when the propargyl acetate 107, derived from geranylacetone, was treated with AuCl3 (Scheme 43), ring closure to 108 was successfully achieved in excellent yield and purity (95%). Further manipulation delivered (-)-sesquicarene, 109.
8. Reactions of Terpene—Palladium(II) Complexes with Nucleophiles
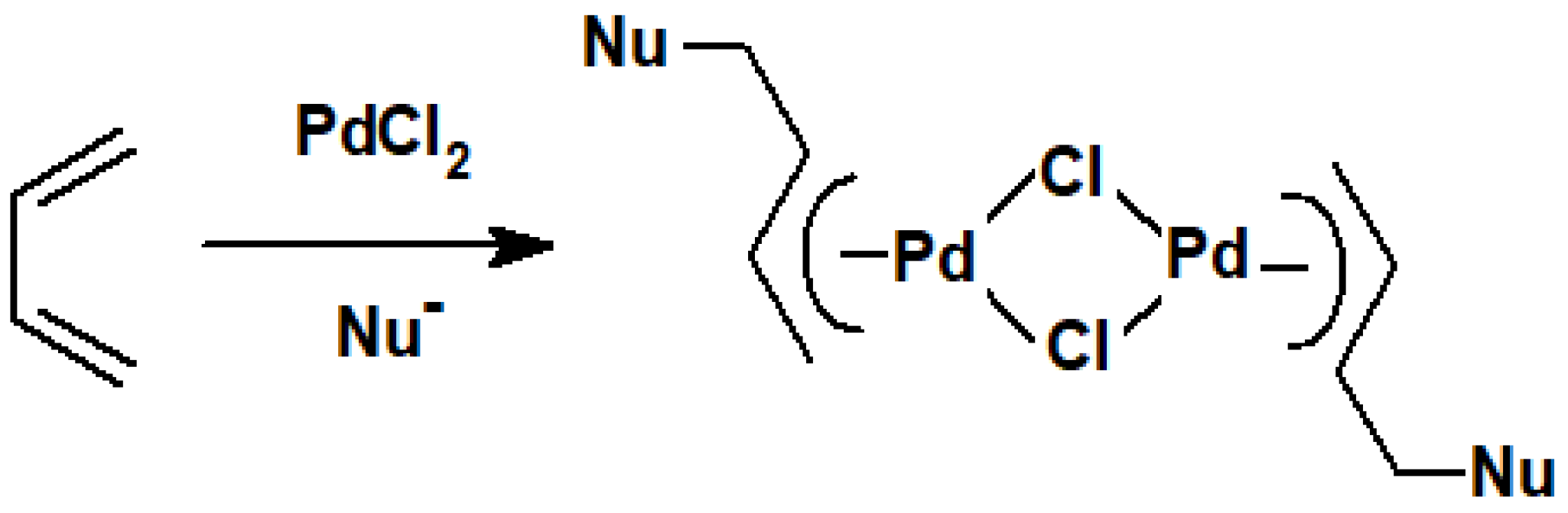
The preponderance of alkene double bonds in terpenoids inevitably suggested that treatment with complexes of palladium(II), iron(0), or nickel(0) would be the most productive areas to be investigated initially. The general reaction with a source of PdCl2 is to form a π-allyl-palladium chloride dimer in which the nucleophile has added to a terminal carbon of the original diene (Scheme 44).
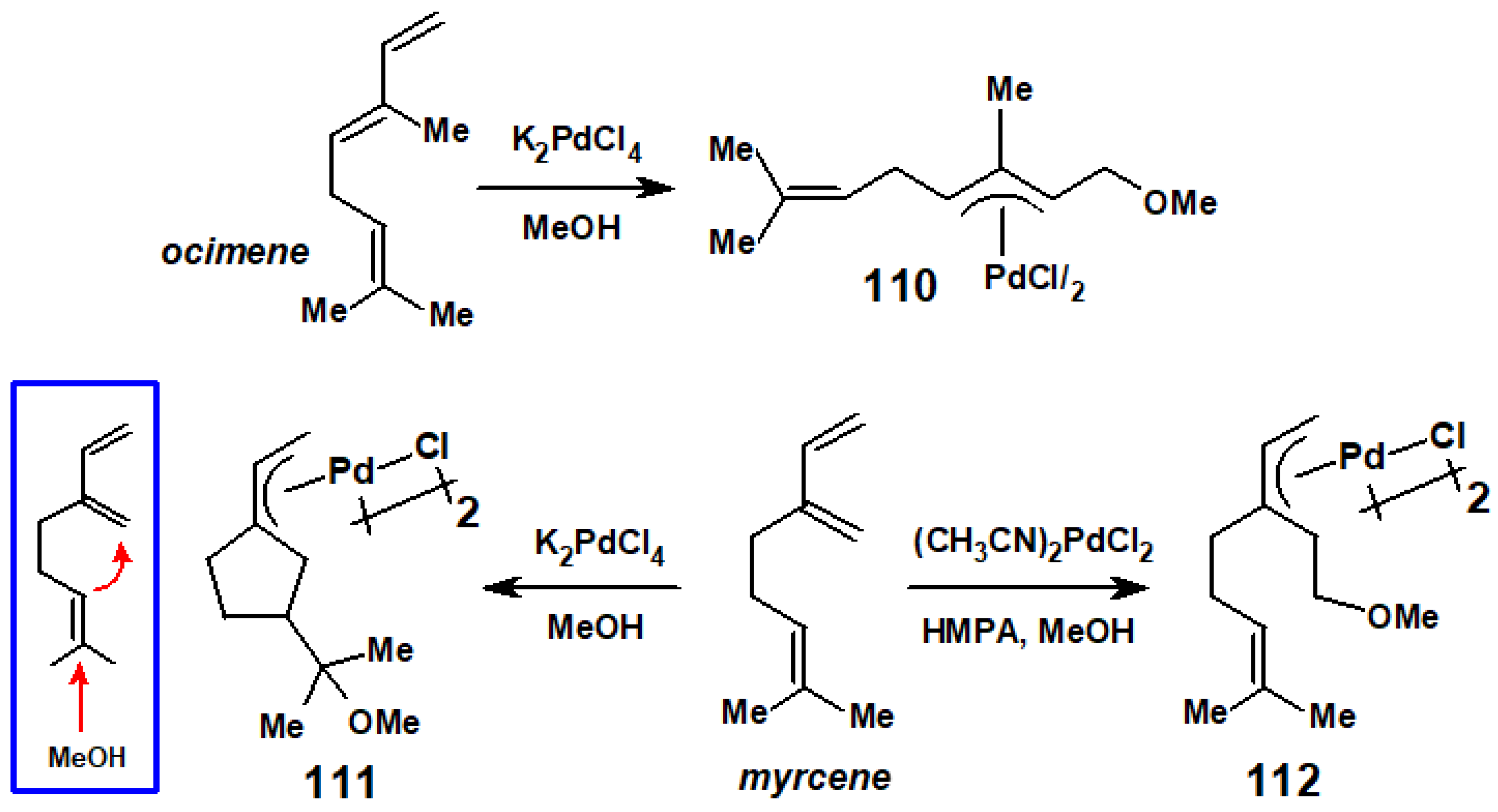
In a series of publications in the 1970s, Francis McQuillin (in Newcastle, UK) and Hiroharu Suzuki (in Tokyo) studied the reactions of palladium chloride with the isomeric monoterpenes ocimene and myrcene that are found in numerous natural sources, such as bay leaves, hops or wild thyme, and are widely used in the perfume industry.
Both cis and trans ocimene react with K2PdCl4 in methanol, bringing about a colour change from red-brown to yellow, to form the η3-palladium complex 110, that was characterised principally by NMR spectroscopy and mass spectrometry. In contrast, the analogous reaction of myrcene in methanol yielded a product, 111, comprising an exo-cyclic allyl-palladium fragment and a cyclopentane ring bearing a CMe2OMe substituent. The overall effect was to bring about palladium-promoted nucleophilic attack on the terminal carbon of the unconjugated double bond with concomitant cyclisation to form the five-membered ring, as depicted in Scheme 45 [55]. Changing the palladium precursor and the solvent to hexamethylphosphoramide led instead to 112, as the primary product [56,57].
The corresponding reaction of limonene in methanol solution proceeded via the exocyclic η3-palladium complex 113 that yielded dihydro-α-dihydroterpinyl methyl ether, 114, upon hydrogenation, while palladation of either α- and β-pinene yielded the same π-allyl complex, 115 (Scheme 46) [58].
McQuillin also probed the products formed when (+)-3,7-dimethylocta-1,6-diene, 116, was treated either with palladium dichloride or mercuric acetate in different solvents (Scheme 47) [59]. This isoprenoid diene is readily available from pyrolysis of (+)-pinane [2,60] and, when the reaction was carried out in acetone at 70 °C, cyclisation in the allyl-palladium complex 117 proceeded in Markovnikov fashion to yield the six-membered ring in α-terpineol, 118.
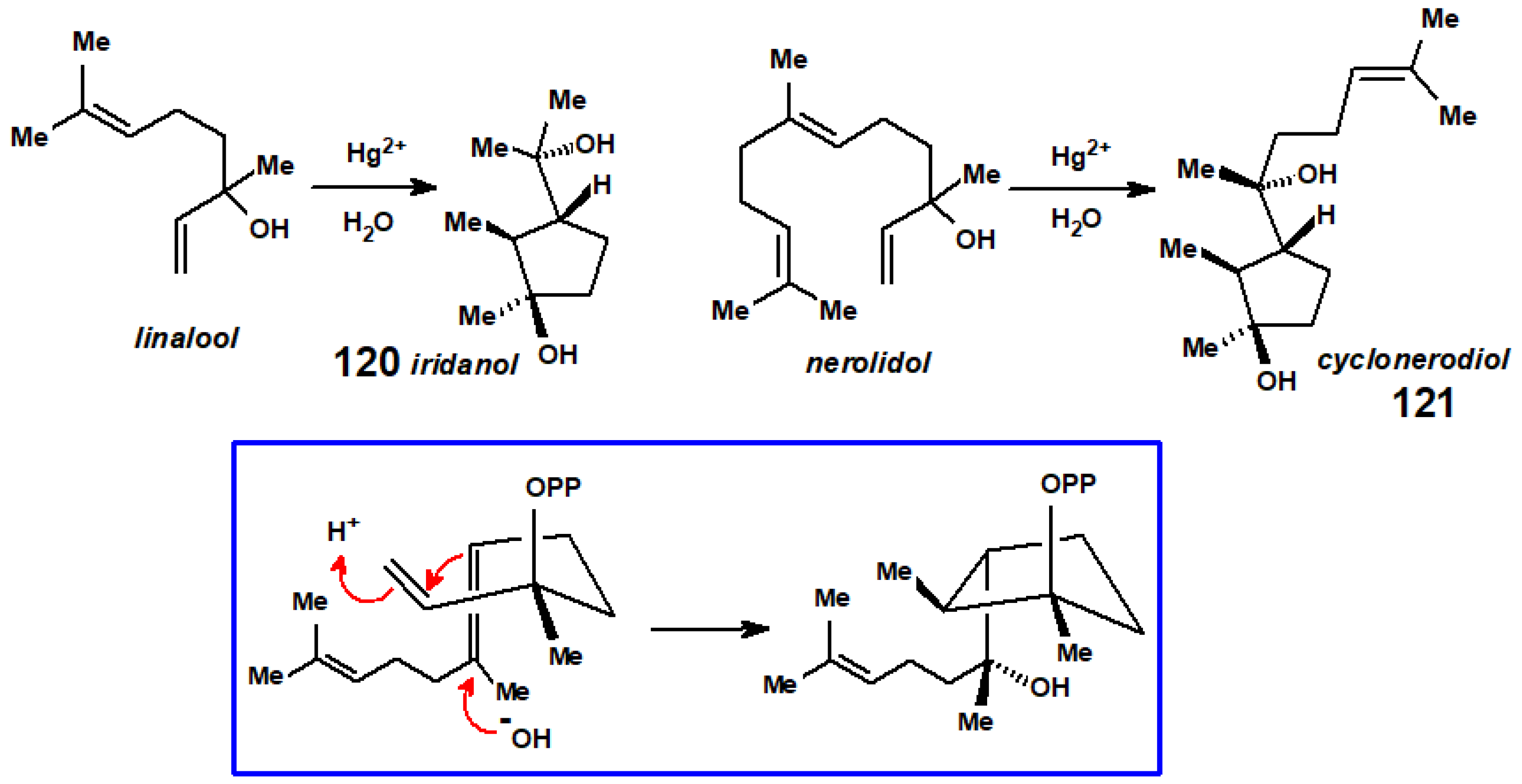
By way of contrast, when 3,7-dimethylocta-1,6-diene was stirred with mercuric acetate in aqueous THF and then reduced with sodium borohydride, the products were reported to be stereoisomers of (2,3-dimethylcyclopentyl)propan-2-ol, 119 [59]. The proposed Hg(II)-promoted mechanism invokes anti-Markovikov addition to the coordinated double bond. As noted below, such molecules had been described previously as products of the thermal cyclisation of linalool, and named as iridanols [61].
In very closely related work by the Itô group in Japan [62], also using a mercury(II) salt, the cyclisation of the monoterpene linalool to iridanol, 120, or of the sesquiterpene (C15) system nerolidol to cyclonerodiol, 121, proceeds with almost exclusive stereospecificity. In many ways, this mimics the biosynthetic pathway elucidated by Cane [63], as shown in the box in Scheme 48.
9. Reactions of Terpene—Iron Carbonyl Complexes with Electrophiles
Myrcene reacts with Fe2(CO)9, in benzene, or better with Fe3(CO)12 in dibutyl ether, to form 122, in which the tricarbonyliron fragment is η4-bonded to the conjugated diene unit. When protonated with HBF4 in ether, it brings about cyclisation and the formation of the allyl cation complex, 123; as shown in Scheme 49, subsequent treatment with sodium borohydride yields 1-ethylidene-3,3-dimethylcyclohexane, 124 [64].
Treatment of 122 with acetyl chloride under Friedel–Crafts conditions brings about the introduction of an acetyl group on the isolated double bond, as in 125. More interestingly, the use of oxalyl chloride led to the acyl chloride, 126, which reacted with AgNO3 to yield the two cyclic ketone complexes, 127 and 128, via carbonylative annulation (Scheme 50) [65].
The π-bonded Fe(CO)3 in 122 can also act as a protecting group whereby a reaction that would normally occur on the diene instead proceeds at the unconjugated alkene terminus. Typically, treatment with diborane reduces the isopropylidene unit, and decomplexation with ceric ammonium nitrate yields the dihydro monoterpene, 129 [66]. Likewise, addition of dichlorocarbene, generated from chloroform and potassium tert-butoxide, forms the dichloro-dimethylcyclopropane 130 after decomplexation (Scheme 51) [67].
The reaction of (ocimene)Fe(CO)3, 131, with trityl fluoroborate, followed by reduction with sodium borohydride, brings about a rearrangement such that the transfer of a hydrogen from the methyl group onto its ethylidene neighbour led to an isomer, 132, bearing a methylene and an ethyl substituent. One can envisage the formation of the allyl cation, 133, and then the bond rotation, thus offering the Fe(CO)3 unit a potential cisoid diene fragment available for complexation after migration as well as the addition of hydride to complete the formation of the new ethyl substituent. In support of this scenario, the original methyl group was doubly labelled with tritium and carbon-14, and the final positions of these labels were elucidated (Scheme 52) [66].
10. Metal-Promoted Terpene Isomerisations
10.1. π-Allyl Metal Hydrides
The observation of an increasing number of hydrogen rearrangement processes in alkenes prompted suggestions of the involvement of π-allyl metal hydrides [68]. Typically, (-)-β-pinene is converted into high optical purity (-)-α-pinene when heated with iron pentacarbonyl at 135 °C [69]. In support of this hypothesis, it was found that when the isotopically labelled allyl alcohol CH2=CH-CD2-OH was treated with Fe(CO)5, the NMR spectrum of the resulting propionaldehyde, CH2D-CH2-CD=O, revealed the presence of deuterium in the methyl but not in the methylene group. Scheme 53 illustrates the mechanism by which the intermediate 134 allows the transfer of deuterium, leading to the vinyl alcohol–iron carbonyl, 135, that tautomerizes upon decomplexation to yield the labelled propionaldehyde, 136 [70].
This work was later extended, whereby endo and exo 1-hydroxy-5,6-dihydrodicyclopentadiene, 137 and 138, respectively, were each heated with Fe(CO)5 in attempts to bring about isomerisation to the ketone, 139. In the event, only the endo-hydroxy epimer, 137, underwent exchange, indicating that hydrogen migration via the exo (π-allyl)tricarbonyliron hydride, 140, was only viable when the iron was complexed to the same face as the migrating hydrogen (Scheme 54) [71]. Examination of models clearly shows that endo coordination of the bulky tricarbonyliron fragment would encounter serious steric problems.
10.2. [3,3]-Sigmatropic Shifts in Terpenoids
As part of his early pioneering work, McQuillin studied the rearrangement behaviour of linalyl and neronidyl acetates, 142 and 58, respectively, when allowed to react with K2PdCl4 in chloroform in the presence of calcium carbonate (acting as a buffer to prevent acid-induced reactions). It was found that in both cases, the acetate underwent a 1,3-migration to form their geranyl and farnesyl isomers, 15 and 143 (Scheme 55), and a mechanistic proposal invoked the formation of a cyclic palladium-linked bridging acetate intermediate, 144 (Scheme 56) [58].
A particularly fine example, also in the terpenoid field, was reported, whereby the 1,3-acetate shift in the prostaglandin precursor 145 proceeded to yield 146 in a suprafacial fashion with a complete transfer of chirality (Scheme 57) [73].
These can now be classified as palladium-initiated [3,3]-sigmatropic shifts, and selected examples of Cope rearrangements in terpenoids [74,75,76,77], including synthetic routes to (10)-epi-elemol, 147 [78], and γ-elemene, 148 [79], are shown in Scheme 58. Many such metal-promoted rearrangements proceed under very mild conditions, presumably via the coupling of π-allyl intermediates, whereas their non-complexed counterparts frequently require prolonged heating.
10.3. Interactions of Ring-Strained Terpenes with Organometallic Reagents
The tendency of many organometallic species to bring about ring-opening in strained systems is well established. Typically, the reaction of bicyclobutane with a platinum(II) complex possessing a readily displaceable ethylene ligand results in the formation of a polymeric intermediate, 149, that reacts with pyridine to yield 150 (Scheme 59) [80].
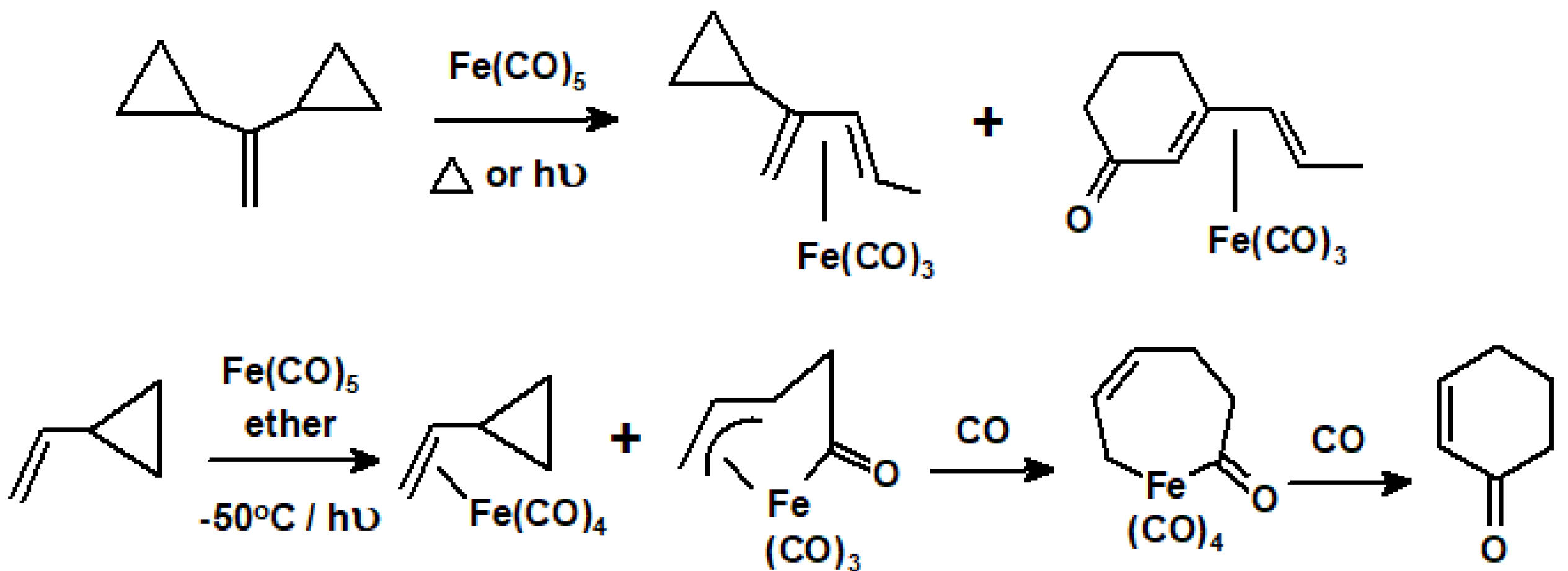
Similarly, when vinylcyclopropanes are thermolyzed or photolyzed with Fe(CO)5, they suffer ring-opening to yield diene-Fe(CO)3 complexes, and can also lead to carbonyl insertion products, as exemplified in Scheme 60 [81,82].
Such behaviour is also found in the terpene field, whereby thermolysis of (+)-2-carene, 151, and Fe(CO)5 yields (-)-(1S)-3,8,8-trimethylbicyclo[4.1.1]oct-3-ene-7-one, 152, and the corresponding alcohol, 153, along with (α-phellandrene)Fe(CO)3, 154 [83]. The proposed mechanism (Scheme 61) invokes cleavage of the bond common to the 3-membered and 6-membered rings with the formation of the allyl-iron intermediate, 155, the migratory insertion of a carbonyl, and finally, the reductive elimination to form the ketone 152. The alcohol 153 is thought to arise from reduction of 152 by H2Fe(CO)4.
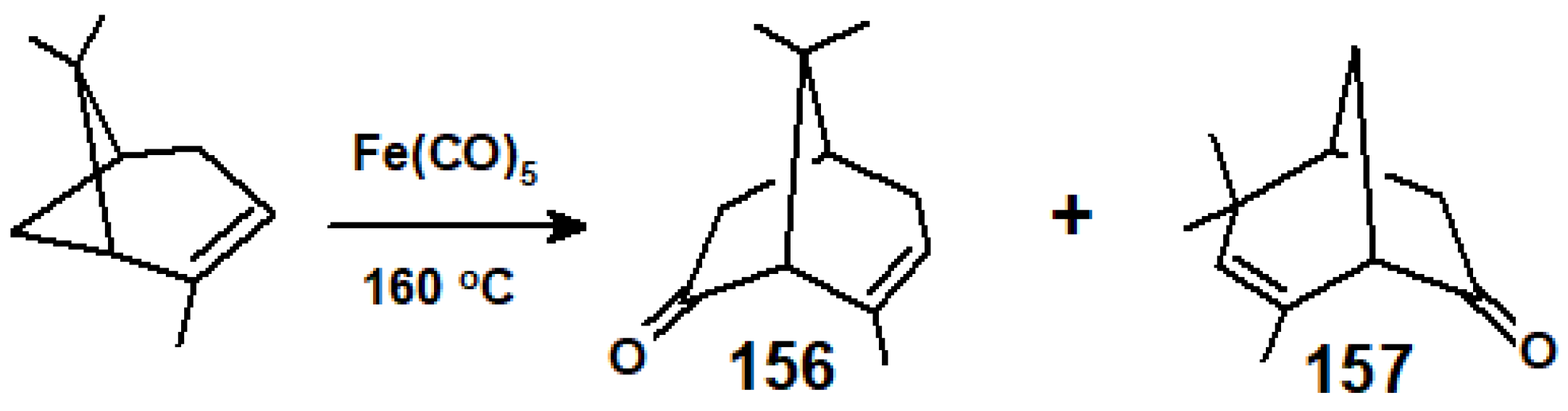
In earlier work on the somewhat less-strained (+)-(α)-pinene system, prolonged thermolysis with Fe(CO)5 in a pressure vessel at 160 °C led to two stereospecific ring expansion products, 156 and 157, in which the bicyclo[3.2.1] molecular frameworks were enantiomeric, but with differently positioned methyl substituents (Scheme 62) [84].
The proposed mechanism involved the cleavage of the cyclobutane ring to form an intermediate, 158, possessing an Fe(CO)3 fragment bonded in both in a π-allyl- and a σ-alkyl fashion. Subsequent carbonyl insertion furnished 159 that was exquisitely poised for reductive elimination to occur with concomitant C-C bond formation at either end of the allyl unit, leading to 156 and 157 (Scheme 63). Moreover, further reaction of the enantiomer of 159, derived from (-)-(α)-pinene, with dimethyl acetylenedicarboxylate (DMAD) brings about an insertion of the alkyne to deliver the bicyclo[4.3.1]octadienone, 160 [85].
Impressively, in 1975, the authors characterised their products and suggested a reaction mechanism solely on the basis of their NMR data (at 60 MHz) and mass spectra [85]. Gratifyingly, their proposals were fully vindicated almost three decades later by the isolation and X-ray crystallographic structural determination of the π-allyl/σ-alkyl intermediate, 161 (Figure 4) [86].
10.4. Terpene Rearrangements Induced by Pt(II) or Rh(I) Salts
Pinene is also ring-opened when left in solution at room temperature with K2PtCl4 in dilute HCl for an extended period [87]. The product isolated and characterised by X-ray crystallography was found to be dichloro[η4-p-mentha-1,8(9)-diene]platinum(II) in which the metal is coordinated not only to the original double bond in pinene, but also to the isopropenyl group formed upon ring cleavage. The mechanism envisaged (Scheme 64) requires initial coordination to pinene, as in 162, ring-opening to form 163, and loss of KCl to yield the observed product 164.
The introduction of a terminal alkyne substituent, thus forming a terpenoid enyne, led to unexpected cyclopropane formation when treated with a Rh(I) or Pt(II) salt. The enynes 165 and 166, derived from perillyl alcohol and geraniol, respectively, reacted to form the cyclopropyl systems 167 and 168 shown in Scheme 65. The favoured mechanism invokes complexation to the alkyne, as in 169, a rearrangement to a metal carbene complex, 170, with subsequent addition to the neighbouring double bond to form the cyclopropane, and is supported by isotopic labelling, as shown in Scheme 66 [88]. In many ways, this parallels the approach towards cyclopropane formation taken by Fürstner in his gold-carbene route to carenes (Section 7).
11. Regiospecific Epoxidation of Terpenes
Epoxidation is frequently accomplished by the oxidisation of an alkene linkage by a peroxy acid. However, when the target molecule has multiple sites of unsaturation, stereo- and regio-selectivity need to be controlled. This was first achieved in the terpene field by K.B. Sharpless (Nobel 2001, 2022), whereby geraniol and linalool were selectively converted into the previously unknown epoxides, 171 and 172, respectively, using vanadyl acetylacetonate as the catalyst in conjunction with tert-butyl hydroperoxide (Scheme 67) [89]. In contrast, reaction with meta-chloro-perbenzoic acid is markedly less selective. The Sharpless approach has since been very widely adopted, and it is interesting to note that almost half a century later, it was reported that, when this catalyst was grafted onto silica nanoparticles, 2,3-epoxygeraniol was obtained with 100% conversion and 99% selectivity [90].
In another early example, the combination of tert-amyl hydroperoxide and either molybdenum hexacarbonyl or molybdenum pentachloride brought about specific and almost quantitative conversion of α- and β-pinenes or 2- or 3-carenes into their corresponding epoxides (Scheme 68) [91].
In closely related later work [92], the reaction of (+)-3-carene with the high oxidation state complex methyltrioxorhenium(VII), in the presence of a Lewis base, led to stereospecific, almost quantitative, formation of α-3,4-epoxycarene, 173α, along with the diol 174. Interestingly, isomeric β-3,4-epoxycarene, 173β, was preparable by the treatment of (+)-3-carene with N-bromosuccinimide and water to deliver the bromohydrin, 175, with subsequent epoxide formation, resulting from base-promoted elimination of HBr (Scheme 69).
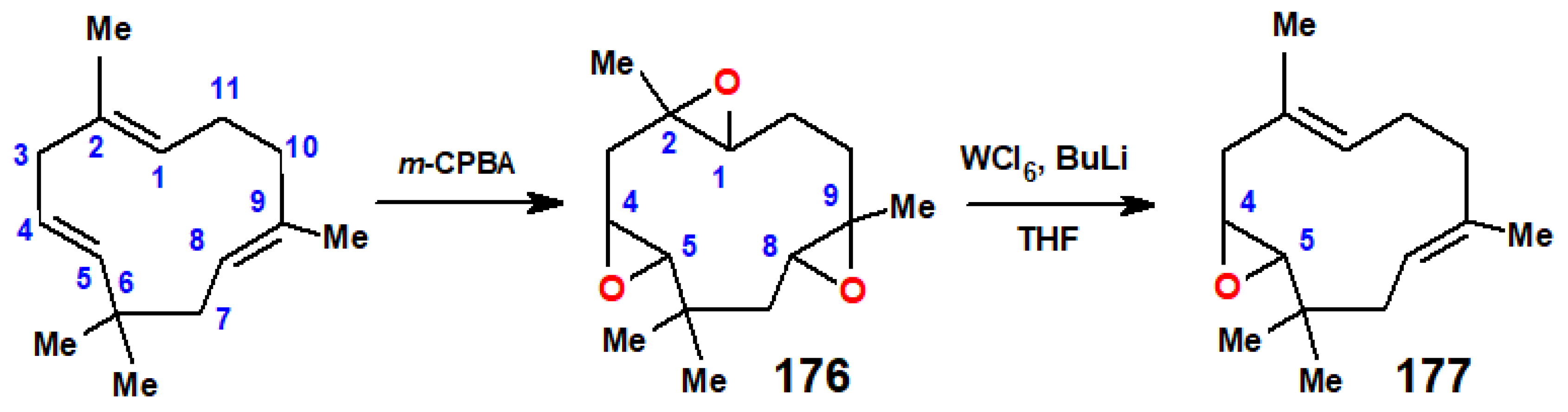
Epoxidation of humulene posed a particular problem when there was a need to functionalise only at C(3)–C(4), the least reactive double bond, because epoxidation occurs preferentially at C(1)–C(2), the most strained alkene linkage, and then at C(8)–C(9), as shown in the X-ray crystal structure of the di-epoxide (Figure 5) [93]. This difficulty was finally overcome [94] by first generating the tri-epoxide, 176, by continued treatment with m-chloroperbenzoic acid, and then removing the oxygens at C(1)–C(2) and C(8)–C(9) by reaction with WCl6 and n-BuLi, a deoxygenation reaction previously discovered by Sharpless [95], to form 177 (Scheme 70).
Attempts to attach organometallic fragments to humulene were generally unsuccessful, although there is a report of the formation of the π complex [(humulene)Fe(CO)2(C5H5)]+ [BF4]-; however, the combination of low yield and thermal instability precluded its unambiguous identification by NMR spectroscopy [96].
12. Organometallic Derivatives of Guaiazulene
Guaiazulene (1,4-dimethyl-7-isopropylazulene) is a dark blue bicyclic sesquiterpene possessing five- and seven-membered rings, found as a constituent in oil of guaiac and in chamomile oil, and is widely used in skin care products. The synthesis and structure of the many metal complexes of azulene itself has been comprehensively reviewed [97]. Early work by Cotton established that guaiazulene forms bimetallic complexes in which the linked metals bind in a pentahapto fashion to the 5-membered ring and in a trihapto mode to the 7-membered ring (Scheme 71) [98]. Unlike azulene with its C2v symmetry, the pattern of substituents in guaiazulene breaks the mirror orthogonal to the molecular plane, thus reducing its symmetry to CS, a single mirror plane. Now, complexation of one metal (or more) to one face of the molecule lowers the symmetry to C1 and renders the system chiral. Moreover, these molecules can adopt two different trihapto coordination sites, as in 178 and 179, which have been synthesised, separated chromatographically and then fully characterised by X-ray crystallography. Variable-temperature NMR data reveal that these coordination isomers interconvert when heated. Later studies established that this interconversion can also be induced photochemically, and the barrier to haptotropic migration was found to be 117 ± 8 kJ mol−1 [99]; in the ruthenium analogues, the barriers were found to be markedly higher [100].
In an excellent very recent publication, it was reported that hydrolithiation of guaiazulene yields the isomerically pure dihydroguaiazulenide anion, a stable and storable cyclopentadienide-type ligand ideally suited for reaction with a wide range of organometallic precursors [101]. A number of half-sandwich complexes shown in Figure 6, in particular the trimethylplatinum complex, exhibit excellent catalytic activity in alkene hydrosilylations.
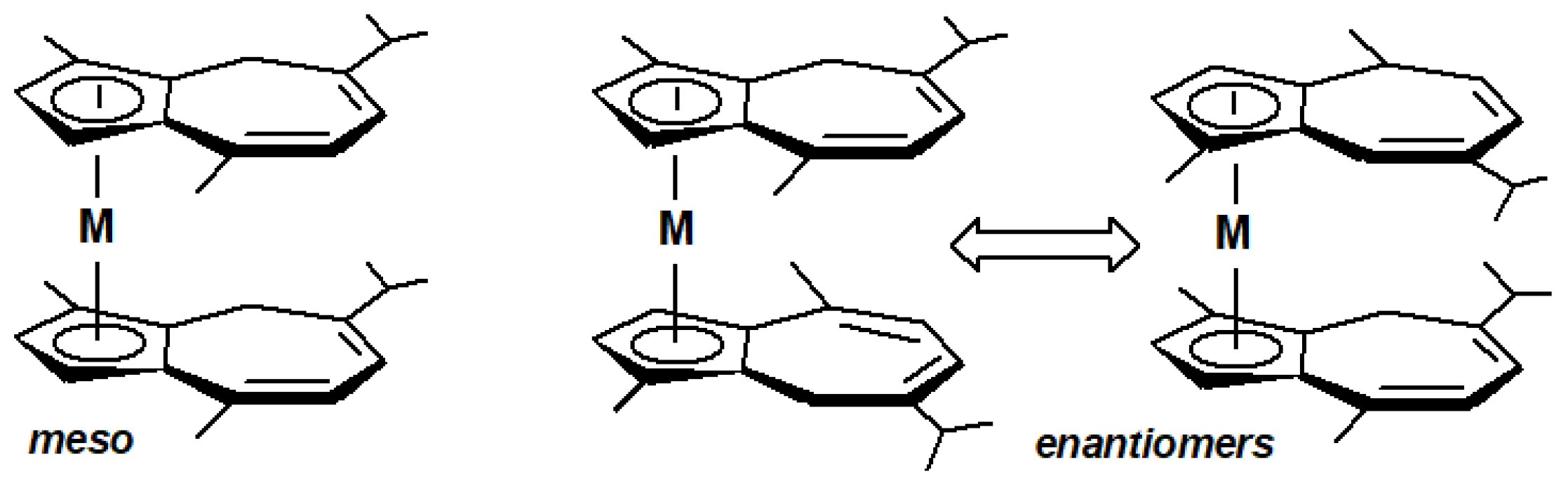
The issue of chirality in these molecules becomes more apparent in the metallocene sandwich compounds shown in Figure 7, for which meso and racemic isomers have been isolated and fully characterised by X-ray crystallography (Figure 8). These include complexes possessing iron, ruthenium, cobalt or rhodium as the central metal.
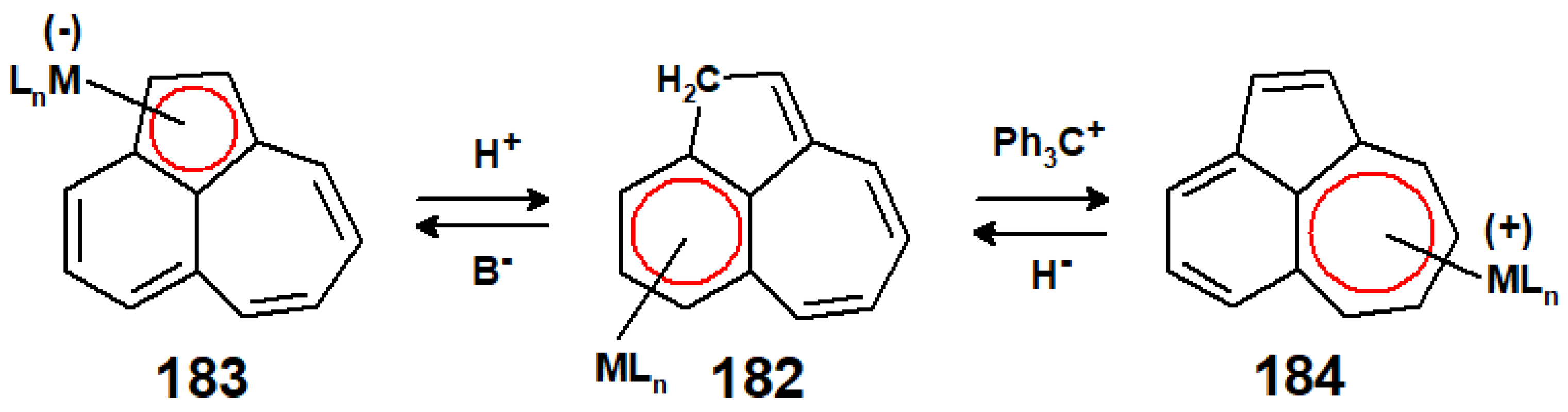
Unlike its parent azulene, guaiazulene is readily functionalised by the ready deprotonation of the 4-methyl group, whereby the negative charge is delocalised into the five-membered ring, thus generating a cyclopentadienide-type anion (Scheme 72). Treatment with 1-chloropinacolone, followed by Friedel–Crafts ring closure with AlCl3, yielded the benz[cd]azulene 180 that was characterised crystallographically as the η6-Cr(CO)3 complex, 181 [102]. This tricyclic system has the potential to exhibit η6 → η5 (182 → 183), or η6 → η7 (182 → 184) haptotropic migrations when deprotonated or treated with a hydride abstractor, such as the trityl cation (Scheme 73).
13. Metal Cluster Complexes of Terpenoids
13.1. Cobalt Cluster-Stabilised Carbocations
The chemistry of metal clusters is voluminous, but we focus here on the syntheses and reactivity of bi- and tri-metallic derivatives of terpenoids, in particular, tetrahedral clusters derived from alkynes and metal carbonyl precursors. The Nicholas reaction [103] is based on the intermediacy of cobalt-stabilised propargyl cations that are susceptible to nucleophilic attack, thus allowing for a controlled enhancement of molecular complexity (Scheme 74).
A typical example from the terpene field (Scheme 75) involves the addition of a zinc acetylide to citronellal in the presence of chiral ligand such as N-methylephredine, followed by the treatment of the alkyne 185 with dicobalt octacarbonyl to yield a tetrahedral alkyne-dicobalt cluster. Upon treatment with a Lewis acid, it forms the metal-stabilised propargyl cation 186, that undergoes intramolecular cyclisation to 187. Addition of ceric ammonium nitrate (CAN) leads to decomposition of the metal cluster and liberates the final product 188 [104]. Another fine example is Marshall’s synthesis of the cyclododecadienynol 189 (Scheme 76) [105].
13.2. X-ray Crystallographic Characterisation of Metal-Stabilised Bornyl Carbocations
Treatment of (1R)-(+)-camphor with an alkynyl anion yields the corresponding endo-2-alkynylborneol, whereby nucleophilic attack at the carbonyl proceeds so as to avoid steric interactions with the gem-dimethyl bridging moiety. As is well-known, complex molecular rearrangements are frequently encountered in terpene chemistry, and the protonation of endo-2-ethynylborneol 190 to form the corresponding propargyl cation, 191, leads to a spectacular series of migrations that culminate with the alkynyl substituent which is no longer adjacent to the carbocationic site (Scheme 77). The initial Wagner–Meerwein (W-M) rearrangement, to form 192, is followed by a 1,2-methyl migration (Nametkin shift) to 193, and then a second W-M and hydrolysis to yield the final product, 194 [106].
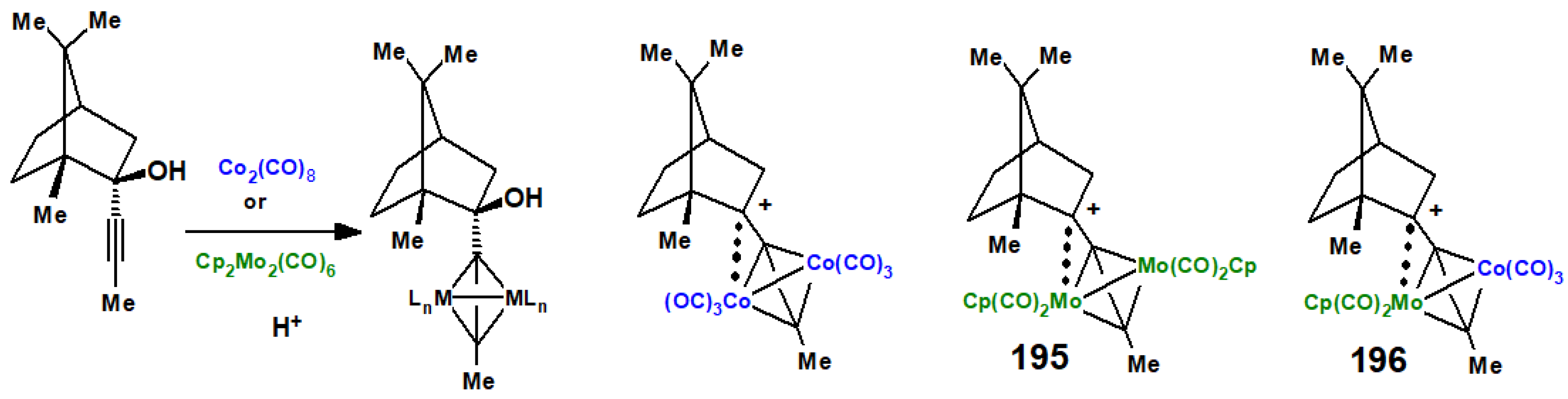
When endo-2-propynylborneol was allowed to react with either dicobalt octacarbonyl or (cyclopentadienyl)dicarbonylmolybdenum dimer (Scheme 78), the resulting tetrahedral alkyne-dimetallic clusters isolated were each characterised by X-ray crystallography [107,108]. However, upon protonation, the metal-stabilised cationic clusters resisted skeletal rearrangement and were unambiguously identified spectroscopically. Moreover, the dimolybdenum cationic complex, 195, was also characterised by X-ray crystallography, which revealed that the bornyl cation clearly leaned toward molybdenum such that the Mo-C(+) distance was only 2.74 Å. In the mixed Co-Mo cation, 196, once again, the Mo-C(+) distance was short (2.91 Å), indicating that the molybdenum centre was the preferred site for cation stabilisation, a conclusion supported by calculations at the EHMO level [109]; this area has been reviewed by Gruselle [110].
13.3. Metal Cluster Complexes in the Fenchyl, Verbenyl, Menthyl and Geranyl Series
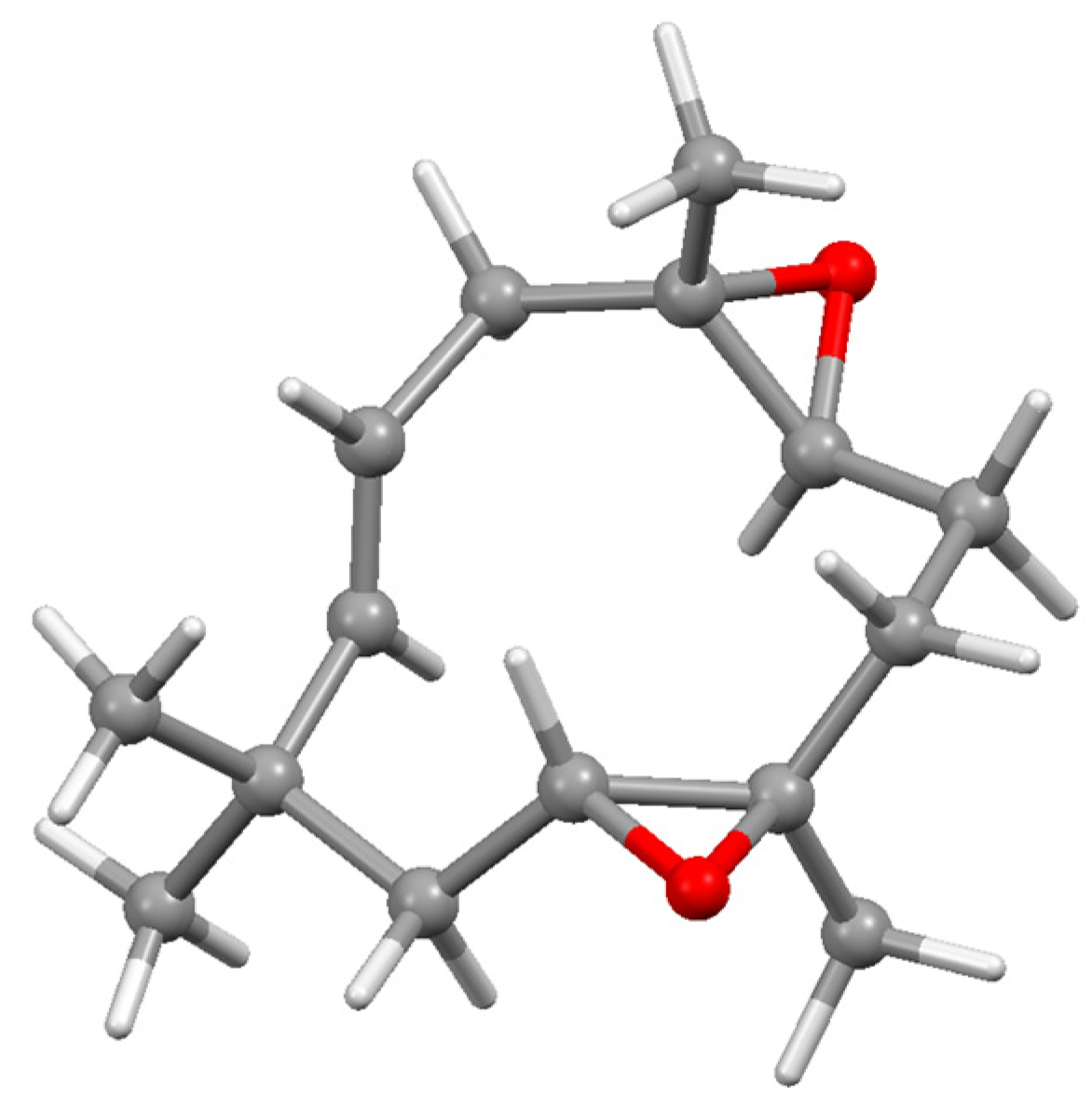
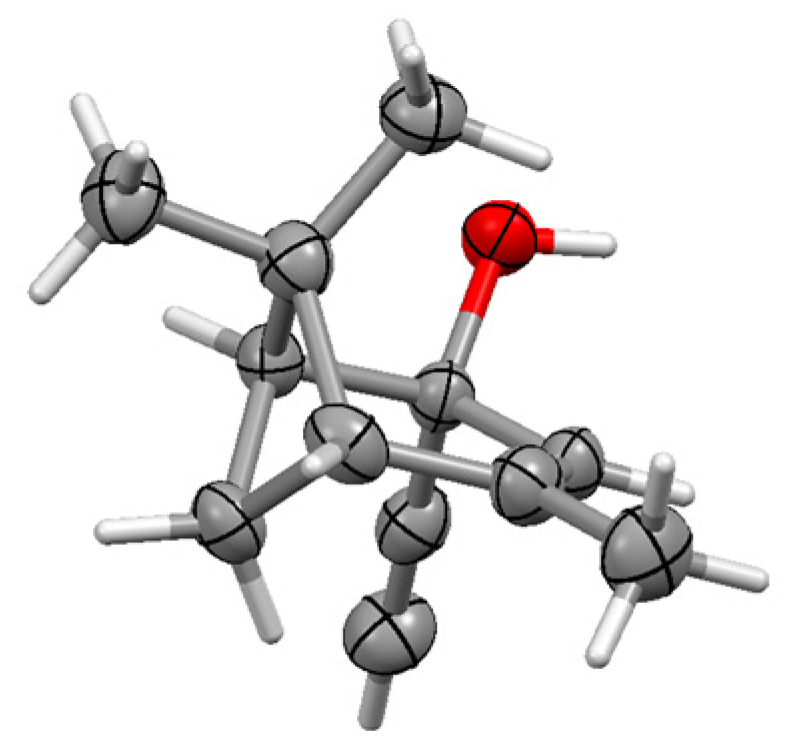
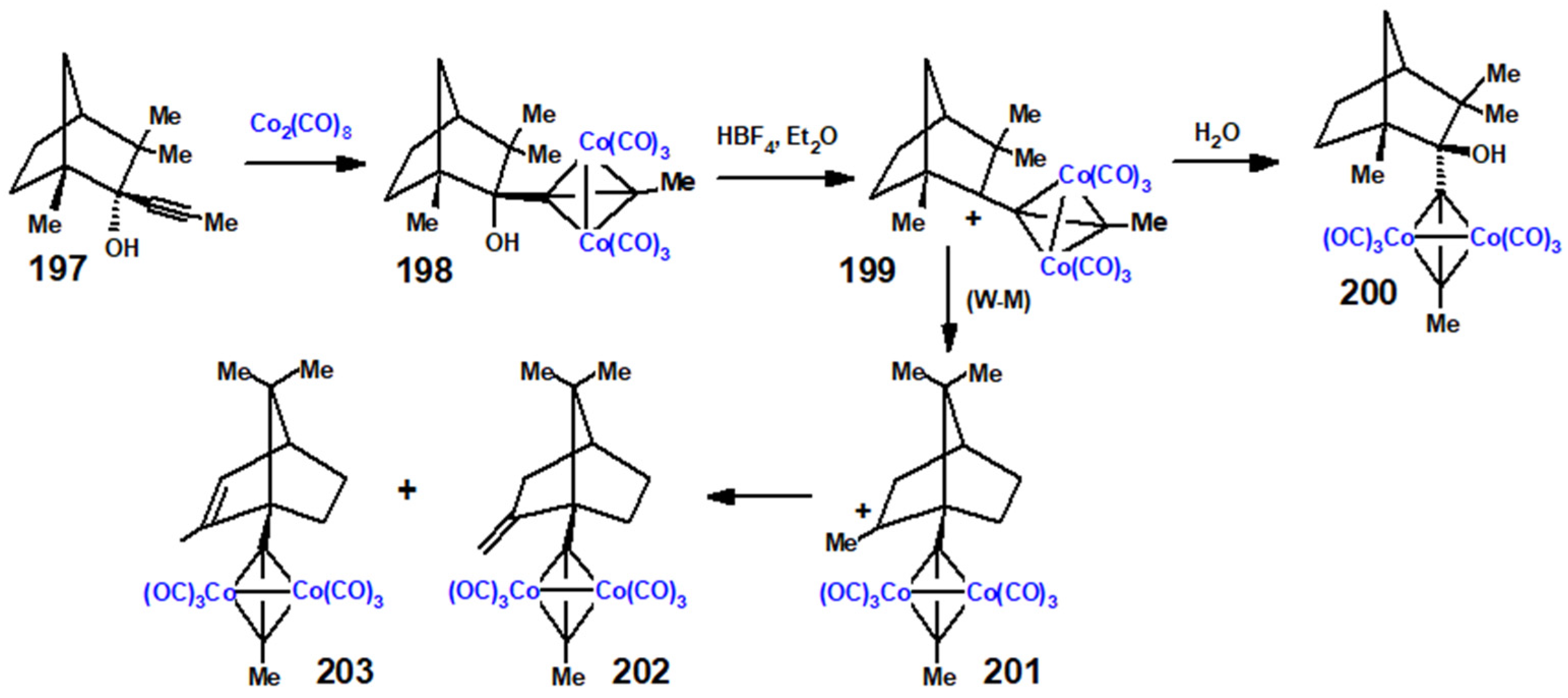
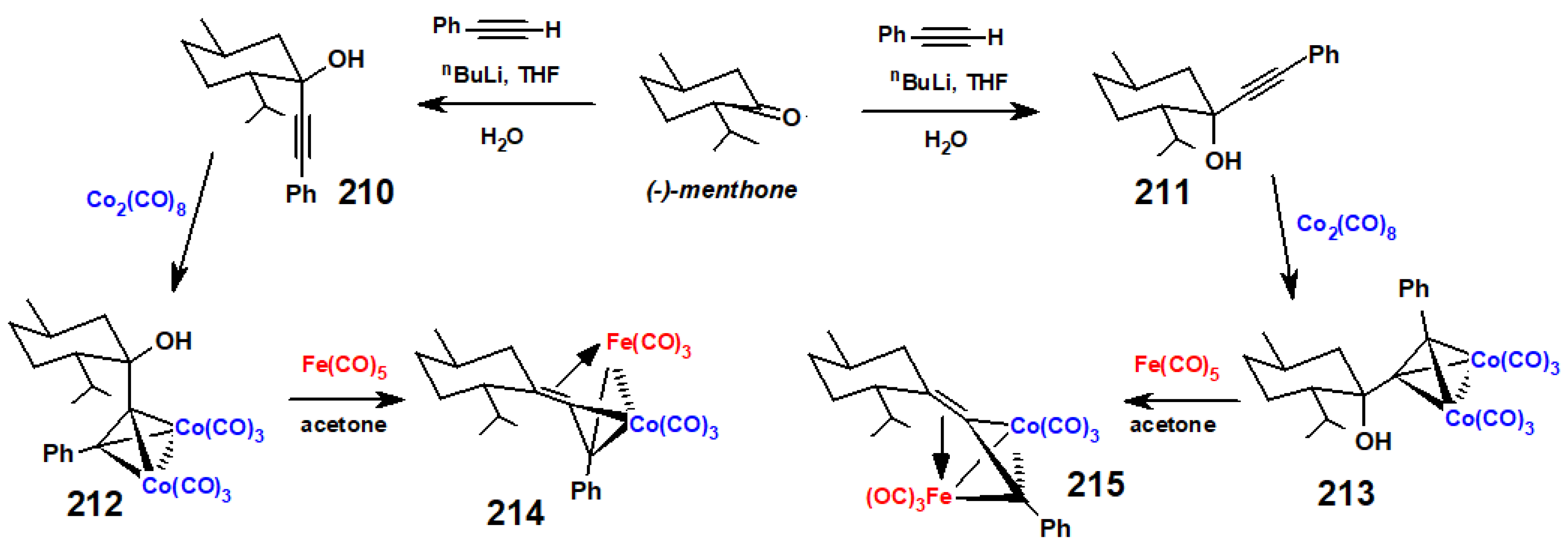
The situation becomes more interesting in the fenchyl case. Initial nucleophilic attack on fenchone by an alkynyl anion proceeds almost exclusively (95:5) to form the 2-exo-alkynol 197. (Strictly speaking, we should perhaps designate this as the endo epimer since OH takes precedence over the C≡C linkage in the Cahn–Ingold–Prelog protocol.) Treatment of 197 with Co2(CO)8 yielded the expected complex, 198, that was relatively unstable until protonated to form the cationic complex 199; however, when rapidly quenched with water, the product was characterised by X-ray crystallography as 200 (Figure 9), the epimer of the original alkynol (Scheme 79). This is presumably attributable to the steric strain engendered between the bulky cluster fragment and the gem-dimethyl group at C(3). However, when the cation 199 was allowed to stand for an extended time, it underwent a W-M shift to form 201, thereby placing the cluster fragment at C(1) with the carbocationic site at C(6), prior to elimination, leading to the alkenes 202 and 203 [111].
Now, displacement of a tricarbonylcobalt vertex in 203 by a (cyclopentadienyl)dicarbonyl molybdenum moiety to form the mixed metal cluster 204, followed by reprotonation, brought about a retro W-M migration to yield 205, thus placing the molybdenum adjacent to the carbocation at C(2) as in Scheme 80. The structure was verified crystallographically (Figure 9) and revealed that the Mo-C(+) interaction was now directed to the exo face of the fenchyl framework, contrary to the previously observed endo interactions seen in the cationic bornyl clusters. The absolute configuration of the chiral cluster moiety in 205 can be designated as R relative to a dummy atom positioned within the tetrahedron (Figure 10) [112]; CIP rules give the order of precedence as Mo > Co > C-fenchyl > C-methyl. Evidently, the differing abilities of the molybdenum and cobalt vertices to alleviate the positive charge on carbon and stabilise the structure ultimately control the direction of the Wagner–Meerwein rearrangements [111].
In related work on other chiral monoterpenoids, we should mention verbenone that possesses a pinene skeleton and is found in a variety of plants, especially rosemary. It has a pleasant odour and is widely used in perfumery, aromatherapy, herbal teas and spices and herbal remedies. The ketone itself plays an important role in the control of the southern Bark Beetle, and cis and trans verbenols are pheromones for Ips paraconfusus, another species of bark beetle. Alkynyl anion attack on verbenone occurs exclusively on the face opposite to the gem-dimethyl bridge to form the alkynol, 206, shown in Figure 11 [113]. The addition of Co2(CO)8 or [CpMo(CO)3]2 delivers the alkyne-dimetallic tetrahedral clusters, 207, and further reaction in the former with bis(diphenylphosphino)methane (dppm) yields the complex 208 that exhibits two 31P NMR signals arising from the diastereotopic character of the phosphorus nuclei (Scheme 81).
2-Ethynyl-verbenol also reacts with the indenyl reagent, (η5-C9H7)(PPh3)2RuCl, leading ultimately to the allenylidene cationic complex, 209 (Scheme 82), that is also susceptible to nucleophilic attack [114], somewhat analogous to the reactions of cobalt-stabilised carbocations.
In contrast to the earlier cases, alkynyl anion attack on menthone yields a mixture of alkynyl-menthols, 210 and 211, in which the predominant isomer is the latter, whereby the alkynyl substituent occupies the equatorial site. The original attribution of these isomers was based on NMR data of their derived allenyl-phosphine oxides [115], and the X-ray crystallographic structure determinations of their phenylethynyl-Co2(CO)6 tetrahedral clusters, 212 and 213, respectively, validated these assignments [116]. As anticipated, protonation of these alkynol clusters yielded cationic complexes but, since they were not amenable to X-ray structural analysis, they were also treated with iron pentacarbonyl in acetone, a reaction previously shown to yield neutral mixed-metal Fe-Co complexes [117] in which Fe(CO)3 replaced the isoelectronic, and isolobal, [Co(CO)3]+ fragment. As depicted in Scheme 83, the structures of the neutral Fe-Co clusters, 214 and 215 (established X-ray crystallographically), provide excellent models for the cobalt-stabilised menthyl carbocations analogous to those found in their bornyl and fenchyl counterparts.
Finally, we note the preparation of tetrahedral clusters 216 and 217, arising from the reaction of Co2(CO)8 with propargyl geranyl ether, or the alkynol derived from geranyl acetone; these complexes may have potential for Pauson–Khand cyclisation, or a Nicholas reaction (Scheme 84).
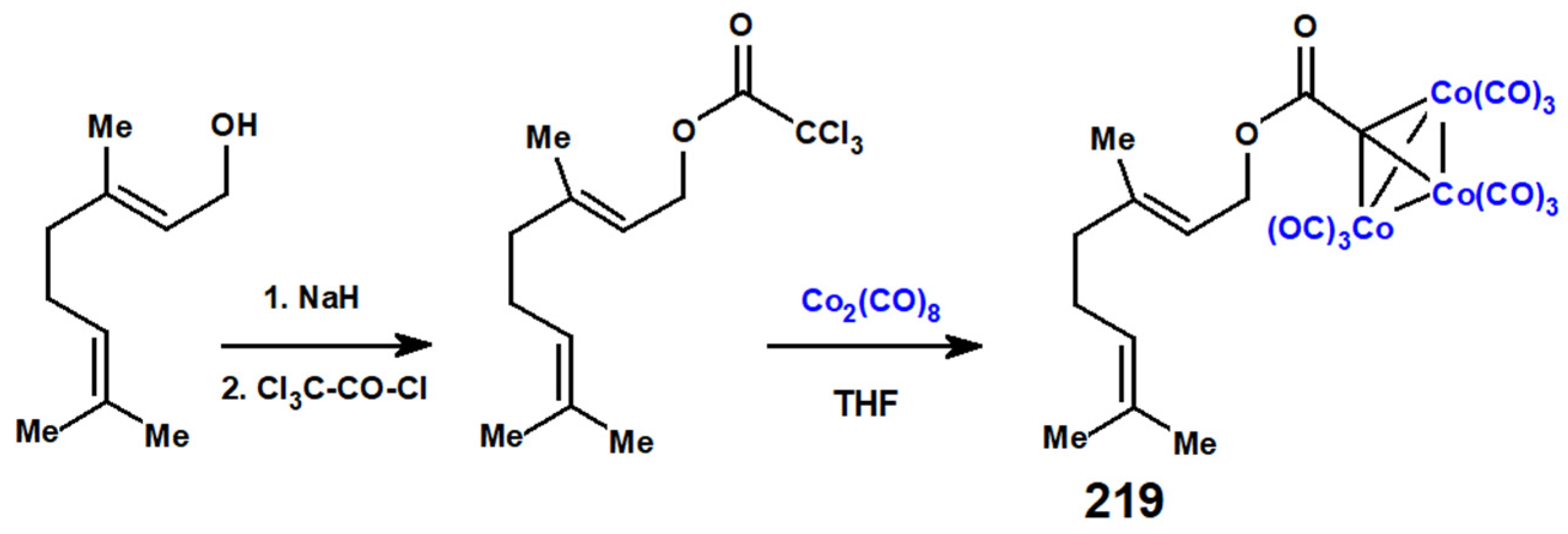
The syntheses of carbynyl-tricobalt-nonacarbonyl clusters, RCCo3(CO)9, are readily achieved either by direct reaction of Co2(CO)8 with the appropriate trichloromethyl precursor (Scheme 85), or by nucleophilic attack on the acylium cation, 218, derived from an ester (Scheme 86). Both of these approaches have been used, thereby leading successfully to tricobalt-nonacarbonyl clusters bearing geranyl, 219, or farnesyl, 220, substituents [118].
14. Concluding Remarks
The evolution of the organometallic chemistry of terpenes and terpenoids has been dramatic, from the early (1960s and 1970s) syntheses of mono- and sesqui-terpenes which involved taking advantage of the newly developed carbon–carbon coupling procedures using nickel or iron carbonyls, or a variety of palladium-based reagents, to the more recently reported efficient routes to industrially or medicinally important molecules. These studies have been complemented by the elucidation of mechanistic details, as well as the discovery of unexpected molecular rearrangements brought about by reactions of terpenes with a wide variety of organometallic species.
The initial burst of publications in this area, with 37 in each period of decades, 1966–1975 and 1976–1985, was followed by a period of retrenchment with 21 reports from 1986–2005, and 28 more since then. However, looking to the future and hoping for enlightenment, we have tried to focus on very recent publications that may stimulate further breakthroughs in the area. These include new industrial applications, enhanced methods of structural determination and the re-evaluation of earlier work by using today’s spectroscopic techniques.
In many countries, access to the vitamins A, E and K, as well as important materials such as geraniol, linalool, farnesol and phytol, is limited, owing to the lack of industrially competitive technologies for their syntheses, thus necessitating reliance on imports. The development of simple and efficient catalytic methods for the production of dimers, trimers and tetramers of isoprene as routes to regular acyclic trans-isoprenoids is a growing area, and is the focus of a 2023 review [119].
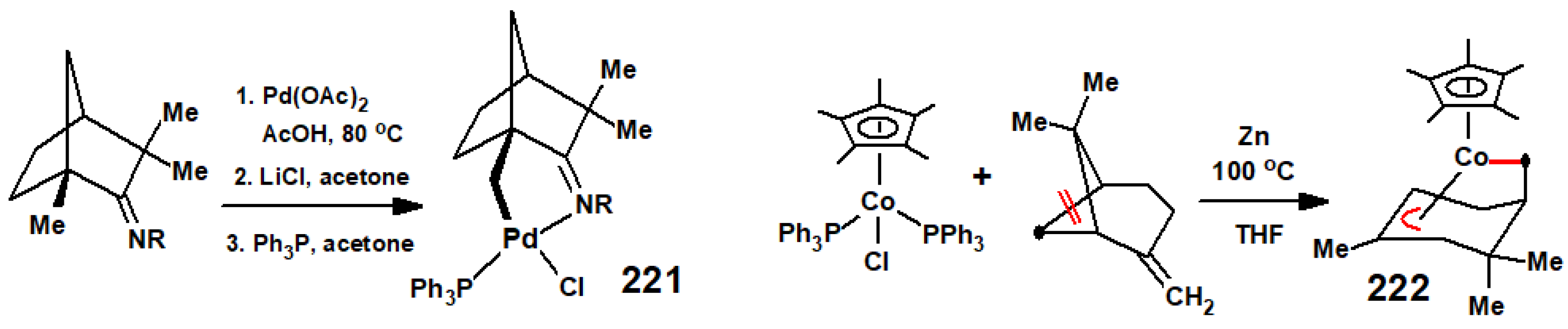
As noted in the introduction, chiral terpenoids have long been incorporated into coordination compounds as part of the search for improved catalysts in asymmetric syntheses, and these have since been modified to possess a metal carbon σ-bond, thereby linking the metal directly to the asymmetric moiety. Typically, readily available and inexpensive (1R)-(-)-fenchone is convertible into an imine that, when cyclopalladated, yields systems such as 221 that can easily be further elaborated (Scheme 87) [120]. A different application of this approach involves the reaction of (C5Me5)Co(PPh3)2 with β-pinene, whereupon bond fission leads to 222 containing both η1- and η3-allyl attachments to cobalt, a structure closely analogous to that of the iron complex 161 shown in Figure 4. However, the goal here is very different; this molecule, and a number of its variants, have been found to be ideal for the low-temperature atomic-layer deposition of cobalt-containing thin films to cover complex 3D structures [121].
One should always respect and admire those pioneers in the chemistry of terpenoids who attempted to determine the structures of complex organic and organometallic molecules without access to the spectroscopic equipment now available. Much of the earlier work is based on 1H NMR data at 60 or 90 MHz, whereas nowadays, access to multi-nuclear, multi-pulse FT instruments operating at 600 MHz and above is routine. In a typical example (Figure 12), the proposed structure, 222, of the sesquiterpene nordine, isolated from the bark or roots of the evergreen shrub Anaxagorea javanica Blume, has since been reassigned as 223, based on the full range of two-dimensional NMR techniques, and then confirmed by X-ray crystallography [122]. We can anticipate that the reinvestigation of contentious structures by taking advantage of the latest analytical techniques will be an ongoing activity.
Perhaps the greatest advances in structural determination techniques have taken place in X-ray crystallography. Of course, there have been enormous technological improvements, such as more powerful rotating anode X-ray sources, more sensitive charge-coupled or area detectors, infinitely better computational facilities, and modern direct method programs, but many common mono- or sesqui-terpenes are low-melting and volatile, characteristics that enhance their role in perfumery. The currently available structures of such materials are frequently based on crystallizable derivatives, such as the humulene complex with silver nitrate [123], or as its di-epoxide (Figure 5) [93]. This difficulty has been largely overcome recently by the application of the crystalline sponge method, whereby the compound of interest is included as a guest into a porous metal organic framework consisting of, for example, ZnI2 and 2,4,6-tri(pyridin-4-yl)-1,3,5-triazine.
An impressive demonstration of the power of this approach is the report by Makoto Fujita at the University of Tokyo, the originator of the crystalline sponge method, describing the identification of a multitude of products arising from the oxidation of α-humulene that yields a number of epoxides, and also aldehydes derived from the single methyls, all of which were structurally characterised [124]. A typical example is shown in Figure 13. Other recently reported structures of low-melting monoterpenes include α- and β-pinene, and camphene, as shown in Figure 14 [125].
In closing, we feature the spectacular report from a Japan–Spain collaboration of the multinuclear binding ability of β-carotene, 224 (Figure 15), to capture as many as ten palladium atoms in a metal-bonded chain sandwich [126]. This exists in both meso (C2h) and racemic (D2) forms, and both have been fully characterised spectroscopically and by X-ray crystallography (Figure 16). Even more impressively, the system can be selectively de-metallated under a CO atmosphere, and palladium atoms can then be replaced by platinum atoms—a quite brilliant achievement!
Funding
This research was funded for many years by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Petroleum Research Fund (PRF), administered by the American Chemical Society, and by Science Foundation Ireland (SFI).
Acknowledgments
The author thanks University College Dublin and the UCD School of Chemistry for additional financial support, and the Centre for Synthesis and Chemical Biology (CSCB) for the use of analytical facilities, and also the Cambridge Crystallographic Data Centre (CCDC) for their efficiency in locating difficult to obtain X-ray structures.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Mann, J. Secondary Metabolism, 2nd ed.; Oxford University Press: Oxford, UK, 1987; Chapter 7; pp. 303–328. [Google Scholar]
- Stolle, A.; Ondruschka, B.; Hopf, H. Thermal rearrangements of monoterpenes and monoterpenoids. Helv. Chim. Acta 2009, 92, 1673–1719. [Google Scholar] [CrossRef]
- Willett, J.D.; Sharpless, K.B.; Lord, K.E.; Van Tamelen, E.E.; Clayton, R.B. Squalene-2,3-oxide an intermediate in enzymatic conversion of squalene to lanosterol and cholesterol. J. Biol. Chem. 1967, 242, 4182–4191. [Google Scholar] [CrossRef] [PubMed]
- Zalevskaya, O.A.; Gur’eva, Y.A.; Kutchin, A.V. Terpene ligands in the coordination chemistry: Synthesis of metal complexes, stereochemistry, catalytic properties and biological activity. Russ. Chem. Rev. 2019, 88, 979–1012. [Google Scholar] [CrossRef]
- Saucy, G.; Marbet, R.; Lindlar, H.; Isler, O. Über eine neue Synthese von Citral und verwandten Verbindungen. Helv. Chim. Acta 1959, 42, 1945–1955. [Google Scholar] [CrossRef]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods utilizing first-row transition metals in natural product total synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef] [PubMed]
- I.G. Farbenindustrie Aktiengesellschaft. Procédé de condensation d’hydrocarbures halogénés. Belgian Patent 448,884. February 27 1943. Chem. Abstr. 1947, 41, 6576. [Google Scholar]
- Webb, I.D.; Borcherdt, G.T. Coupling of allylic halides by nickel carbonyl. J. Am. Chem. Soc. 1951, 73, 2654–2655. [Google Scholar] [CrossRef]
- Corey, E.J.; Hamanaka, E. A new synthetic approach to medium-size carbocyclic systems. J. Am. Chem. Soc. 1964, 85, 1641–1642. [Google Scholar] [CrossRef]
- Corey, E.J.; Semmelhack, M.F. Organonickel compounds as reagents for selective carbon-carbon bond formation between unlike groups. J. Am. Chem. Soc. 1967, 89, 2755–2757. [Google Scholar] [CrossRef]
- Corey, E.J.; Watt, E.W.K. The synthesis of large-ring 1,5-dienes by cyclization of allylic dibromides with nickel carbonyl. J. Am. Chem. Soc. 1967, 89, 2757–2758. [Google Scholar] [CrossRef]
- Corey, E.J.; Hamanaka, E. Total synthesis of humulene. J. Am. Chem. Soc. 1967, 89, 2758–2759. [Google Scholar] [CrossRef]
- Vig, O.P.; Ram, B.; Atwal, K.S.; Bari, S.S. Terpenoids 125. New synthesis of humulene (2,6,6,9-tetramethylcycloundeca-1,4,8-triene). Indian J. Chem. 1976, 14, 855–857. [Google Scholar] [CrossRef]
- Dauben, W.G.; Beasley, G.H.; Broadhurst Muller, B.; Peppard, D.J.; Pesnelle, P.; Suter, C. A synthesis of cembrene. A 14-membered ring diterpene. J. Am. Chem. Soc. 1974, 96, 4724–4726. [Google Scholar] [CrossRef]
- Drew, M.G.B.; Templeton, D.H.; Zalkin, A. The crystal and molecular structure of cembrene. Acta Cryst. 1969, B25, 261–267. [Google Scholar] [CrossRef]
- Sato, K.; Inoue, S.; Ota, S.; Fujita, Y. Reactions of π-allylic nickel(II) bromide with organic halides. A novel synthesis of monoterpenoid compounds. J. Org. Chem. 1972, 37, 462–466. [Google Scholar] [CrossRef]
- Wilke, G. Contributions to organo-nickel chemistry. Angew. Chem. Int. Ed. 1988, 27, 185–206. [Google Scholar] [CrossRef]
- Billups, W.E.; Cross, J.H.; Smith, C.V. Synthesis of (+/−)-grandisol. J. Am. Chem. Soc. 1973, 95, 3438–3439. [Google Scholar] [CrossRef]
- Baker, R.; Blackett, B.N.; Cookson, R.C. Reaction of dodecatrienylnickel; with allene—Formation of muscone. J. Chem. Soc. Chem. Commun. 1972, 802–803. [Google Scholar] [CrossRef]
- Baker, R.; Cookson, R.C.; Vinson, J.R. Insertion of isocyanides into bis-pi-allylnickel complexes and formation of (+/−)-muscone. J. Chem. Soc. Chem. Commun. 1974, 515–516. [Google Scholar] [CrossRef]
- Momose, D.-I.; Iguchi, K.; Sugiyama, T.; Yamada, Y. Reductive coupling of allylic halides by chlorotris(triphenylphosphine)cobalt(I). Tetrahedron Lett. 1983, 24, 921–924. [Google Scholar] [CrossRef]
- Hegedus, L.S.; Perry, R.J. Phosphinecarbonylnitrosylacylcobaltate complexes as acyl transfer reagents. Acylation of allylic halides, conjugated enones, and quinones. J. Org. Chem. 1985, 50, 4960–4995. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Yokoyama, K.; Noyori, R. Carbon-carbon bond formation promoted by transition metal carbonyls. 20. Iron carbonyl promoted reaction of α,α′-dibromo ketones and aromatic olefins leading to 3-arylcyclopentanones. The [3+2) cycloaddition involving an allylic cation. J. Am. Chem. Soc. 1978, 100, 1791–1799. [Google Scholar] [CrossRef]
- Noyori, R.; Hayakawa, Y.; Takaya, H.; Murai, S.; Kobayashi, R.; Sonoda, N. Reaction of α,α′-dibromo ketones and iron carbonyls. Mechanistic aspects. J. Am. Chem. Soc. 1978, 100, 1759–1765. [Google Scholar] [CrossRef]
- Noyori, R.; Yokoyama, K.; Hayakawa, Y. Reaction of α,α′-dibromo ketones and aromatic olefins promoted by iron carbonyl. A cationic 3 + 2 → 5 cycloaddition. J. Am. Chem. Soc. 1973, 95, 2722–2723. [Google Scholar] [CrossRef]
- Noyori, R.; Hayakawa, Y. Natural product syntheses via the polybromo ketone-iron carbonyl reaction. Tetrahedron 1985, 41, 5879–5886. [Google Scholar] [CrossRef]
- Celebuski, J.; Rosenblum, M. Carbon-carbon bond formation employing organoiron reagents: Syntheses of lavandulol and red scale pheromone. Tetrahedron 1985, 41, 5741–5746. [Google Scholar] [CrossRef]
- Heck, R.F. The allylation of aromatic compounds with organopalladium salts. J. Am. Chem. Soc. 1968, 90, 5531–5534. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part III. Dimerisation by means of tetrakis(triphenylphosphine)palladium. J. Chem. Soc. C 1970, 2203–2206. [Google Scholar] [CrossRef]
- Neilan, J.P.; Laine, R.M.; Cortese, N.; Heck, R.F. Monoterpene syntheses via a palladium catalyzed isoprene dimerization. J. Org. Chem. 1976, 41, 3455–3460. [Google Scholar] [CrossRef]
- Patel, B.A.; Kao, L.-C.; Cortese, N.A.; Minkiewicz, J.V.; Heck, R.F. Palladium-catalyzed vinylation of conjugated dienes. J. Org. Chem. 1979, 44, 918–921. [Google Scholar] [CrossRef]
- Tsuji, J. Carbon-carbon bond formation via palladium complexes. Acc. Chem. Res. 1969, 2, 144–152. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L. New synthetic reactions. Stereochemistry of allylic alkylation. J. Am. Chem. Soc. 1975, 97, 1611–1612. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L.; Strege, P.; Fullerton, T.J.; Dietsche, T.J. Allylic alkylation: Nucleophilic attack on π-allylpalladium complexes. J. Am. Chem. Soc. 1978, 100, 3416–3426. [Google Scholar] [CrossRef]
- Ronson, T.O.; Taylor, R.J.K.; Fairlamb, I.J.S. Palladium-catalysed macrocyclisations in the total synthesis of natural products. Tetrahedron 2015, 71, 989–1009. [Google Scholar] [CrossRef]
- Trost, B.M. Cyclization via palladium-catalyzed allylic alkylations. Angew. Chem. Int. Ed. 1989, 28, 1173–1192. [Google Scholar] [CrossRef]
- Trost, B.M.; Min, C. Total synthesis of terpenes via palladium-catalysed cyclisation strategy. Nature Chem. 2020, 12, 568–573. [Google Scholar] [CrossRef]
- Trost, B.M.; Weber, L.; Strege, P.; Fullerton, T.J.; Dietsche, T.J. Allylic alkylation: Nature of the nucleophile and application to prenylation. J. Am. Chem. Soc. 1978, 100, 3426–3435. [Google Scholar] [CrossRef]
- Trost, B.M.; Malhotra, S.; Chan, W.H. Exercising regiocontrol in palladium-catalyzed asymmetric prenylations and geranylation: Unifying strategy toward flustramines A and B. J. Am. Chem. Soc. 2011, 133, 7328–7331. [Google Scholar] [CrossRef] [PubMed]
- Goliaszewski, A.; Schwartz, J. Specific allylic-allylic coupling procedures effected by ligand-induced elimination from di(allylic)palladium species. Tetrahedron 1985, 41, 5779–5789. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Itoh, A.; Hashimoto, S.; Yamamoto, H.; Nozaki, H. Total synthesis of humulene. A stereoselective approach. J. Am. Chem. Soc. 1977, 99, 3864–3867. [Google Scholar] [CrossRef]
- Miyaura, N.; Suginome, H.; Suzuki, A. New stereo- and regiospecific synthesis of humulene by means of the palladium-catalyzed cyclization of haloalkenylboranes. Tetrahedron Lett. 1984, 25, 761–764. [Google Scholar] [CrossRef]
- Hu, T.; Corey, E.J. Short syntheses of (±)-δ-araneosene and humulene utilizing a combination of four-component assembly and palladium-mediated cyclization. Org. Lett. 2002, 4, 2441–2443. [Google Scholar] [CrossRef]
- Han, Y.T.; Kim, N.-J.; Jung, J.-W.; Yun, H.; Lee, S.; Suh, Y.-G. A versatile synthetic approach to grandisol monoterpene pheromone. Arch. Pharm. Res. 2011, 34, 1437–1442. [Google Scholar] [CrossRef]
- Manchand, P.S.; Wong, H.S.; Blount, J.F. Synthesis of vitamin A and related compounds via a π-allyl palladium complex. J. Org. Chem. 1978, 43, 4669–4774. [Google Scholar] [CrossRef]
- Negishi, E.; Valente, L.F.; Kobayashi, M. Palladium-catalyzed cross-coupling reaction of homoallylic or homopropargylic organozincs with alkenyl halides as a new selective route to 1,5-dienes and 1,5-enynes. J. Am. Chem. Soc. 1980, 102, 3298–3299. [Google Scholar] [CrossRef]
- Sum, F.W.; Weiler, L. Synthesis of mokupalide. J. Am. Chem. Soc. 1979, 101, 4401–4403. [Google Scholar] [CrossRef]
- Hart, D.W.; Blackburn, T.F.; Schwartz, J. Hydrozirconation. III Stereospecific and regioselective functionalization of alkylacetylenes via vinylzirconium(IV) intermediates. J. Am. Chem. Soc. 1975, 97, 679–680. [Google Scholar] [CrossRef]
- McMurry, J.E. Titanium-induced dicarbonyl-coupling reactions. Acc. Chem. Res. 1983, 16, 405–411. [Google Scholar] [CrossRef]
- McMurry, J.E.; Matz, J.R. Stereospecific synthesis of humulene by titanium-induced dicarbonyl coupling. Tetrahedron Lett. 1982, 23, 2723–2724. [Google Scholar] [CrossRef]
- McMurry, J.E.; Matz, J.R.; Kees, K.L.; Bock, P.A. Synthesis of flexibilene, a naturally-occurring 15-membered-ring diterpene. Tetrahedron Lett. 1982, 23, 1777–1780. [Google Scholar] [CrossRef]
- Kamat, V.P.; Hagiwara, H.; Katsumi, T.; Susuki, T.; Ando, M. Ring closing metathesis directed synthesis of (R)-(−)-muscone from (+)-citronellal. Tetrahedron 2000, 56, 4397–4403. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Quilez del Moral, J.F.; Arteaga, P.; Arteaga, J.F.; Diéguez, H.R.; Sánchez, E.M. Mild Ti(III)- and Mn/Zr(IV)-catalytic reductive coupling of allylic halides: Efficient synthesis of symmetric terpenes. J. Org. Chem. 2007, 72, 2988–2995. [Google Scholar] [CrossRef]
- Fürstner, A.; Hannen, P. Carene terpenoids by gold-catalyzed cycloisomerization reactions. Chem. Commun. 2004, 2546–2547. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part I. The reactions of cis-ocimene and trans-ocimene and of myrcene with palladium(II). J. Chem. Soc. C 1970, 2196–2200. [Google Scholar] [CrossRef]
- Takahashi, M.; Suzuki, H.; Moro-Oka, Y.; Ikawa, T. Regioselective hydroxylation of β-myrcene using palladium(II) complexes. Chem. Lett. 1979, 8, 53–56. [Google Scholar] [CrossRef]
- Takahashi, M.; Urata, H.; Suzuki, H.; Moro-Oka, Y.; Ikawa, T. Regioselective introduction of O-nucleophiles into β-myrcene and trans-ocimene using palladium(II) complexes. J. Organomet. Chem. 1984, 266, 327–336. [Google Scholar] [CrossRef]
- Dunne, K.; McQuillin, F.J. Complexes of terpenes with transition metals. Part II. Reactions of linalyl and nerolidyl acetates, limonene and the pinenes with disodium tetrachloropalladate(II). J. Chem. Soc. C 1970, 2200–2203. [Google Scholar] [CrossRef]
- McQuillin, F.J.; Parker, D.G. Complexing of terpenes with transition metals. Part IV. A comparison of the reactions of (+)-3,7-dimethylocta-1,6-diene with palladium(II) and mercury(II). J. Chem. Soc. Perkin Trans. 1 1974, 809–815. [Google Scholar] [CrossRef]
- Rienacker, R.; Ohloff, G. Optisch aktives β-Citronellol aus (+) or (−) pinane. Angew. Chemie 1961, 73, 240. [Google Scholar] [CrossRef]
- Strickler, H.; Ohloff, G.; Kováts, E. The thermal cyclisation of (−)-(R)-linalool. The structure of the plinoles and some derivates with an iridan framework. Helv. Chim. Acta 1967, 50, 759–797. [Google Scholar] [CrossRef]
- Matsuki, Y.; Kodama, M.; Ito, S. Regioselective and stereoselective cyclization of linalool and nerolidol with mercuric salts. Synthesis of iridanols and cyclonerodiol. Tetrahedron Lett. 1979, 20, 2901–2904. [Google Scholar] [CrossRef]
- Cane, D.; Shiao, M.-S. Biosynthesis of cyclonerodiol. J. Am. Chem. Soc. 1978, 100, 3203–3207. [Google Scholar] [CrossRef]
- Pearson, A.J. Protonation of tricarbonyliron complexes: Acid-catalyzed cyclization of tricarbonylmyrceneiron. Aust. J. Chem. 1976, 29, 1841–1844. [Google Scholar] [CrossRef]
- Birch, A.J.; Pearson, A.J. Friedel-Crafts chemistry of tricarbonyldieneiron complexes: Carbonylative annulation of tricarbonylmyrceneiron. J. Chem. Soc. Chem. Commun. 1976, 601–602. [Google Scholar] [CrossRef]
- Banthorpe, D.V.; Fitton, H.; Lewis, J. Isomerisation and addition reactions of some monoterpene-tricarbonyliron complexes. J. Chem. Soc. Perkin Trans. 1 1973, 2051–2057. [Google Scholar] [CrossRef]
- Taylor, G.A. Dichlorocarbene addition to tricarbonyliron complexes of polyenes. J. Chem. Soc. Perkin I 1979, 1716–1719. [Google Scholar] [CrossRef]
- Hubert, A.J.; Georis, A.; Warin, R.; Tessié, P. Base-catalysed prototropic rearrangement. Part 1. Comparison of the base-catalysed and the metal carbonyl-catalysed isomerisation of allyl ethers. J. Chem. Soc. Perkin II 1972, 366–370. [Google Scholar] [CrossRef]
- Spanninger, P.A.; von Rosenberg, J.L. Isomerization of (−)-β-pinene to high optical purity (−)-α-pinene. J. Org. Chem. 1969, 34, 3658–3659. [Google Scholar] [CrossRef]
- Hendrix, W.Y.; Cowherd, F.G.; von Rosenberg, J.L. Mechanism of the rearrangement of allyl alcohol with iron carbonyl: Evidence for a π-allyl-hydroirontricarbonyl complex. Chem. Commun. 1968, 97–99. [Google Scholar] [CrossRef]
- Cowherd, F.G.; von Rosenberg, J.L. Mechanism of iron pentacarbonyl-catalyzed 1,3-hydrogen shifts. J. Am. Chem. Soc. 1969, 91, 2157–2158. [Google Scholar] [CrossRef]
- Tulip, T.H.; Ibers, J.A. η3-Allyl metal hydride complexes. Oxidative addition of cyclopropane and olefin substrates to iridium(I) complexes. Structure of IrClH[η3-C3H4(1-C6H5)][P(C6H5)3]2. J. Am. Chem. Soc. 1979, 101, 4201–4211. [Google Scholar] [CrossRef]
- Grieco, P.A.; Takigawa, T.; Bongers, S.L.; Tanaka, H. Complete transfer of chirality in the [3.3]-sigmatropic rearrangement of allylic acetates catalyzed by palladium(II). Application to stereocontrolled syntheses of prostaglandins possessing either the C-15(S) or C-15(R) configuration. J. Am. Chem. Soc. 1980, 102, 7588–7590. [Google Scholar] [CrossRef]
- Trebellas, J.C.; Olechowski, J.R.; Jonassen, H.B. Metal olefin complexes: II. Palladium(II) and platinum(II) complexes of 1,2-divinylcyclohexane by the metal- catalyzed isomerization of 1,5-cyclodecadiene. J. Organomet. Chem. 1966, 6, 412–420. [Google Scholar] [CrossRef]
- Heimbach, P.; Molin, M. PdCl2-induced rearrangement of substituted cis,trans-cyclodeca-1,5-diene. J. Organomet. Chem. 1973, 49, 477–482. [Google Scholar] [CrossRef]
- Overman, L.E. Mercury(II) and palladium(II)-catalyzed [3,3]-sigmatropic rearrangements. Angew. Chem. Int. Ed. Engl. 1984, 23, 579–598. [Google Scholar] [CrossRef]
- Overman, L.E.; Renaldo, A.F. Palladium dichloride catalyzed Cope rearrangements of functionalized acyclic 1,5-dienes. Tetrahedron Lett. 1983, 24, 3757–3760. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Gomez-Bengoa, E.; Mendez, M.; Echavarren, A.M. Synthesis of (±)-10-epi-elemol by a highly stereoselective intramolecular palladium-catalyzed coupling of an allylstannane with an allyl acetate. J. Org. Chem. 1997, 62, 7540–7541. [Google Scholar] [CrossRef]
- Brown, E.D.; Sam, T.W.; Sutherland, J.K.; Torre, A. Medium-ring 1,5-dienes. Part II. The radical and electrophile-induced cyclisation of germacra-1(10),4,7(11)-triene. J. Chem. Soc. Perkin Trans. 1 1975, 2326–2332. [Google Scholar] [CrossRef]
- Miyashita, A.; Takahashi, M.; Takaya, H. Reaction of bicyclo[1.1.0]butanes with platinum(II) complexes. Isolation and characterization of new platinacycle compounds. J. Am. Chem. Soc. 1981, 103, 5257–6259. [Google Scholar] [CrossRef]
- Victor, R.; Ben-Shoshan, R.; Sarel, S. Photochemically induced 1,5-insertion of carbon monoxide into vinylcyclopropane systems. A novel synthesis of cyclohexenones mediated by iron carbonyl. Tetrahedron Lett. 1970, 11, 4253–4256. [Google Scholar] [CrossRef]
- Aumann, R. Iron carbonyl complexes from vinylcyclopropane. J. Am. Chem. Soc. 1974, 96, 2631–2632. [Google Scholar] [CrossRef]
- Santelli-Rouvier, C.; Santelli, M.; Zahra, J.P. Carbonylation of (+)-2-carene induced by iron pentacarbonyl. Tetrahedron Lett. 1985, 26, 1213–1216. [Google Scholar] [CrossRef]
- Stockis, A.; Weissberger, E. Metal assisted ring expansions. The stereospecific expansion of pinene induced by Fe(CO)5. J. Am. Chem. Soc. 1975, 97, 4288–4292. [Google Scholar] [CrossRef]
- Jenny, T.A.; Ma, L. Synthetic use of a σ-alkyl-π-allyl iron complex obtained by stereospecific ring opening of α-pinene. Tetrahedron Lett. 1991, 32, 6101–6104. [Google Scholar] [CrossRef]
- Jenny, T.A.; Huber, V.; Ma, L.; Raemy, J.; Zeller, D.; Stoeckli-Evans, H. Synthesis, X-ray structure, and reactivity of phosphine-substituted iron carbonyl complexes containing σ-alkyl—π-allyl ligands. Organometallics 2002, 21, 2504–2513. [Google Scholar] [CrossRef]
- De Vekki, D.A.; Uvarov, V.I.; Bel’skii, V.K.; Skvortsov, N.K. Reaction of platinum complexes with (+)-α-pinene and (+)-limonene. Synthesis, molecular structure, and catalytic activity of dichloro(η4-[p-mentha-1,8{9}-diene])platinum(II). Russ. J. Gen. Chem. 2006, 76, 1288–1294. [Google Scholar] [CrossRef]
- Costes, P.; Weckesser, J.; Dechy-Cabaret, O.; Urrutigoïty, M.; Kalck, P. Rh-and Pt-catalyzed cycloisomerization of enynes derived from terpenes. Appl. Organomet. Chem. 2008, 22, 211–214. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Michaelson, R.C. High stereoselectivities in the transition metal catalyzed epoxidations of olefinic alcohols by tert-butyl hydroperoxide. J. Am. Chem. Soc. 1973, 95, 6136–6137. [Google Scholar] [CrossRef]
- Pereira, C.; Silva, J.F.; Pereira, A.M.; Araújo, J.P.; Blanco, G.; Pintado, J.M.; Freire, C. [VO(acac)2] hybrid catalyst: From complex immobilization onto silica nanoparticles to catalytic application in the epoxidation of geraniol. Catal. Sci. Technol. 2011, 1, 784–793. [Google Scholar] [CrossRef]
- Yur′ev, V.P.; Gailyunas, I.A.; Isaeva, Z.G.; Tolstikov, G.A. Stereoselective epoxidation of bicyclic monoterpenes with tert-amyl hydroperoxide in presence of molybdenum compounds. Bull Acad. Sci. USSR 1974, 23, 885–886. [Google Scholar] [CrossRef]
- Curlat, S. Recent studies of (+)-3-carene transformations with the retention of the native framework. Chem. J. Mold. 2019, 14, 32–55. [Google Scholar] [CrossRef]
- Cradwick, M.E.; Cradwick, P.D.; Sim, G.A. Sesquiterpenoids. Part XV. Conformation of humulene: X-ray analysis of the crystal structure of humulene diepoxide. J. Chem. Soc. Perkin Trans. 2 1973, 4040407. [Google Scholar] [CrossRef]
- Sattar, A.; Forrester, J.; Moir, M.; Roberts, J.S.; Parker, W. Regiospecific functionalisation of humulene. Tetrahedron Lett. 1976, 17, 1403–1406. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Umbreit, M.A.; Nieh, M.T.; Flood, T.C. Low valent tungsten halides. A new class of reagents for deoxygenation of organic molecules. J. Am. Chem. Soc. 1972, 94, 6538–6540. [Google Scholar] [CrossRef]
- Mlokiewicz, J.A. Chemistry of Humulene Epoxides. Ph.D. Thesis, University of Stirling, Scotland, UK, 1979; p. 101. [Google Scholar]
- Churchill, M.R. Transition metal complexes of azulene and related ligands. Prog. Inorg. Chem. 1970, 11, 53–98. [Google Scholar] [CrossRef]
- Cotton, F.A.; Hanson, B.E.; Kolb, J.R.; Lahuerta, P.; Stanley, G.G.; Stults, B.R.; White, A.W. The carbonyl scrambling processes in the isomeric pentacarbonylguaiazuleneiron and homologous ruthenium molecules; a novel mechanism for the internuclear processes. J. Am. Chem. Soc. 1977, 99, 3673–3683. [Google Scholar] [CrossRef]
- Nagashima, H.; Fukahori, T.; Nobata, M.; Suzuki, A.; Nakazawa, M.; Itoh, K. Haptotropic rearrangement between two isomers of (μ2:η3:η5-guaiazulene)Fe2(CO)5 revisited: Both thermal and photochemical processes induce haptotropic interconversion. Organometallics 1994, 13, 3427–3433. [Google Scholar] [CrossRef]
- Matsubara, K.; Oda, T.; Nagashima, H. Diruthenium carbonyl complexes bound to guaiazulene: Preparation and thermally reversible photoisomerisation studies of phosphine and phosphite derivatives of (μ2:η3:η5-guaiazulene)Ru2(CO)5 and iron homologues. Organometallics 2001, 20, 881–892. [Google Scholar] [CrossRef]
- Vollgraff, T.; Doppiu, A.; Sundermeyer, J. Dihydroguaiazulenide complexes and catalysts of Group 8-12 transition metals: Ligands from renewable feedstock replace, even outmatch petrochemical bases cyclopentadienyl chemistry. Chem. Eur. J. 2023, 30, e202302994. [Google Scholar] [CrossRef]
- Balduzzi, S.; Müller-Bunz, H.; McGlinchey, M.J. A convenient synthetic route to benz[cd]azulenes: Versatile ligands with the potential to bind to metal in an η5, η6, or η7 fashion. Chem. Eur. J. 2004, 10, 5398–5405. [Google Scholar] [CrossRef]
- Teobald, B.J. The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis. Tetrahedron 2002, 58, 4133–4170. [Google Scholar] [CrossRef]
- Tyrrell, E.; Millet, J.; Tesfa, K.H.; Williams, N.; Mann, A.; Tillett, C.; Muller, C. A study into asymmetric Nicholas cyclisation reactions. Tetrahedron 2007, 63, 12769–122778. [Google Scholar] [CrossRef]
- Marshall, J.A.; Gung, B.W. Stereoselective cyclization of alpha-alkoxy allylstannane alkynals and their Co-complexes—A new route to cyclododecyne-1,2-diol derivatives. Tetrahedron Lett. 1989, 30, 309–312. [Google Scholar] [CrossRef]
- Morris, D.G.; Shepherd, A.G.; Walker, M.F.; Jemison, R.W. Anisochrony induced by transmission across a triple bond. Aust. J. Chem. 1982, 35, 1061–1064. [Google Scholar] [CrossRef]
- D’Agostino, M.F.; Frampton, C.S.; McGlinchey, M.J. Diastereoselective ligand and vertex substitutions in bimetallic bridged alkyne clusters: X-ray crystal structure of (µ2-endo-2-propynylborneol)hexacarbonyldicobalt. Organometallics 1990, 9, 2972–2984. [Google Scholar] [CrossRef]
- D’Agostino, M.F.; Frampton, C.S.; McGlinchey, M.J. An NMR spectroscopic and EHMO investigation of cationic mixed metal clusters: X-ray crystal structure of (1,7,7-trimethyl-µ2-2-propynylnorbornadiene)-bis(cyclopentadienyl)tetracarbonyldimolybdenum. J. Organomet. Chem. 1990, 394, 145–166. [Google Scholar] [CrossRef]
- Gruselle, M.; El Hafa, H.; Nikolski, M.; Jaouen, G.; Vaissermann, J.; Li, L.; McGlinchey, M.J. Metal cluster stabilized 2-norbornyl cations: A synthetic X-ray crystallographic and EHMO study. Organometallics 1993, 12, 4917–4925. [Google Scholar] [CrossRef]
- Gruselle, M. 2-Bornyl cations stabilized by metal-clusters—Synthetic and X-ray structural and quantum-chemical studies. Russ. Chem. Bull. 1994, 43, 538–542. [Google Scholar] [CrossRef]
- Kondratenko, M.; El Hafa, H.; Gruselle, M.; Vaissermann, J.; Jaouen, G.; McGlinchey, M.J. A synthetic and X-ray crystallographic study of cobalt and molybdenum complexes of the 2-fenchyl cation: Metal-mediated Wagner-Meerwein rearrangements. J. Am. Chem. Soc. 1995, 117, 6907–6913. [Google Scholar] [CrossRef]
- El Hafa, H.; Cordier, C.; Gruselle, M.; Besace, Y.; Jaouen, G.; McGlinchey, M.J. NMR study of the dynamic behavior of [2-propynylbornyl]Mo2(CO)4Cp2]+BF4−: Nonfluxional molybdenum-cobalt clusters as the key to understanding the mechanism of the formation of metal-stabilized cations. Organometallics 1994, 13, 5149–5156. [Google Scholar] [CrossRef]
- Moore, A.; Ostermann, J.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Metal-stabilized carbocations derived from monoterpenes: Dicobalt hexacarbonyl complexes of alkynyl-verbenols. Tetrahedron 2016, 72, 4186–4193. [Google Scholar] [CrossRef]
- Cadierno, V.; Conejero, S.; Gamasa, M.P.; Gimeno, J. Indenyl-ruthenium(II) allenylidene complexes containing terpenic substituents as precursors of optically active terminal alkynes: Scope and limitations. Dalton Trans. 2003, 3060–3066. [Google Scholar] [CrossRef]
- Phillipe, J.; Capmau, M.-L.; Chodkiewicz, W. Stereochemistry of addition of organometallic derivatives to terpenoid ketones. Bull. Soc. Chim. Fr. 1971, 2248–2255. [Google Scholar]
- Moore, A.; Ortin, Y.; Müller-Bunz, H.; McGlinchey, M.J. Alkynyl-dicobalt hexacarbonyl complexes of menthyl cations: Isolobal substitution of [Co(CO)3]+ by Fe(CO)3 as a structural model. Organometallics 2010, 29, 4882–4892. [Google Scholar] [CrossRef]
- Dunn, J.A.; Britten, J.F.; Daran, J.-C.; McGlinchey, M.J. Toward an understanding of the mechanism of metal exchange in (propargyl alcohol)Co2(CO)6 complexes: Syntheses and structures of (η5-C5Ph2R2-C≡C-TMS)(Fe(CO)2(µ-H)Co2(CO)6, R = Ph or Et. Organometallics 2001, 20, 4690–4694. [Google Scholar] [CrossRef]
- Moore, A.; Ostermann, J.; Ortin, Y.; McGlinchey, M.J. Organometallic derivatives of natural products: Dicobalt hexacarbonyl complexes of geranyl-alkynes. New J. Chem. 2016, 40, 7881–7888. [Google Scholar] [CrossRef]
- Dzhemilev, U.M.; Dzhelmileva, L.U.; D′yakonov, V.A. Metallacomplex catalysis in the synthesis of regular isoprenoids. Russ. Chem. Bull. 2023, 72, 404–414. [Google Scholar] [CrossRef]
- Dickmu, G.C.; Smoliakova, I.P. Preparation and characterization of cyclopalladated complexes derived from L-(−)-fenchone. J. Organomet. Chem. 2014, 772–773, 42–48. [Google Scholar] [CrossRef]
- Lu, K.; Zhu, J.; Wang, J.; Wa, Q.; Guo, Z.; Xiong, W.; Gao, Y.; Lei, R.; Cheng, B.; Wang, X. Syntheses of η1-alkyl-η3-allyl-η5-cyclopentadienyl cobalt(III) complexes and their use in low-temperature atomic layer deposition of cobalt-containing thin films. Chem. Eur. J. 2023, 29, e202203656. [Google Scholar] [CrossRef] [PubMed]
- Silva de Andrade, R.; Sales, K.A.; Abreu, L.S.; Campos, V.R.; Martins dos Santos, F., Jr.; Braz-Filho, R.; Scotti, M.T.; Tavares, J.F.; Sobral da Silva, M. Structure revision of the sesquiterpene Nordine based on NMR spectroscopic analysis and X-ray crystallography. J. Nat. Prod. 2020, 85, 2480–2483. [Google Scholar] [CrossRef] [PubMed]
- McPhail, A.T.; Sim, G.A. Sesquiterpenoids. Part IV. The stereochemistry of humulene: X-ray analysis of the humulene-silver nitrate adduct. J. Chem. Soc. B 1966, 112–120. [Google Scholar] [CrossRef]
- Zigon, N.; Hoshino, M.; Yoshioka, S.; Inokuma, Y.; Fujita, M. Where is the oxygen? Structural analysis of α-humulene oxidation products by the crystalline sponge method. Angew. Chem. Int. Ed. 2015, 54, 9033–9037. [Google Scholar] [CrossRef] [PubMed]
- De Poel, W.; Tinnemans, P.T.; Duchateau, A.L.L.; Honing, M.; Rutjes, F.P.J.T.; Vlieg, E.; de Gelder, R. Racemic and Enantiopure Camphene and Pinene Studied by the Crystalline Sponge Method. Cryst. Growth Des. 2018, 18, 126–132. [Google Scholar] [CrossRef]
- Horiuchi, S.; Tachibana, Y.; Yamashita, M.; Yamamoto, K.; Masai, K.; Takase, K.; Matsutani, T.; Kawamata, S.; Kurashige, Y.; Yanai, T.; et al. Multinuclear metal-binding ability of a carotene. Nat. Commun. 2015, 6, 6742. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Biosynthetic route to monoterpenes and sesquiterpenes.

Scheme 2.
Rearrangement of 2,3-squalene epoxide to form lanosterol.

Figure 1.
Examples of coordination compounds containing a terpenoid substituent.

Scheme 3.
Synthesis of citral mediated by silver carbonate.

Scheme 4.
First reported coupling of allylic halides by nickel tetracarbonyl.

Scheme 5.
Intermediates in the coupling of allyl halides with nickel tetracarbonyl.

Scheme 6.
Formation of large-ring 1,5-dienes using Ni(CO)4 as a coupling reagent.

Scheme 7.
Final stages in the synthesis of humulene by Corey and Hamanaka.

Scheme 8.
Ring-closing sequence in Dauben’s route to cembrene.

Figure 2.
Molecular structure of cembrene.

Scheme 9.
Synthesis of geranyl acetate from the allyl-nickel bromide dimer, 14.

Scheme 10.
Final steps in the syntheses of α- and epi-β-santalenes using the allyl-nickel bromide dimer, 14.
Scheme 10.
Final steps in the syntheses of α- and epi-β-santalenes using the allyl-nickel bromide dimer, 14.

Scheme 11.
Nickel-promoted dimerisation and trimerisation of 1,3-butadiene.

Scheme 12.
Billups’s synthesis of (±)-grandisol.

Scheme 13.
Synthesis of (±)-muscone from dodecatrienylnickel.

Scheme 14.
Coupling of geranyl and farnesyl bromides using (Ph3P)3CoCl.

Scheme 15.
Use of (Ph3P)Co(CO)2NO as a replacement for Ni(CO)4.

Scheme 16.
Formation of oxaallyl-Fe(II) intermediates from α,α′-dibromo ketones.

Scheme 17.
Electrocyclisation of the oxaallyl-Fe(II) species to form 1-phenyl-2-indanone.

Scheme 18.
Proposed iron carbonyl-mediated route to camphor.

Scheme 19.
Noyori’s syntheses of (±)-camphor, 41, and (±)-campherenones 44 and 45.

Scheme 20.
Interchange of σ- and π-bonded Fp environments.

Scheme 21.
Synthesis of (±)-lavandulol mediated by the changeable hapticity of the (C5H5)Fe(CO)2, (Fp) group.
Scheme 21.
Synthesis of (±)-lavandulol mediated by the changeable hapticity of the (C5H5)Fe(CO)2, (Fp) group.

Scheme 22.
Synthesis of the (R,S)-California red scale pheromone, 55.

Scheme 23.
Dimerisation of 1-vinylcyclohexyl acetate and nerolidyl acetate by (Ph3P)4Pd.

Scheme 24.
Palladium-catalysed dimerisation of isoprene.

Scheme 25.
Vinylation of 4-methyl-1,3-pentadiene using Heck’s catalytic methodology.

Scheme 26.
Trost’s prenylation route to (E,E)-farnesol: (i) LiI.3H2O, NaCN,DMF, 120 °C; (ii) DIBAL, toluene, −40 °C, then Na2SO4; (iii) Li, BrCH2CH2Br, −78 °C.
Scheme 26.
Trost’s prenylation route to (E,E)-farnesol: (i) LiI.3H2O, NaCN,DMF, 120 °C; (ii) DIBAL, toluene, −40 °C, then Na2SO4; (iii) Li, BrCH2CH2Br, −78 °C.

Scheme 27.
Trost’s prenylation route to (E,E,E)-geranylgeraniol.

Scheme 28.
Geranylation, and linear-versus-branched prenylations.

Scheme 29.
Head-to-head coupling induced by maleic anhydride.

Scheme 30.
Biological route to humulene from farnesyl pyrophosphate.

Scheme 31.
Principal steps in Nozaki’s route to humulene, 10.

Scheme 32.
Suzuki’s route to humulene: (i) ClMg-CMe2-C≡C-SiMe3, (ii) KF, (iii) SeO2, EtOH, (iv) PBr3, Et2O, (v) HB(Sia)2, and (vi) Pd(PPh3)4, NaOH, benzene, reflux.
Scheme 32.
Suzuki’s route to humulene: (i) ClMg-CMe2-C≡C-SiMe3, (ii) KF, (iii) SeO2, EtOH, (iv) PBr3, Et2O, (v) HB(Sia)2, and (vi) Pd(PPh3)4, NaOH, benzene, reflux.

Scheme 33.
Synthesis of humulene by Hu and Corey: (i) Pd2(dba)3, dppf, THF 70 °C, 52% (ii) NaOH, i-PrOH, then MeSO2Cl, Et3N, 94%, and (iii) SiO2, CH2Cl2, 93%.
Scheme 33.
Synthesis of humulene by Hu and Corey: (i) Pd2(dba)3, dppf, THF 70 °C, 52% (ii) NaOH, i-PrOH, then MeSO2Cl, Et3N, 94%, and (iii) SiO2, CH2Cl2, 93%.

Scheme 34.
An allyl-palladium route to grandisol, 23.

Scheme 35.
Allyl-palladium route to vitamin A acetate: (i) PBr3, (ii) PhSO2Na, (iii) NaH, PPh3, (iv) NaOEt, EtOH.
Scheme 35.
Allyl-palladium route to vitamin A acetate: (i) PBr3, (ii) PhSO2Na, (iii) NaH, PPh3, (iv) NaOEt, EtOH.

Scheme 36.
Negishi’s synthetic route to the tetraenol, 90, and its subsequent conversion into mokupalide: (i) Me3Al—Cp2ZrCl2, CH2Cl2, (ii) I2, THF 0 °C, (iii) Me3SiC≡C-(CH2)2-ZnCl, Pd(PPh3)4, THF (iv) KF·2H2O, (v) Me3Al—Cp2ZrCl2, CH2Cl2, (vi) I2, THF 0 °C, (vii) n-BuLi, (CH2O)n.
Scheme 36.
Negishi’s synthetic route to the tetraenol, 90, and its subsequent conversion into mokupalide: (i) Me3Al—Cp2ZrCl2, CH2Cl2, (ii) I2, THF 0 °C, (iii) Me3SiC≡C-(CH2)2-ZnCl, Pd(PPh3)4, THF (iv) KF·2H2O, (v) Me3Al—Cp2ZrCl2, CH2Cl2, (vi) I2, THF 0 °C, (vii) n-BuLi, (CH2O)n.

Scheme 37.
McMurry’s route to humulene.

Scheme 38.
McMurry’s synthesis of flexibilene: (i) Pd(OCOCF3)2, acetone, (ii) Bu4NCl, (iii) HZrCp2Cl, CH2Cl2.
Scheme 38.
McMurry’s synthesis of flexibilene: (i) Pd(OCOCF3)2, acetone, (ii) Bu4NCl, (iii) HZrCp2Cl, CH2Cl2.

Scheme 39.
Final steps in the synthesis of (±)-δ-araneosene: (i) Pd2(dba)3, dppf, THF 70 °C, 48%; (ii) O3, CH2Cl2, Me2S, 54%; (iii) TiCl3, Zn-Cu, DME, 90%.
Scheme 39.
Final steps in the synthesis of (±)-δ-araneosene: (i) Pd2(dba)3, dppf, THF 70 °C, 48%; (ii) O3, CH2Cl2, Me2S, 54%; (iii) TiCl3, Zn-Cu, DME, 90%.

Scheme 40.
Ring-closing metathesis route to (R)-(-)-muscone, 26, from (+)-citronellal: (i) NaBH4, MeOH, (ii) t-BuMe2SiCl, imidazole, DMF, (ii) OsO4, NaIO4, dioxane, (iii) Ph3PMeBr, n-BuLi, THF, −78 °C, (iv) SO2-pyridine, CH2Cl2, DMSO, Et3N, (v) Jones reagent, (vi) Grubbs II catalyst, (vii) H2, Pd-C.
Scheme 40.
Ring-closing metathesis route to (R)-(-)-muscone, 26, from (+)-citronellal: (i) NaBH4, MeOH, (ii) t-BuMe2SiCl, imidazole, DMF, (ii) OsO4, NaIO4, dioxane, (iii) Ph3PMeBr, n-BuLi, THF, −78 °C, (iv) SO2-pyridine, CH2Cl2, DMSO, Et3N, (v) Jones reagent, (vi) Grubbs II catalyst, (vii) H2, Pd-C.

Scheme 41.
Terpene dimerisations induced by Ti(III) or Zr(III) catalysts.

Scheme 42.
Proposed route to carenes via gold(III)-catalysed rearrangement of propargyl acetates.

Scheme 43.
Gold-catalysed synthesis of sesquicarene, 109.

Scheme 44.
Addition of a nucleophile to a diene to form an allyl-palladium chloride dimer.

Scheme 45.
Reactions of ocimene and myrcene with methanol, induced by L2PdCl2.

Scheme 46.
Reactions of limonene or pinene with potassium tetrachloropalladate(II).

Scheme 47.
Reactions of 3,7-dimethylocta-1,6-diene, 116, with palladium or mercury salts: (i) PdCl2-aqueous acetone, (ii) Hg(OAc)2, (iii) H2O-THF-NaBH4.
Scheme 47.
Reactions of 3,7-dimethylocta-1,6-diene, 116, with palladium or mercury salts: (i) PdCl2-aqueous acetone, (ii) Hg(OAc)2, (iii) H2O-THF-NaBH4.

Scheme 48.
Cyclisation of linalool and of nerolidol mediated by a mercury(II) salt.

Scheme 49.
Protonation of (myrcene)Fe(CO)3 followed by reduction with NaBH4.

Scheme 50.
Carbonylative annulation of (myrcene)Fe(CO)3.

Scheme 51.
Reduction in the isopropylidene unit or addition of dichlorocarbene to (myrcene)Fe(CO)3.

Scheme 52.
Proposed rearrangement mechanism of (ocimene)Fe(CO)3.

Scheme 53.
Evidence for a (π-allyl)tricarbonyliron hydride in an isomerisation process.

Scheme 54.
Reactions of exo and endo 1-hydroxydicyclopentadiene with Fe(CO)5.

Figure 3.
Molecular structure of the (π-allyl)iridium hydride, 141; for clarity, only the ipso carbons of the phenyl rings are shown.
Figure 3.
Molecular structure of the (π-allyl)iridium hydride, 141; for clarity, only the ipso carbons of the phenyl rings are shown.

Scheme 55.
1,3-Migrations in linalyl and nerolidyl acetates.

Scheme 56.
Proposed mechanism of a PdCl2-initiated 1,3-acetyl rearrangement.

Scheme 57.
A 1,3-acetate migration with complete transfer of chirality.

Scheme 58.
Examples of Cope rearrangements in terpenoids.

Scheme 59.
Ring-opening of bicyclobutane by a Pt(II) complex.

Scheme 60.
Ring-opening of cyclopropyl substituents by reaction with Fe(CO)5.

Scheme 61.
Ring-opening of the cyclopropyl moiety in (+)-2-carene by Fe(CO)5.

Scheme 62.
Reaction of pinene with Fe(CO)5 to bring about carbonyl insertion.

Scheme 63.
Proposed mechanism of ring expansion of pinene induced by Fe(CO)3.

Figure 4.
Molecular structure of 161, showing the π-allyl/σ-alkyl bonding to iron; for clarity, only the ipso carbons of the Ph3P ligand are shown.
Figure 4.
Molecular structure of 161, showing the π-allyl/σ-alkyl bonding to iron; for clarity, only the ipso carbons of the Ph3P ligand are shown.

Scheme 64.
Ring-opening of pinene by K2PtCl4.

Scheme 65.
Cyclopropane formation induced by Rh(I) or Pt(II) salts.

Scheme 66.
Proposed mechanism for cyclopropane formation via a metal carbene intermediate.

Scheme 67.
Catalytic selective epoxidation of (left) geraniol and (right) linalool.

Scheme 68.
Catalytic selective epoxidation of pinenes and carenes.

Scheme 69.
Stereospecific epoxidation of (+)-3-carene with methyltrioxorhenium(VII).

Scheme 70.
Synthetic route to humulene-3,4-epoxide, 177.

Figure 5.
Molecular structure of humulene-(1,2)(8,9)-di-epoxide.

Scheme 71.
Migration of one component of a bimetallic fragment across the guaiazulene framework.

Figure 6.
Selected organometallic derivatives of the dihydroguaiazulenide anion.

Figure 7.
Meso and racemic isomers of bis(dihydroguaiazulenyl)metal complexes.

Figure 8.
Molecular structures of (left) the meso and (right) a racemic isomer of bis(dihydroguaiazulenyl)iron.
Figure 8.
Molecular structures of (left) the meso and (right) a racemic isomer of bis(dihydroguaiazulenyl)iron.

Scheme 72.
Synthesis of the (benz[cd]azulene)Cr(CO)3, 181, derived from guaiazulene.

Scheme 73.
Possible haptotropic shifts over the benz[cd]azulene skeleton.

Scheme 74.
The Nicholas reaction, depicting overlap of a filled d orbital with the vacant p orbital on carbon.
Scheme 74.
The Nicholas reaction, depicting overlap of a filled d orbital with the vacant p orbital on carbon.

Scheme 75.
A metal cluster-promoted cyclisation reaction.

Scheme 76.
Cobalt-mediated cyclisation to form a cyclododecadienynol.

Scheme 77.
Sequential molecular rearrangements of 2-ethynylborneol.

Scheme 78.
Characterisation of 2-bornyl cations preferentially stabilised by interaction with molybdenum.
Scheme 78.
Characterisation of 2-bornyl cations preferentially stabilised by interaction with molybdenum.

Scheme 79.
Rearrangement behaviour of the [(fenchyl)Co2(CO)6]+ cluster cation.

Scheme 80.
Rearrangement behaviour of the [(fenchyl)(C5H5)MoCo(CO)5]+ cluster cation.

Figure 9.
Structures of (left) the dicobalt-fenchyl cluster, 200, and (right) the cobalt–molybdenum cation, 205.
Figure 9.
Structures of (left) the dicobalt-fenchyl cluster, 200, and (right) the cobalt–molybdenum cation, 205.

Figure 10.
Designation of the absolute configuration of a tetrahedral organometallic cluster.

Figure 11.
Molecular structure of 2-ethynyl-verbenol, 206.

Scheme 81.
Reactions of verbenone with alkynyl anions leading to tetrahedral bimetallic clusters.

Scheme 82.
Formation of the allenylidene–ruthenium complex, 209, from 2-ethynyl-verbenol.

Scheme 83.
Alkynyl-menthol dicobalt and mixed iron–cobalt tetrahedral cluster complexes.

Scheme 84.
Dicobalt hexacarbonyl complexes from alkynyl derivatives of geraniol.

Scheme 85.
Synthesis of a geranyl-carbynyltricobalt-nonacarbonyl cluster, 219.

Scheme 86.
Synthesis of geranyl- and farnesyl-carbynyltricobalt-nonacarbonyl clusters.

Scheme 87.
Complexes possessing a direct terpenoid metal σ-bond.

Figure 12.
Proposed, 222, and revised structure, 223, of nordine.

Figure 13.
Molecular structure of humulene-2,9-di-aldehyde.

Figure 14.
Molecular structures of (left) α-pinene, (centre) β-pinene, and (right) camphene, acquired using the crystalline sponge X-ray crystallographic method.
Figure 14.
Molecular structures of (left) α-pinene, (centre) β-pinene, and (right) camphene, acquired using the crystalline sponge X-ray crystallographic method.

Figure 15.
Structure of β-carotene, 224.

Figure 16.
X-ray crystal structures of the carotene-Pd10 chain complexes: (a) rac-side view, (b) rac-top view, (c) meso-top view.
Figure 16.
X-ray crystal structures of the carotene-Pd10 chain complexes: (a) rac-side view, (b) rac-top view, (c) meso-top view.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McGlinchey, M.J. Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements. Molecules 2024, 29, 1409. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29061409
AMA Style
McGlinchey MJ. Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements. Molecules. 2024; 29(6):1409. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29061409
Chicago/Turabian StyleMcGlinchey, Michael J. 2024. "Organotransition Metal Chemistry of Terpenes: Syntheses, Structures, Reactivity and Molecular Rearrangements" Molecules 29, no. 6: 1409. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules29061409