QSAR Studies on Andrographolide Derivatives as α-Glucosidase Inhibitors

Abstract
:1. Introduction
2. Computational Methods
2.1. Database and Software
2.2. Training Set Selection
2.3. Generation and Validation of the 2D QSAR Model
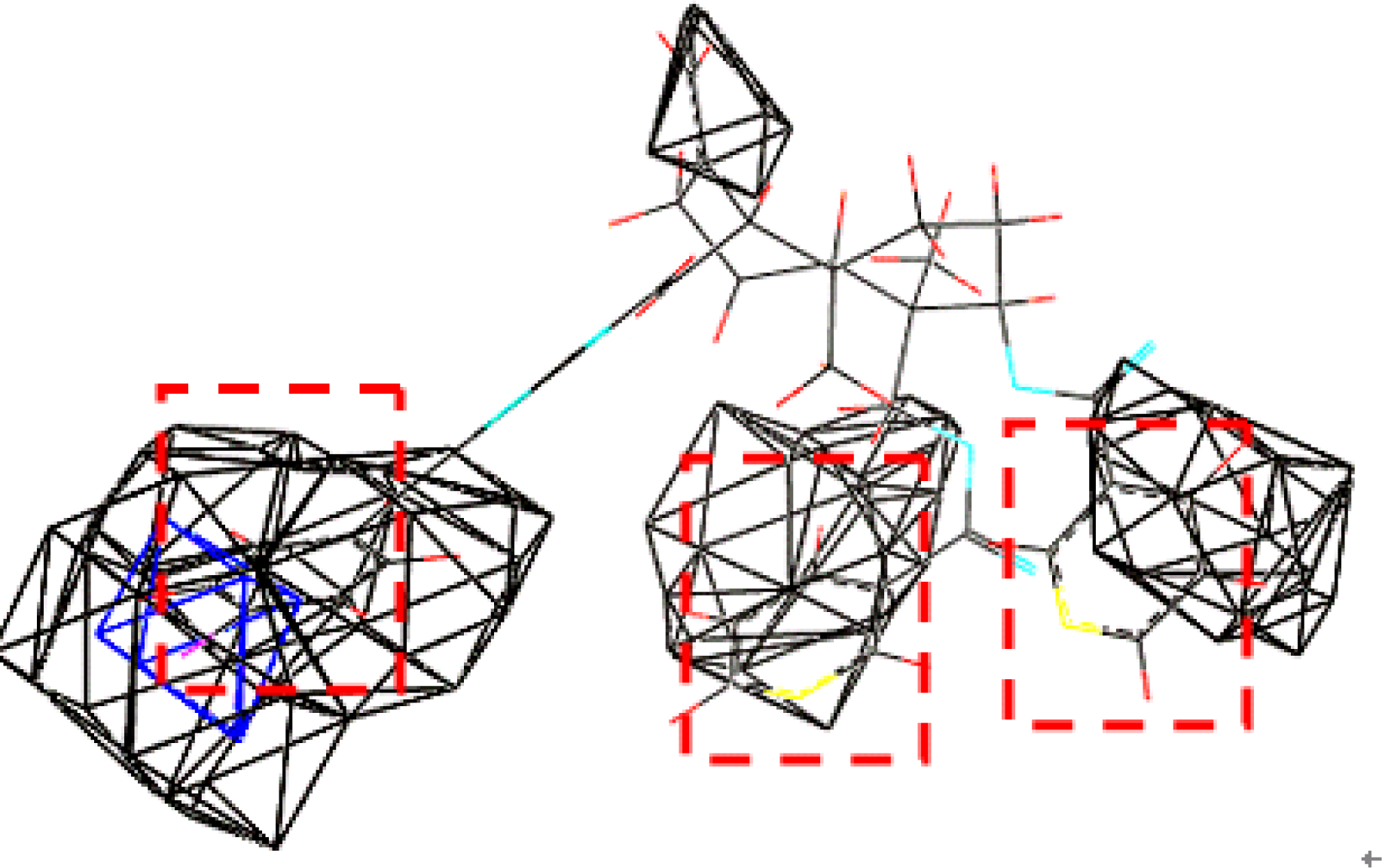
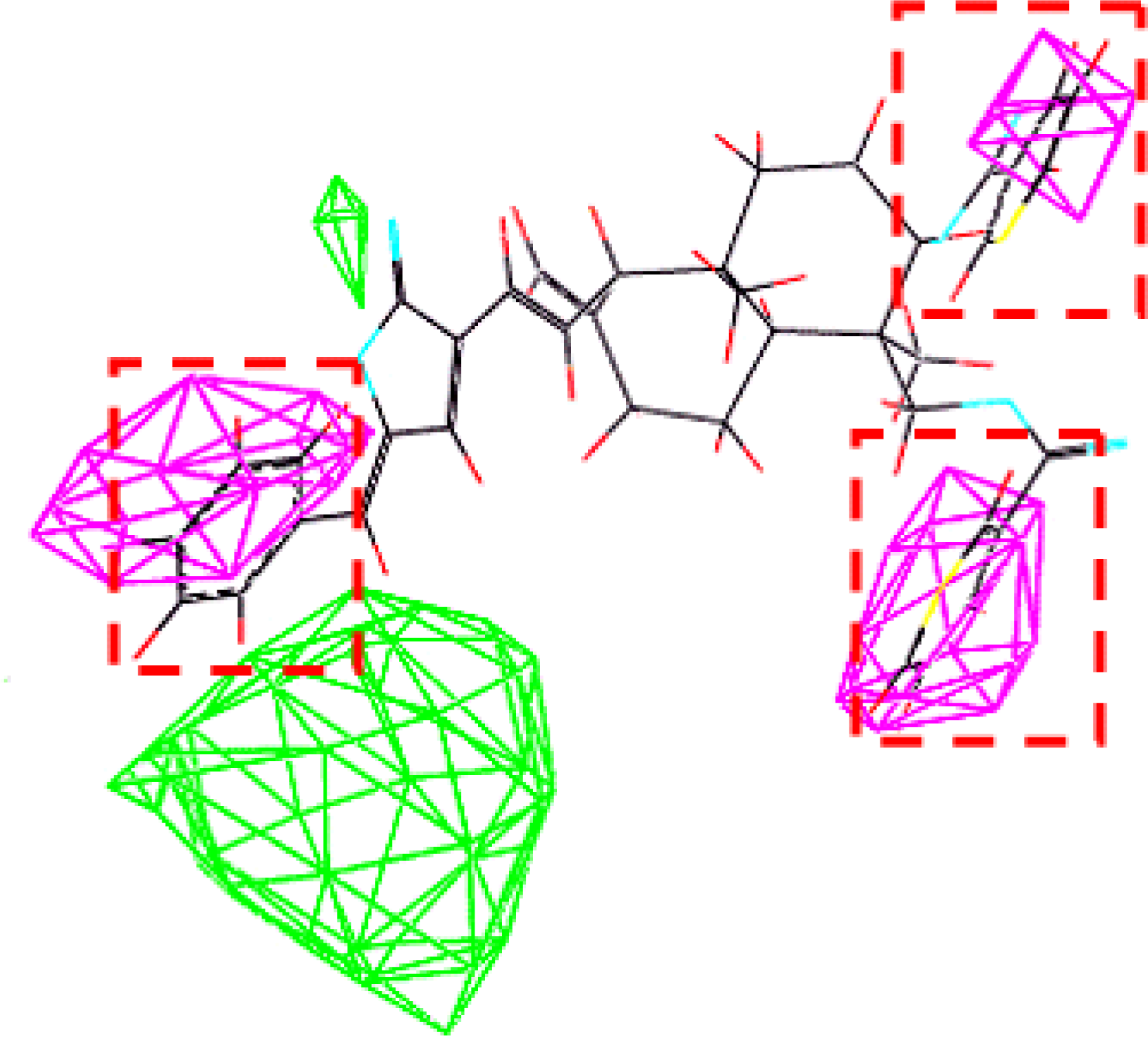
2.4. Generation and Validation of the 3D QSAR Model
3. Result and Discussion
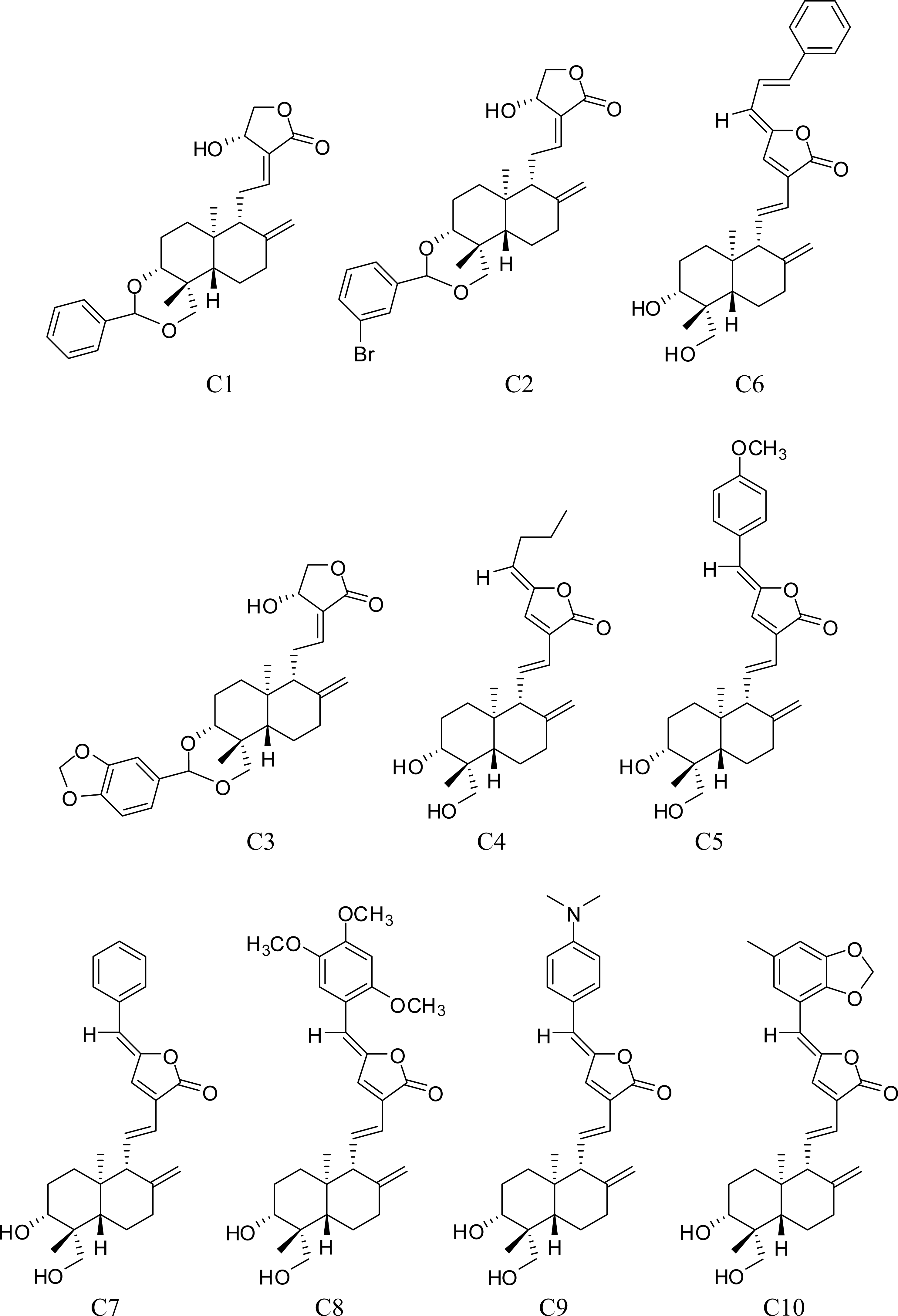
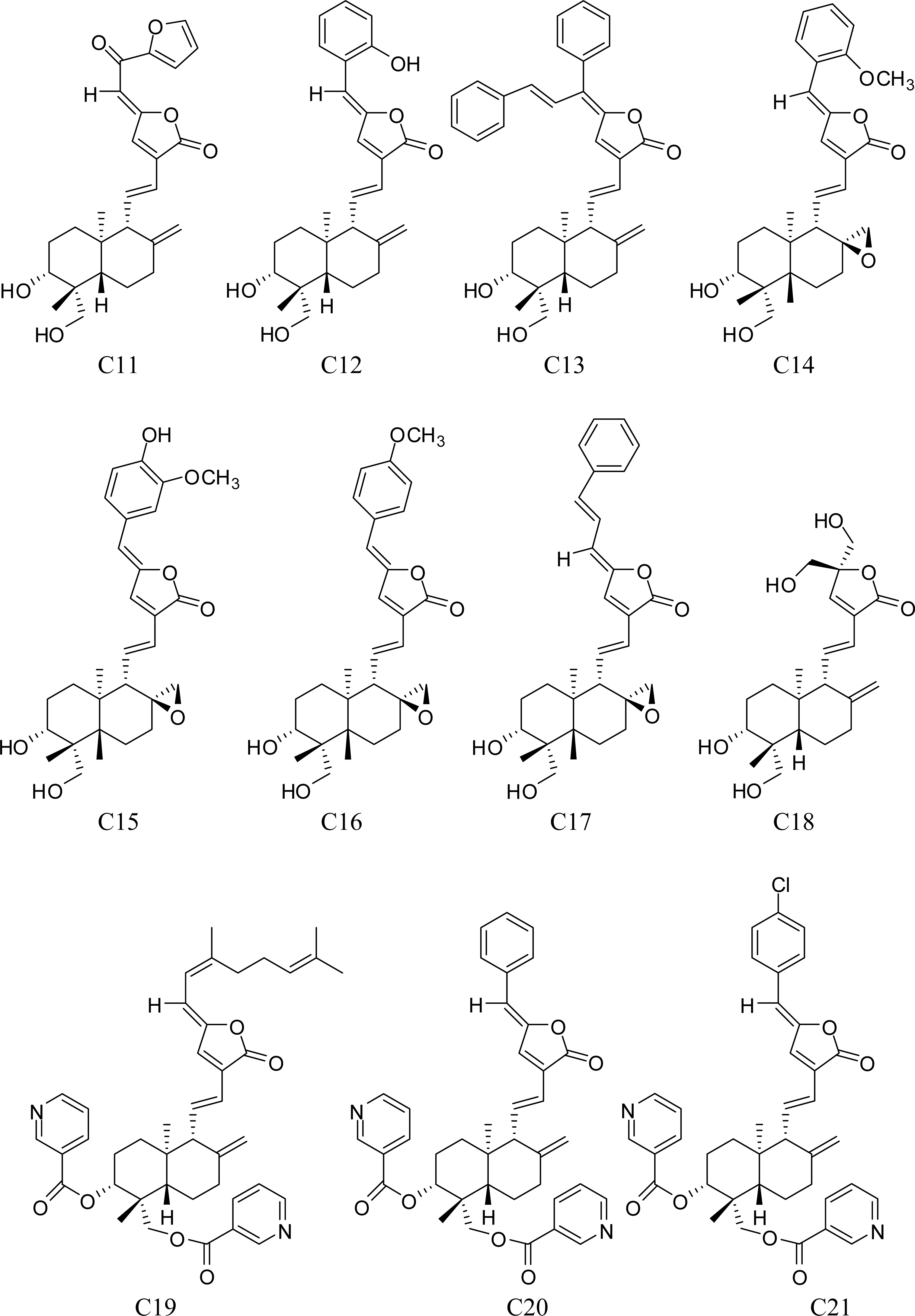
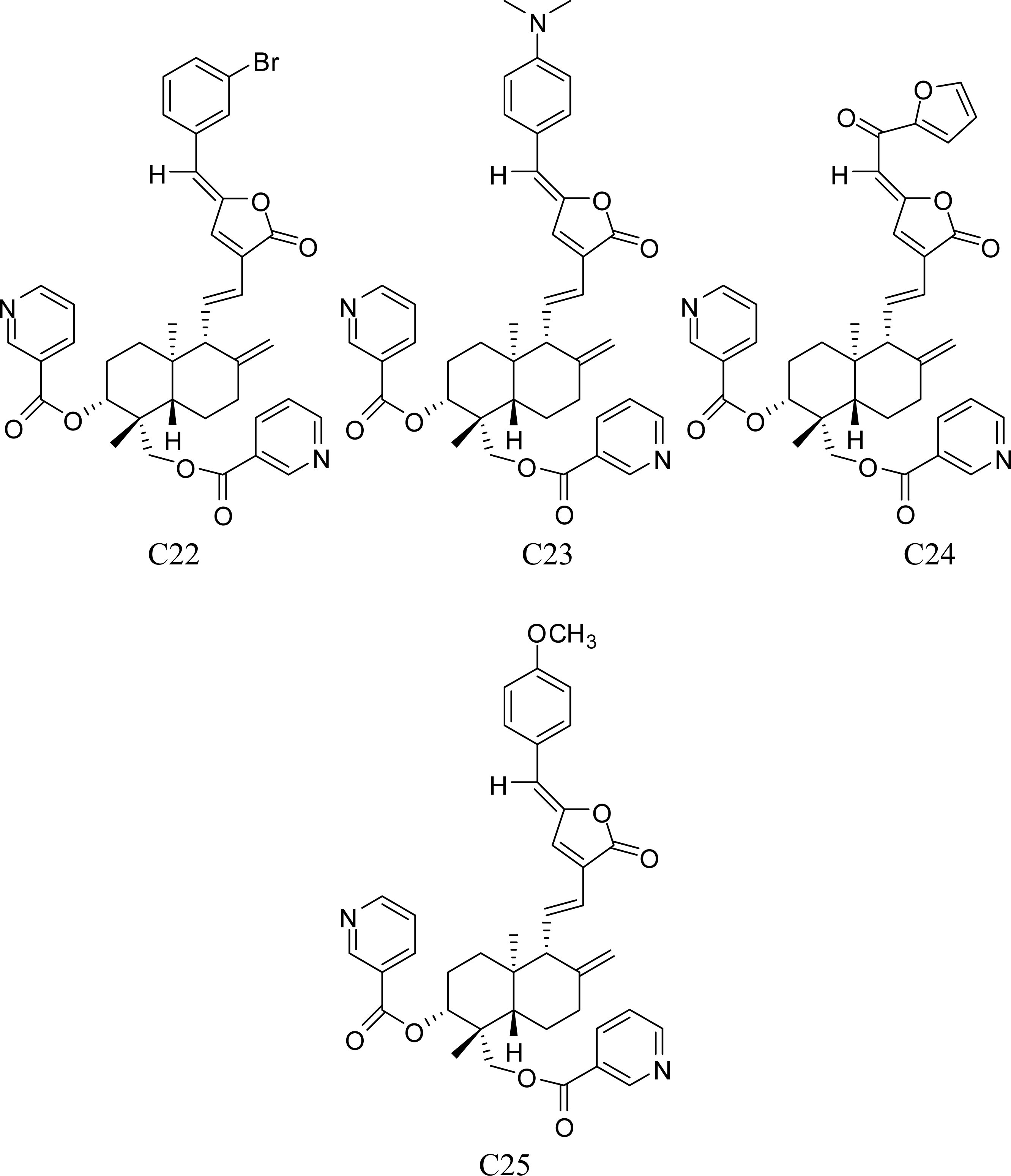
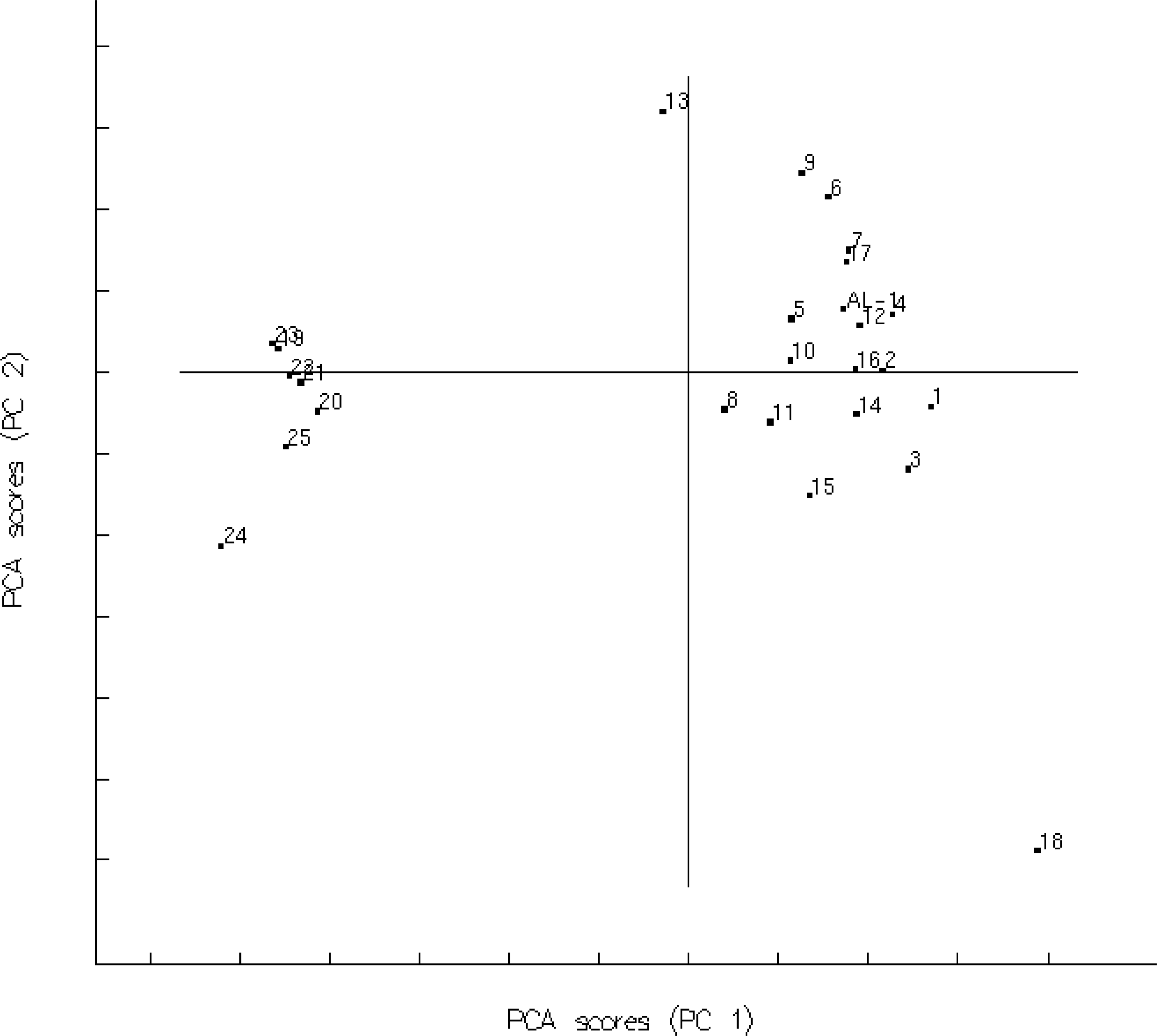
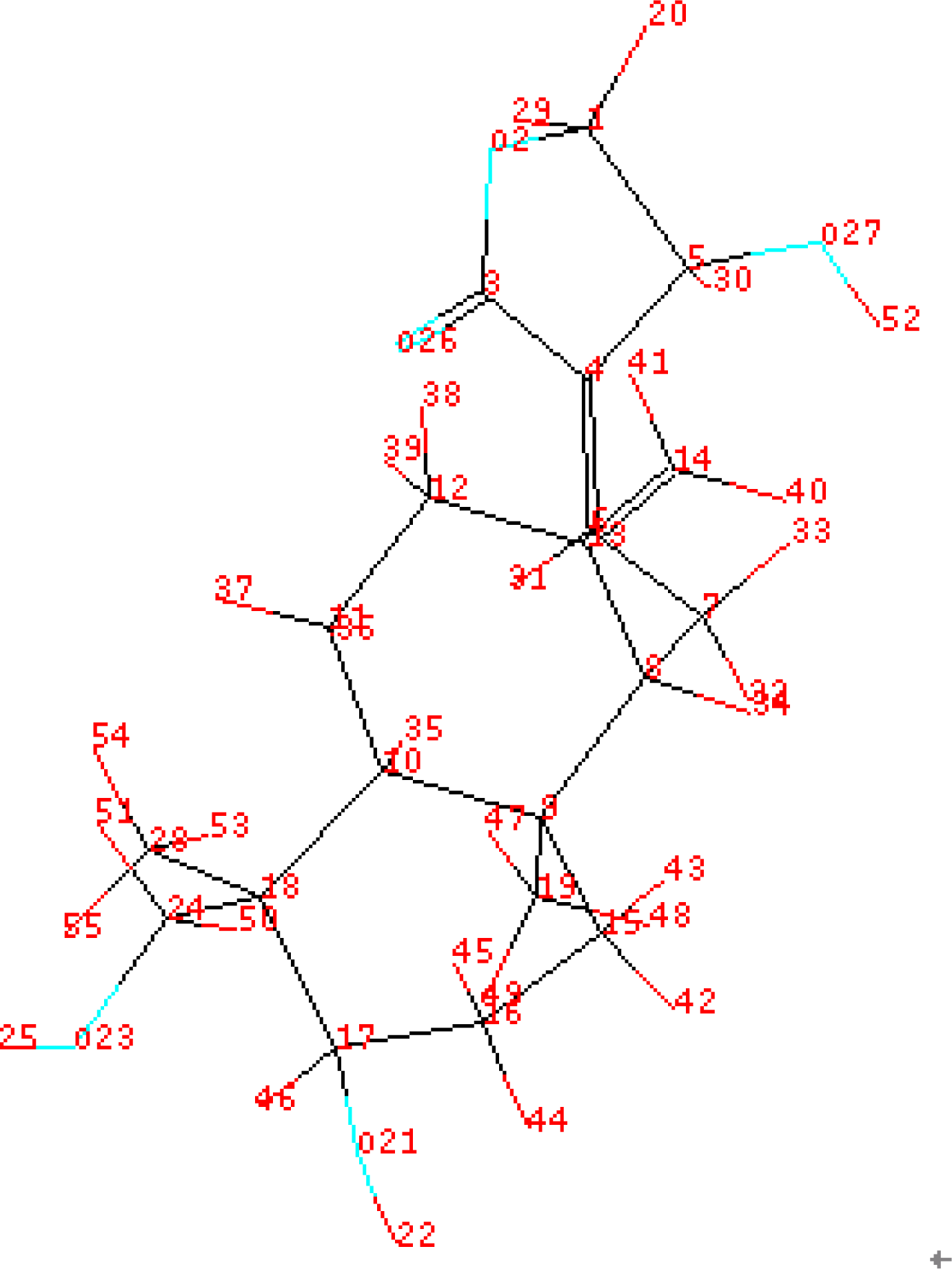
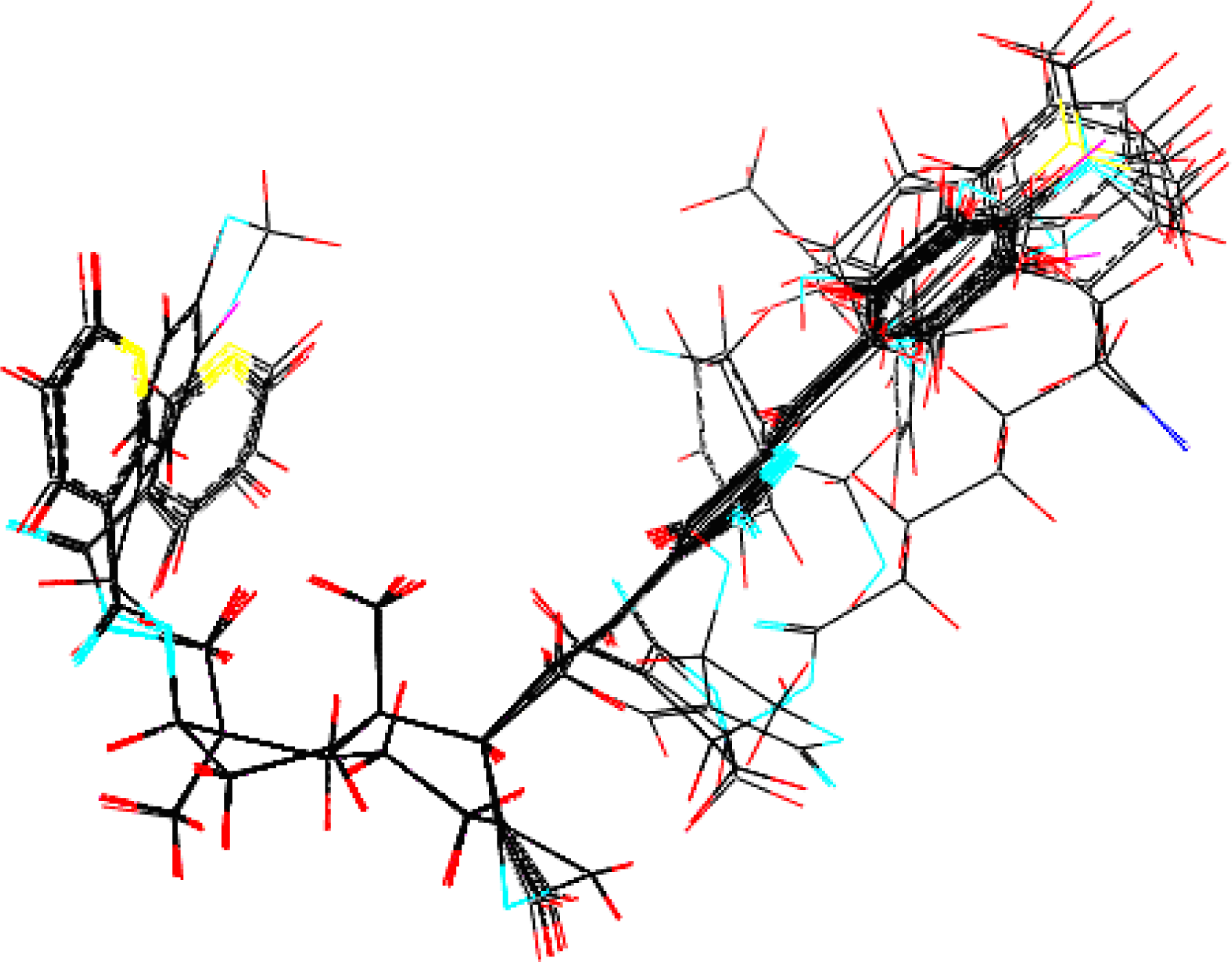
3.1. Training Set Selection
3.2. Establishment and Validation of 2D-QSAR Model
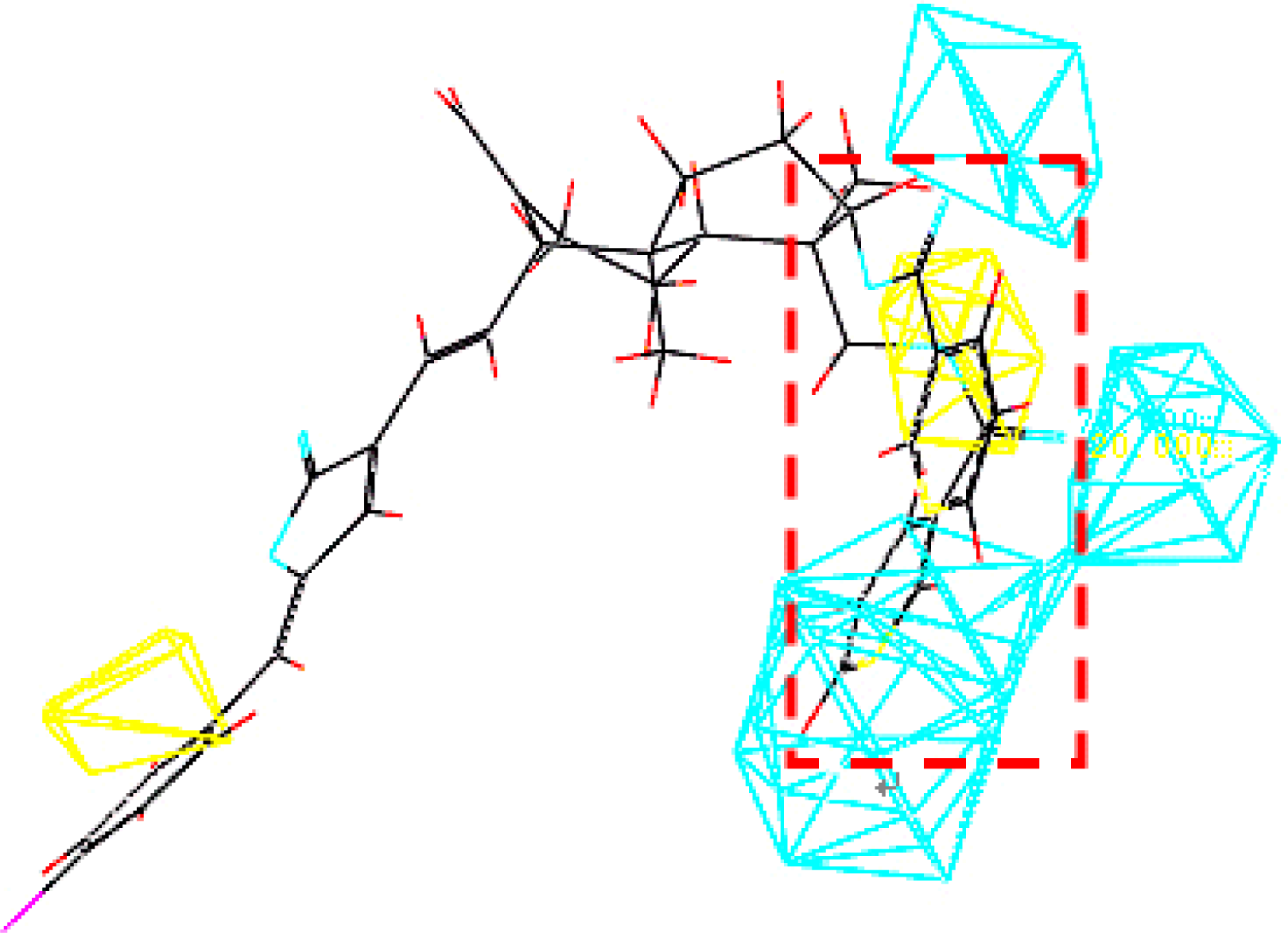
3.3. Establishment and Validation of the 3D-QSAR Model
4. Conclusions
Acknowledgments
References and Notes
- Zhang, CY; Tan, BK. Effects of 14-deoxyandrographolide and 14-deoxy-11,12-didehydroandrographolide on nitric oxide production in cultured human endothelial cells. Phytother. Res 1999, 13, 157–159. [Google Scholar]
- Sabu, KK; Padmesh, P; Seeni, SJ. Intraspecific variation in active principle content and isozymes of Andrographis paniculata (kalmegh): A traditional hepatoprotective medicinal herb of India. Med. Aromat. Plant Sci 2001, 23, 637–647. [Google Scholar]
- Bernacki, RJ; Niedbala, MJ; Korytnyk, W. Glycosidases in cancer and invasion. Cancer Metastasis Rev 1985, 4, 81–101. [Google Scholar]
- Pili, R; Chang, J; Partis, RA; Mueller, RA; Chrest, FJ; Passaniti, A. The alpha-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res 1995, 55, 2920–2926. [Google Scholar]
- Humphries, MJ; Matsumoto, K; White, SL; Olden, K. Inhibition of experimental metastasis by castanospermine in mice: Blockage of two distinct stages of tumor colonization by oligosaccharide processing inhibitors. Cancer Res 1986, 46, 5215–5222. [Google Scholar]
- Papandreou, MJ; Barbouche, R; Guieu, R; Kieny, MP; Fenouillet, E. The alpha-glucosidase inhibitor 1-deoxynojirimycin blocks human immunodeficiency virus envelope glycoprotein-mediated membrane fusion at the CXCR4 binding step. Mol. Pharmacol 2002, 61, 186–193. [Google Scholar]
- Ouzounov, S; Mehta, A; Dwek, RA; Block, TM; Jordan, R. The combination of interferon alpha-2b and n-butyl deoxynojirimycin has a greater than additive antiviral effect upon production of infectious bovine viral diarrhea virus (BVDV) in vitro: Implications for hepatitis C virus (HCV) therapy. Antiviral Res 2002, 55, 425–435. [Google Scholar]
- Schmidt, DD; Frommer, W; Junge, B; Muller, L; Wingender, W; Truschei, E; Schafer, D. Alpha-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977, 64, 535–536. [Google Scholar]
- Kameda, Y; Asano, N; Yoshikawa, M; Takeucki, M; Yamaguchi, T; Matsui, K; Horii, S; Fukase, HJ. Valiolamine, a new alpha-glucosidase inhibiting aminocyclitol produced by Streptomyces hygroscopicus. J.Antibiot 1984, 37, 1301–1307. [Google Scholar]
- Robinson, KM; Begovic, ME; Rhinehart, BL; Heineke, EW; Ducep, JB; Kastner, PR; Marshall, FN; Danzin, C. New potent alpha-glucohydrolase inhibitor MDL 73945 with long duration of action in rats. Diabetes 1991, 40, 825–830. [Google Scholar]
- Fujisawa, T; Ikegami, H; Inoue, K; Kawabata, Y; Ogihara, T. Effect of two alpha-glucosidase inhibitors, voglibose and acarbose, on postprandial hyperglycemia correlates with subjective abdominal symptoms. Metabolism 2005, 54, 387–390. [Google Scholar]
- van den Broek, LAGM; Kat-van den Nieuwenhof, MW; Butters, TD; van Boeckel, CA. Synthesis of alpha-glucosidase I inhibitors showing antiviral (HIV-1) and immunosuppressive activity. J. Pharm. Pharmacol 1996, 48, 172–178. [Google Scholar]
- Dai, GF; Xu, HW; Wang, JF; Liu, FW; Liu, HM. Studies on the novel alpha-glucosidase inhibitory activity and structure-activity relationships for andrographolide analogues. Bioorgan. Med. Chem 2006, 16, 2710–2713. [Google Scholar]
- Xu, HW; Dai, GF; Liu, GZ; Wang, JF; Liu, HM. Synthesis of andrographolide derivatives: A new family of alpha-glucosidase inhibitors. Bioorgan. Med. Chem 2007, 15, 4247–4255. [Google Scholar]
- Truscheit, E; Frommer, W; Junge, B; Muller, L; Schmidt, DD; Wingender, W. Chemistry and biochemistry of microbial alpha-glucosidase inhibitors. Angew. Chem 1981, 93, 738–755. [Google Scholar]
- Madariaga, H; Lee, PC; Heitlinger, LA; Lenenthal, M. Effects of graded alpha-glucosidase inhibition on sugar absorption in vivo. Dig. Dis. Sci 1988, 33, 1020–1024. [Google Scholar]
- Lee, D-S; Lee, S-H. Genistein, a soy isoflavone, is a potent alpha-glucosidase inhibitor. FEBS Lett 2001, 501, 84–86. [Google Scholar]
- McCulloch, DK; Kurtz, AB; Tattersall, RB. A new approach to the treatment of nocturnal hypoglycemia using alpha-glucosidase inhibition. Diabetes Care 1983, 6, 483–487. [Google Scholar]
- Sou, S; Takahashi, H; Yamasaki, R; Kagechika, H; Endo, Y; Hashimoto, Y. Alpha-glucosidase inhibitors with a 4,5,6,7-tetrachlorophthalimide skeleton pendanted with a cycloalkyl or dicarba-closo-dodecaborane group. Chem. Pharm. Bull 2001, 49, 791–793. [Google Scholar]
- Node, K. Alpha-glucosidase inhibitors: New therapeutic agents for chronic heart failure. Hypertens. Res 2006, 29, 741–42. [Google Scholar]
- Hansch, C; Mahoney, PP; Fujita, T; Muir, RM. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients. Nature 1962, 194, 178–180. [Google Scholar]
- Itzstein, VM; Wu, WY; Kok, GB. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar]
- Melnick, M; Reich, SH; Lewis, KK. Bis tertiary amide inhibitors of the HIV-1 protease generated via protein structure-based iterative design. J. Med. Chem 1996, 39, 2795–2811. [Google Scholar]
- Ring, CS; Sun, E; McKerrow, JH. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc.Natl. Acad. Sci. USA 1993, 90, 3583–3587. [Google Scholar]
- Hibert, MF; Hoffmann, R; Miller, RC. Conformation-activity relationship study of 5-HT3 receptor antagonists and a definition of a model for this receptor site. J. Med. Chem 1990, 33, 1594–1600. [Google Scholar]
- Motoc, I; Sit, SY; Harte, WE. 3-Hydroxy-3-methylglutaryl-coenzyme. A reductase: Molecular modeling, three-dimensional structure-activity relationships, inhibitor design. Quant. Struct-Act. Relat 1991, 10, 30–35. [Google Scholar]
- Xiong, B; Gui, CS; Xu, XY. Acta. A 3D model of SARS_CoV 3CL proteinase and its inhibitors design by virtual screening. Pharmacol. Sin 2003, 24, 497–504. [Google Scholar]
- Pastor, M; Cruciani, G. A novel strategy for improving ligand selectivity in receptor-based drug design. J. Med. Chem 1995, 38, 4637–4647. [Google Scholar]
- Anand, K; Ziebuhr, J; Wadhwani, P; Mesters, JR; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science (Sciencexpress) 2003, 300, 1763–1767. [Google Scholar]
- Carlton, AT; Vinicius, BDS; Carlos, HTD. Current topics in computer-aided drug design. J. Pharm. Sci 2008, 97, 1089–1098. [Google Scholar]
- Xu, S. . The 3D-QSAR Studies on Andrographolide Derivatives Inhibiting α-Glucosidase. Ph.D. Dissertation. Zhengzhou University: Zhengzhou, China, 2006.
- Wolfgang, H; Léopold, S. Applied Multivariate Statistical Analysis, 2nd ed; Springer Press: Berlin, Heidelberg, Germany, 2007; pp. 233–272. [Google Scholar]
- Ash, S; Cline, MA; Homer, RW; Hurst, T; Smith, GB. SYBYL line notation (SLN): A versatile language for chemical structure representation. J Chem Inf Comput Sci 1997, 37, 71–79. [Google Scholar]
- Timothy, EL. VAX Architecture Reference Manual; Digital Press: Newton, MA, USA, 1987; pp. 288–326. [Google Scholar]
- Frank, IE; Feikama, J; Constantine, N; Kowalski, BR. Prediction of Product Quality from Spectral Data Using the Partial Least-Squares Method. J. Chem. Inf. Comput. Sci 1984, 24, 20–24. [Google Scholar]
- Golbraikh, A; Tropsha, A. Beware of q2! J. Mol. Graph. Model 2002, 20, 269–276. [Google Scholar]
- Cramer, RD; Patterson, DE; Bunce, JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Klebe, G; Abraham, U. Comparative Molecular Similarity Index Analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aided Mol. Design 1999, 13, 1–10. [Google Scholar]
- van de Waterbeemd, H. Chemometric Methods in Molecular Design (Methods and Principles in Medicinal Chemistry); Wiley-VCH Press: Weinheim, Germany, 1995; pp. 309–318. [Google Scholar]
- Dixit, A; Kashaw, SK; Gaur, S; Saxena, AK. Development of CoMFA, advance CoMFA and CoMSIA models in pyrroloquinazolines as thrombin receptor antagonist. Bioorgan. Med. Chem 2004, 12, 3591–3598. [Google Scholar]
- Narayanan, R; Gunturi, SB. In silico ADME modelling: Prediction models for blood-brain barrier permeation using a systematic variable selection method. Bioorgan. Med. Chem 2005, 13, 3017–3028. [Google Scholar]
- Gunturi, SB; Narayanan, R; Khandelwal, A. In silico ADME modelling: Computational models to predict human serum albumin binding affinity using ant colony systems. Bioorgan. Med. Chem 2006, 14, 4118–4129. [Google Scholar]
- Gunturi, SB; Narayanan, R. In silico ADME modeling 3: Computational models to predict human intestinal absorption using sphere exclusion and kNN QSAR methods. QSAR Comb. Sci 2007, 26, 653–668. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ACTa | PREb | |Δ|c | Compound | ACT | PRE | |Δ| |
|---|---|---|---|---|---|---|---|
| 1 | 4.000 | 3.933 | 0.067 | 2 | 4.000 | 3.995 | 0.05 |
| 3 | 3.959 | 3.876 | 0.109 | 4 | 3.959 | 4.054 | 0.095 |
| 5 | - | - | -d | 6 | 4.237 | 4.139 | 0.098 |
| 7 | 4.237 | 4.159 | 0.078 | 8 | 4.076 | 4.087 | 0.011 |
| 9 | 4.155 | 4.061 | 0.094 | 10 | 4.000 | 4.099 | 0.099 |
| 11 | 4.000 | 4.089 | 0.089 | 12 | - | - | -d |
| 13 | 3.959 | 4.176 | 0.217 | 14 | 4.000 | 3.946 | 0.054 |
| 15 | 3.983 | 3.924 | 0.059 | 16 | 3.921 | 3.961 | 0.040 |
| 17 | 3.996 | 3.954 | 0.042 | 18 | 3.971 | 3.902 | 0.069 |
| 19 | 4.553 | 4.686 | 0.133 | 20 | 4.796 | 4.813 | 0.017 |
| 21 | 5.222 | 4.806 | 0.416 | 22 | 4.854 | 4.798 | 0.056 |
| 23 | 4.602 | 4.715 | 0.113 | 24 | 4.444 | 4.745 | 0.301 |
| 25 | 4.959 | 4.698 | 0.261 |
| No. | Method | Fielda | OCb | (q2)c | SEd | (R2)e | F |
|---|---|---|---|---|---|---|---|
| 1 | CoMFA | S+E | 1 | 0.741 | 0.178 | 0.819 | 67.905 |
| 2 | S | 2 | 0.748 | 0.159 | 0.866 | 45.280 | |
| 3 | E | 1 | 0.710 | 0.187 | 0.802 | 60.592 | |
| 4 | H | 2 | 0.771 | 0.132 | 0.907 | 68.505 | |
| 5 | D | 1 | 0.313 | 0.297 | 0.498 | 14.876 | |
| 6 | A | 1 | 0.724 | 0.184 | 0.807 | 62.902 | |
| 7 | S+E | 1 | 0.732 | 0.182 | 0.812 | 64.778 | |
| 8 | CoMSIA | S+H | 1 | 0.774 | 0.148 | 0.875 | 105.050 |
| 9 | S+A | 2 | 0.738 | 0.159 | 0.866 | 45.251 | |
| 10 | S+E+H | 1 | 0.755 | 0.169 | 0.838 | 77.788 | |
| 11 | S+H+A | 2 | 0.759 | 0.130 | 0.910 | 70.509 | |
| 12 | S+E+H+A | 1 | 0.747 | 0.174 | 0.829 | 72.588 | |
| 13f | H(Focus) | 1 | 0.776 | 0.144 | 0.882 | 112.028 | |
| 14f | S+H(Focus) | 2 | 0.772 | 0.1.43 | 0.891 | 57.188 | |
| 15f | S+E+H(Focus) | 2 | 0.763 | 0.148 | 0.884 | 53.422 | |
| 16f | S+H+A(Focus) | 2 | 0.794 | 0.127 | 0.915 | 75.093 | |
| Y-Random | S+H+A(Focus) | 1 | 0.199 | - | - | - | |
| No. | Models | R2 | Slope | SE |
|---|---|---|---|---|
| 13 | H(Focus) | 0.906 | 1.007 | 0.143 |
| 8 | S+H | 0.927 | 0.974 | 0.121 |
| 15 | S+E+H(Focus) | 0.895 | 0.937 | 0.142 |
| 16 | S+H+A(Focus) | 0.941 | 0.933 | 0.104 |
| Compound | ACTa | PREb | |Δ|c | Compound | ACT | PRE | |Δ| |
|---|---|---|---|---|---|---|---|
| 1 | 3.996 | 3.960 | 0.04 | 2 | 4.000 | 3.960 | 0.04 |
| 3 | 3.959 | 3.970 | 0.011 | 4 | 3.959 | 3.999 | 0.04 |
| 5 | - | - | -d | 6 | 4.237 | 4.238 | 0.001 |
| 7 | 4.237 | 4.204 | 0.033 | 8 | 4.076 | 4.016 | 0.06 |
| 9 | 4.155 | 4.179 | 0.029 | 10 | 4.000 | 4.119 | 0.119 |
| 11 | 4.000 | 3.935 | 0.065 | 12 | - | - | - |
| 13 | 3.959 | 4.111 | 0.152 | 14 | 4.000 | 4.150 | 0.150 |
| 15 | 3.983 | 4.112 | 0.129 | 16 | 3.921 | 4.075 | 0.154 |
| 17 | 3.996 | 3.916 | 0.08 | 18 | 3.971 | 3.903 | 0.068 |
| 19 | 4.553 | 4.621 | 0.068 | 20 | 4.796 | 4.863 | 0.068 |
| 21 | 5.222 | 5.067 | 0.155 | 22 | 4.854 | 4.886 | 0.032 |
| 23 | 4.602 | 4.831 | 0.229 | 24 | 4.444 | 4.481 | 0.037 |
| 25 | 4.959 | 4.698 | 0.261 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, J.; Huang, S.; Luo, H.; Li, G.; Bao, J.; Cai, S.; Wang, Y. QSAR Studies on Andrographolide Derivatives as α-Glucosidase Inhibitors. Int. J. Mol. Sci. 2010, 11, 880-895. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms11030880
Xu J, Huang S, Luo H, Li G, Bao J, Cai S, Wang Y. QSAR Studies on Andrographolide Derivatives as α-Glucosidase Inhibitors. International Journal of Molecular Sciences. 2010; 11(3):880-895. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms11030880
Chicago/Turabian StyleXu, Jun, Sichao Huang, Haibin Luo, Guoji Li, Jiaolin Bao, Shaohui Cai, and Yuqiang Wang. 2010. "QSAR Studies on Andrographolide Derivatives as α-Glucosidase Inhibitors" International Journal of Molecular Sciences 11, no. 3: 880-895. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms11030880