Human Interleukin 23 Receptor Induces Cell Apoptosis in Mammalian Cells by Intrinsic Mitochondrial Pathway Associated with the Down-Regulation of RAS/Mitogen-Activated Protein Kinase and Signal Transducers and Activators of Transcription factor 3 Signaling Pathways

Abstract
:1. Introduction
2. Results and Discussion
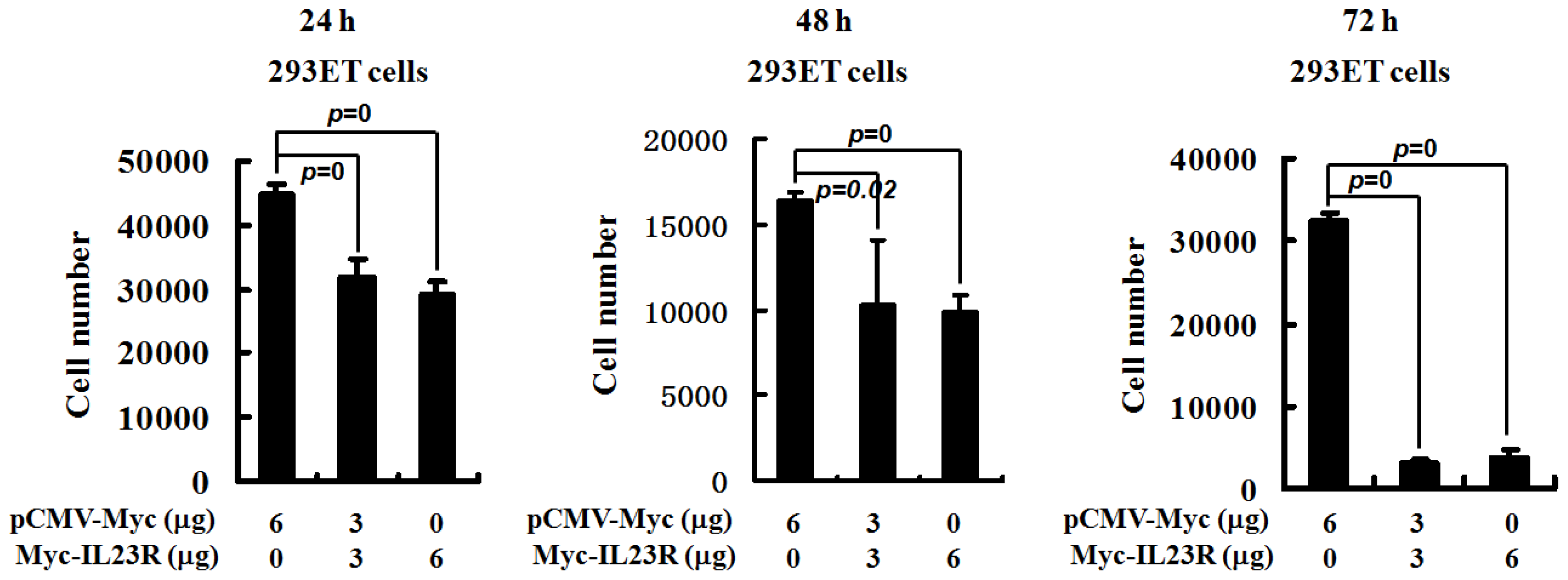
2.1. Overexpression of Human IL-23R Inhibits Cell Proliferation
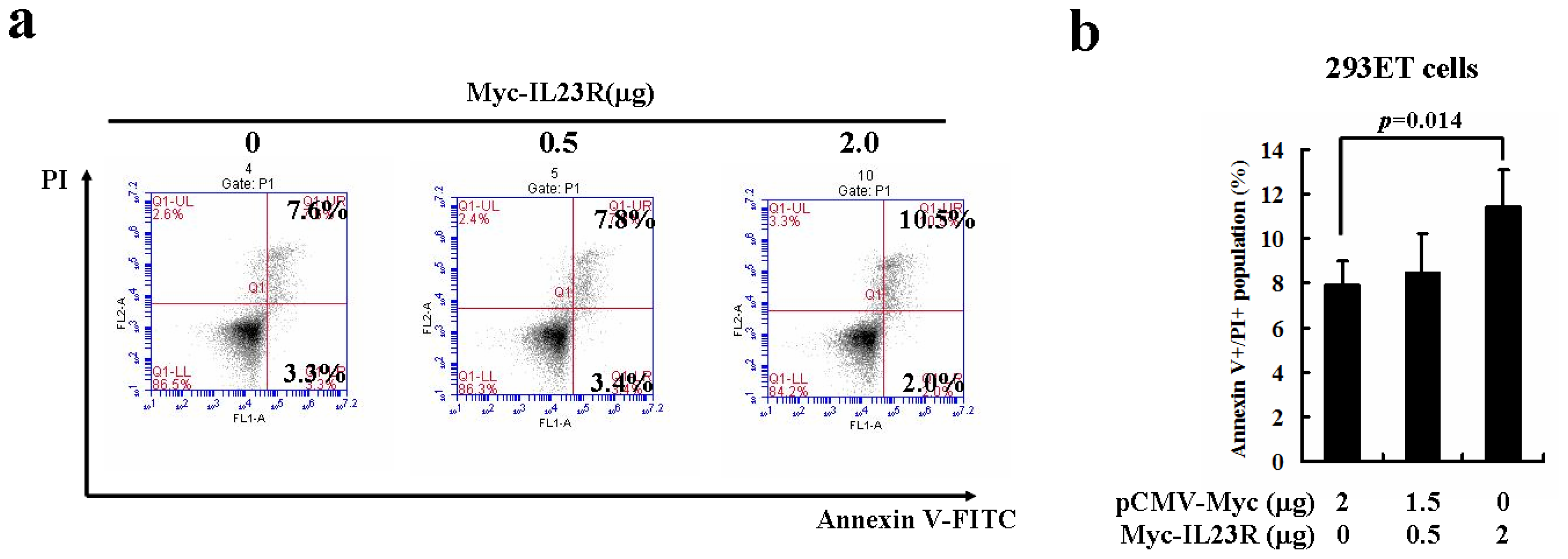
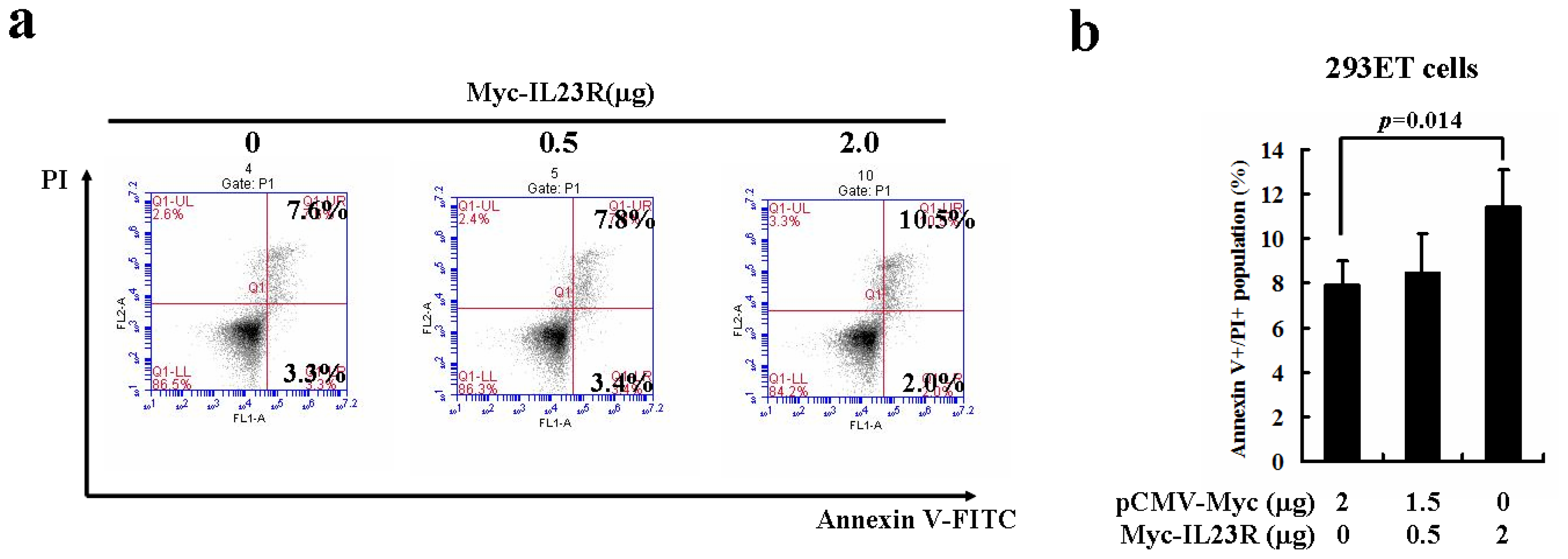
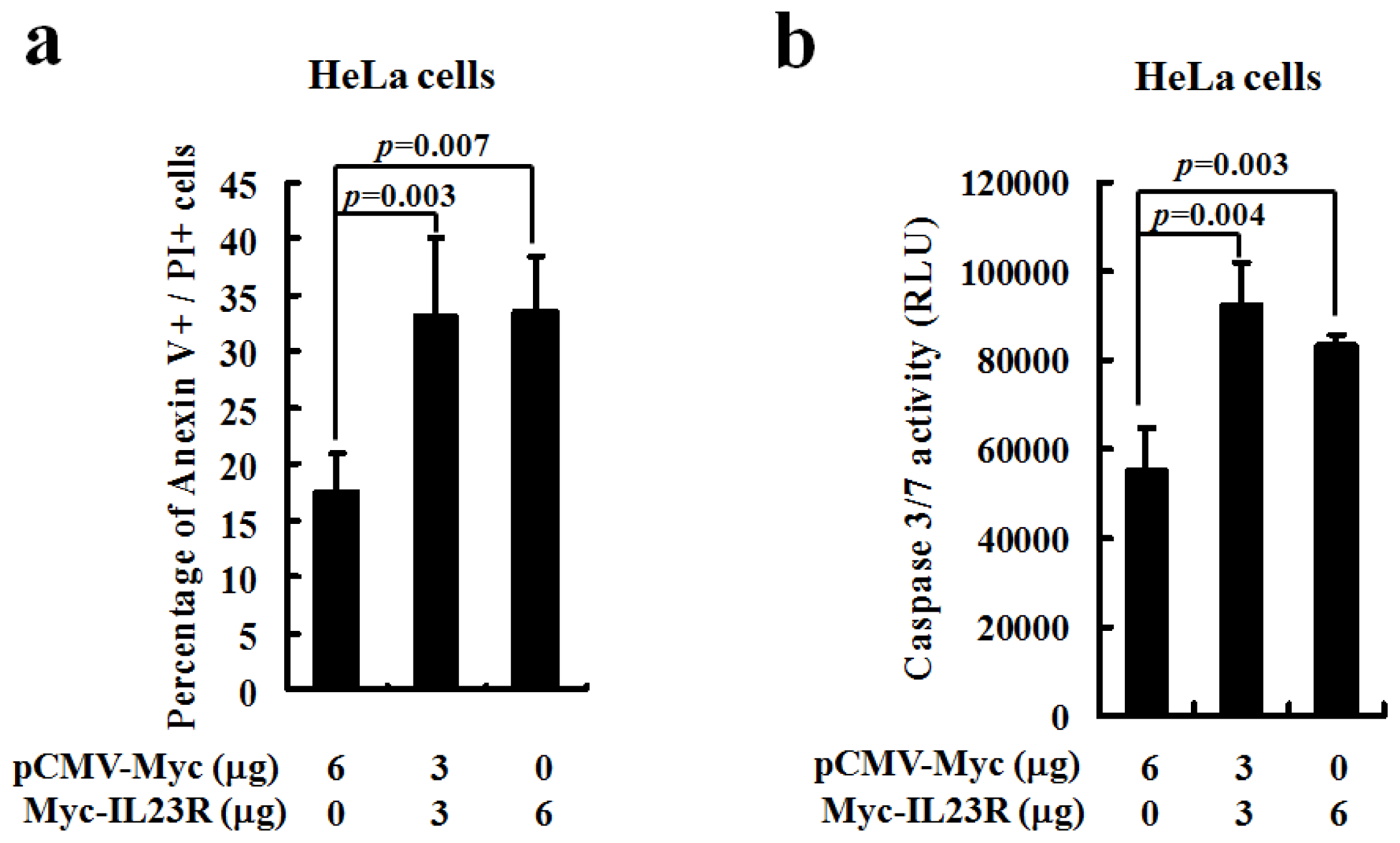
2.2. Over-Expression of Human IL-23R Induces Apoptosis in both 293ET and HeLa Cells
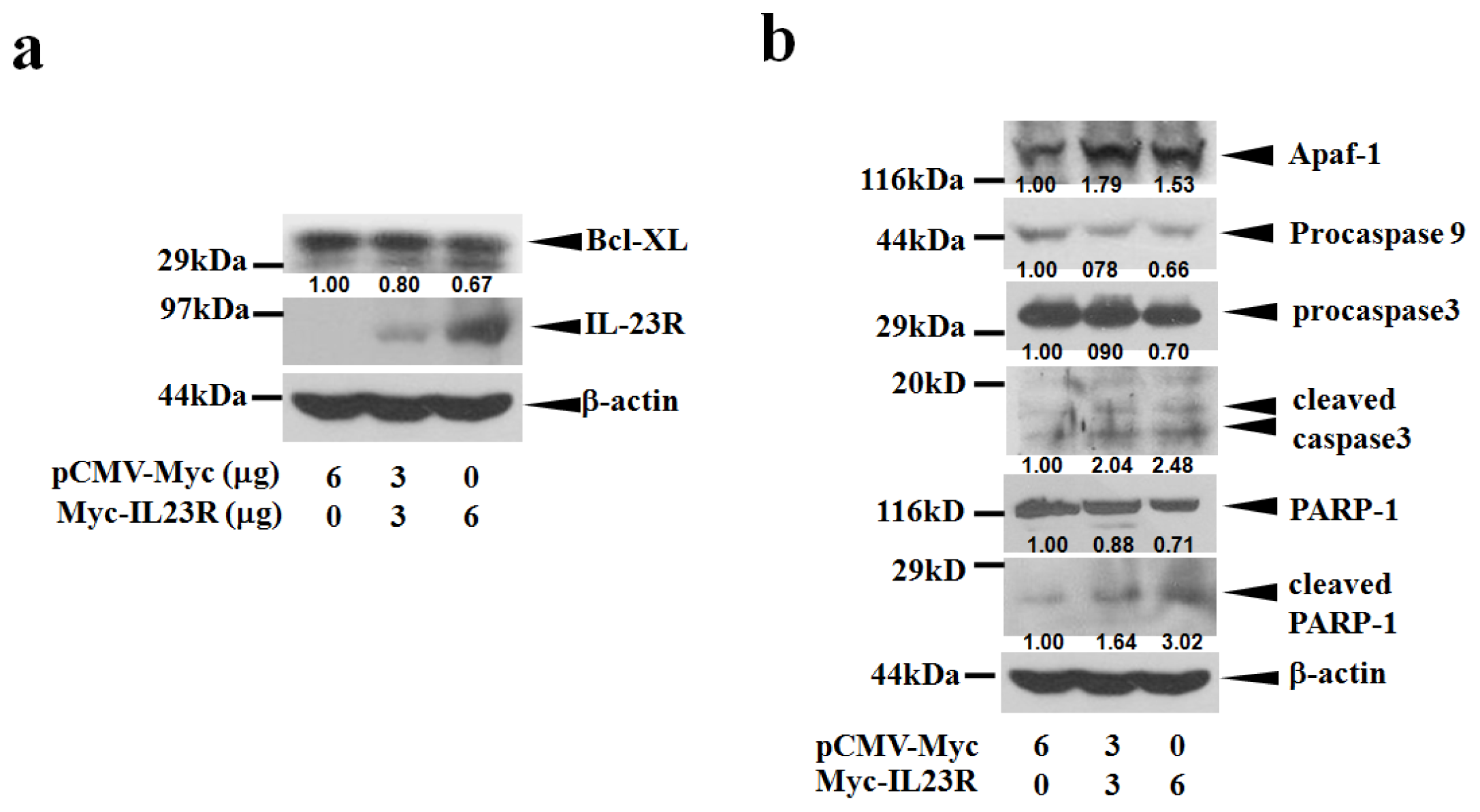
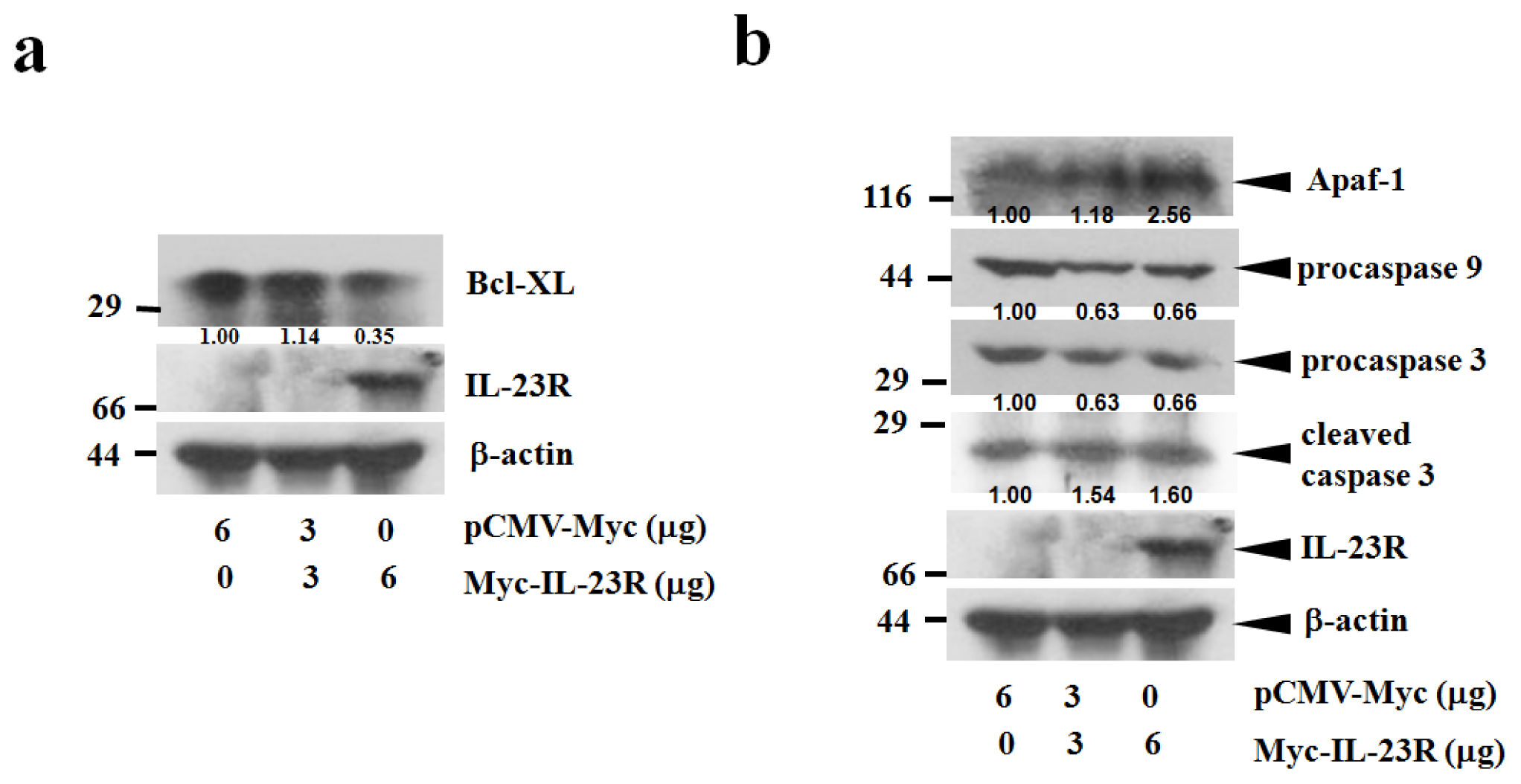
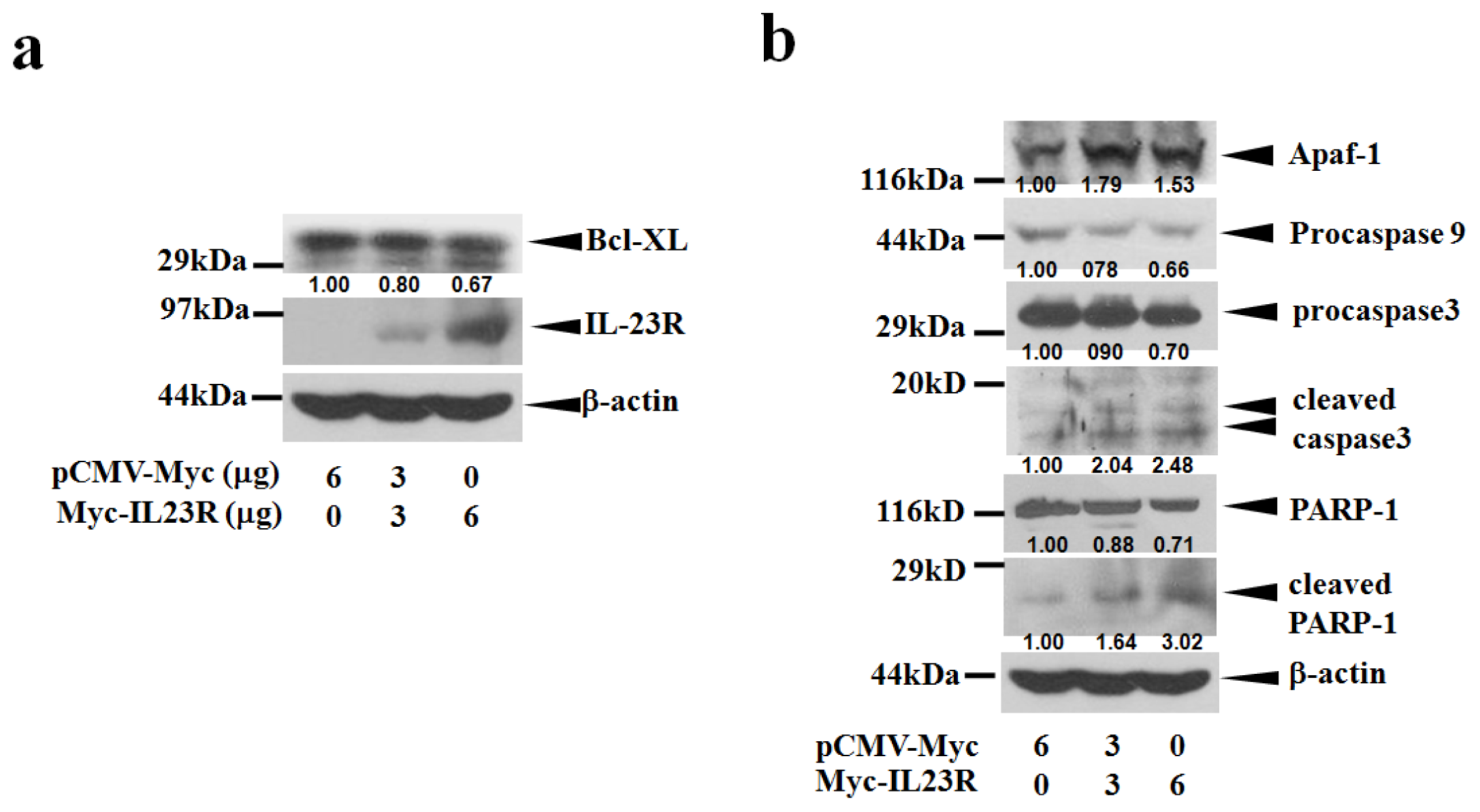
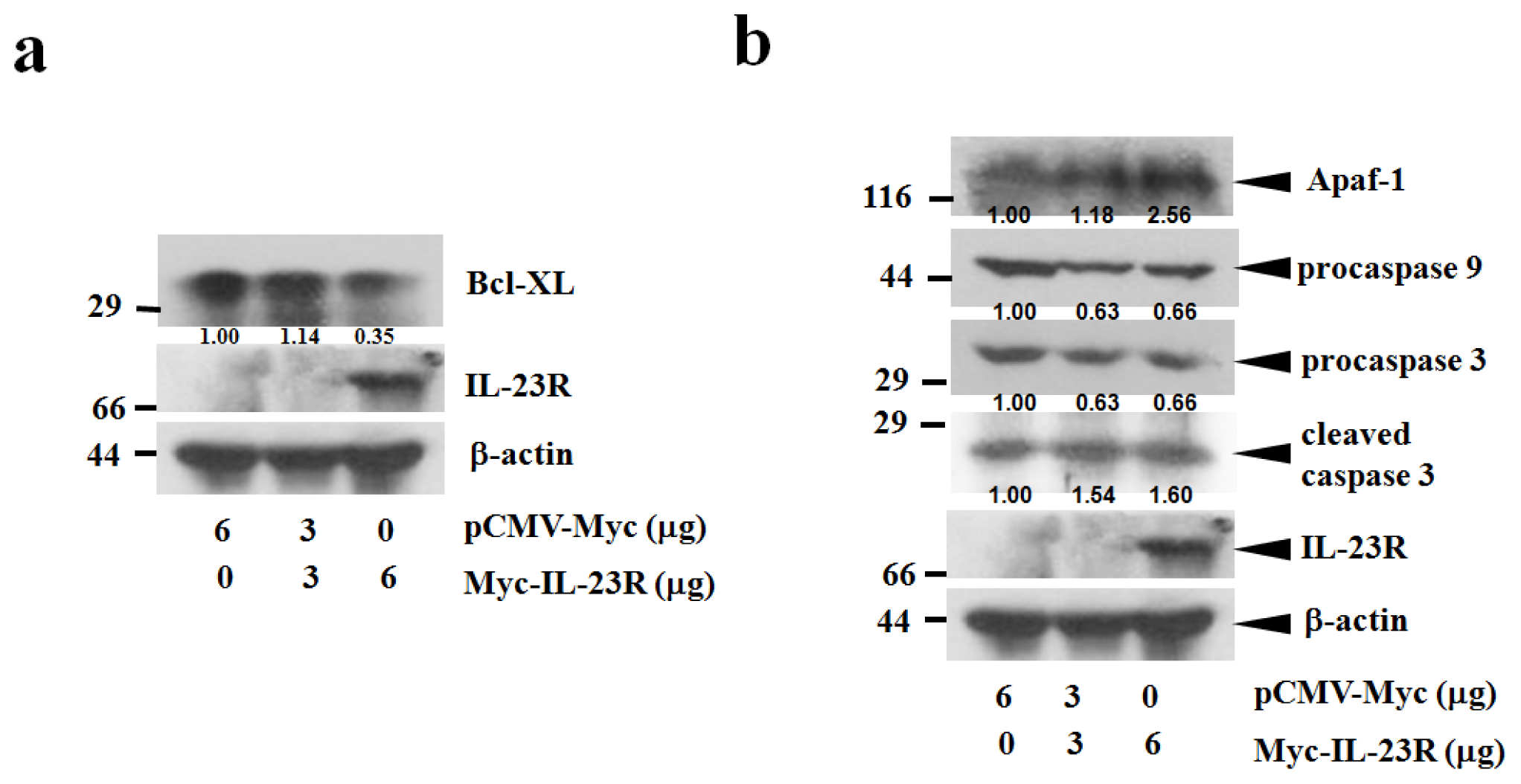
2.3. IL-23R Activates Cell Apoptosis through Intrinsic Pathway
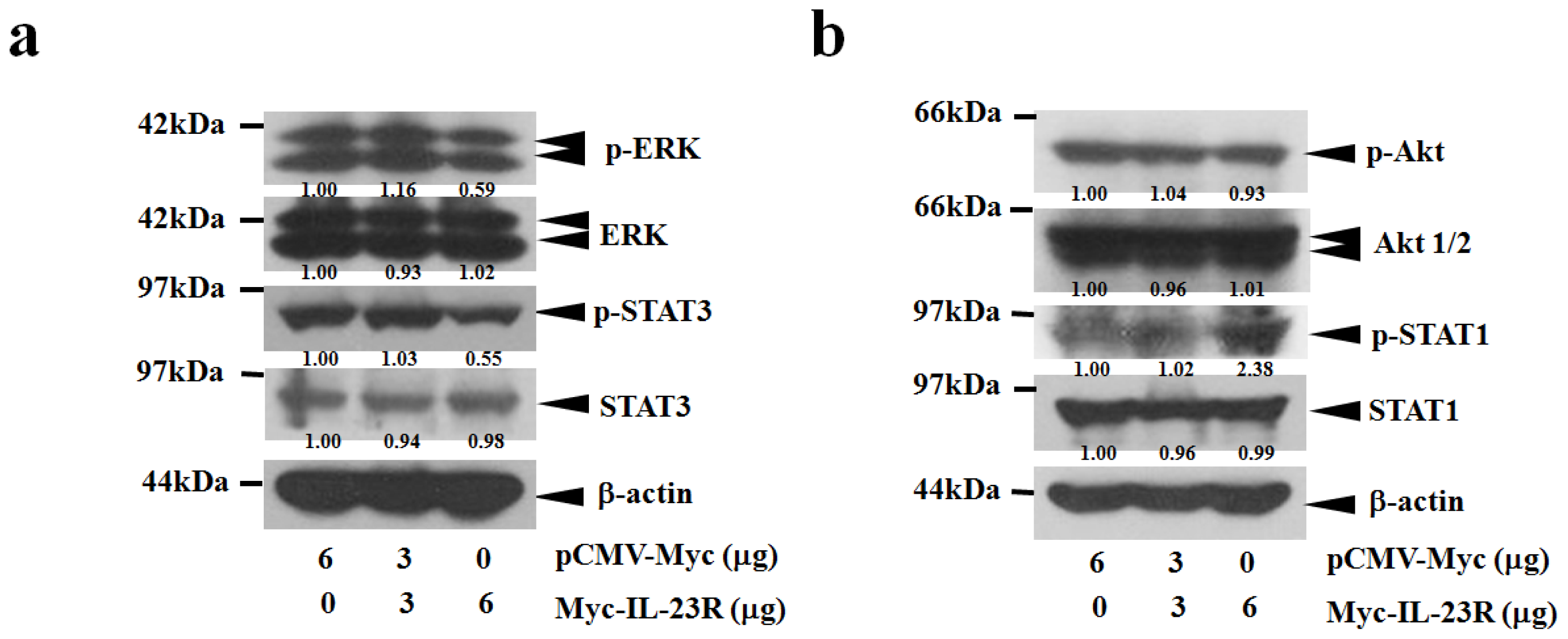
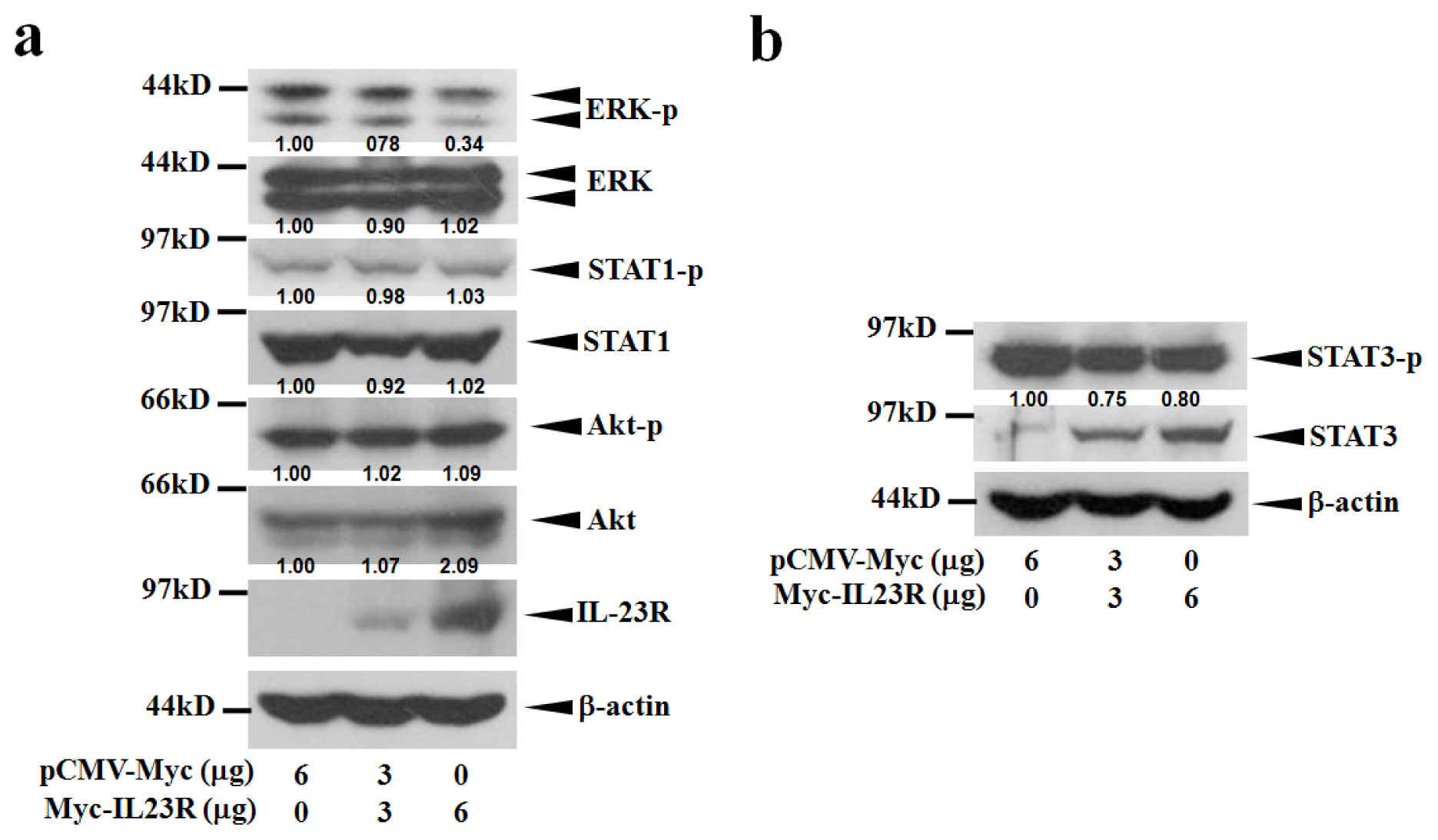
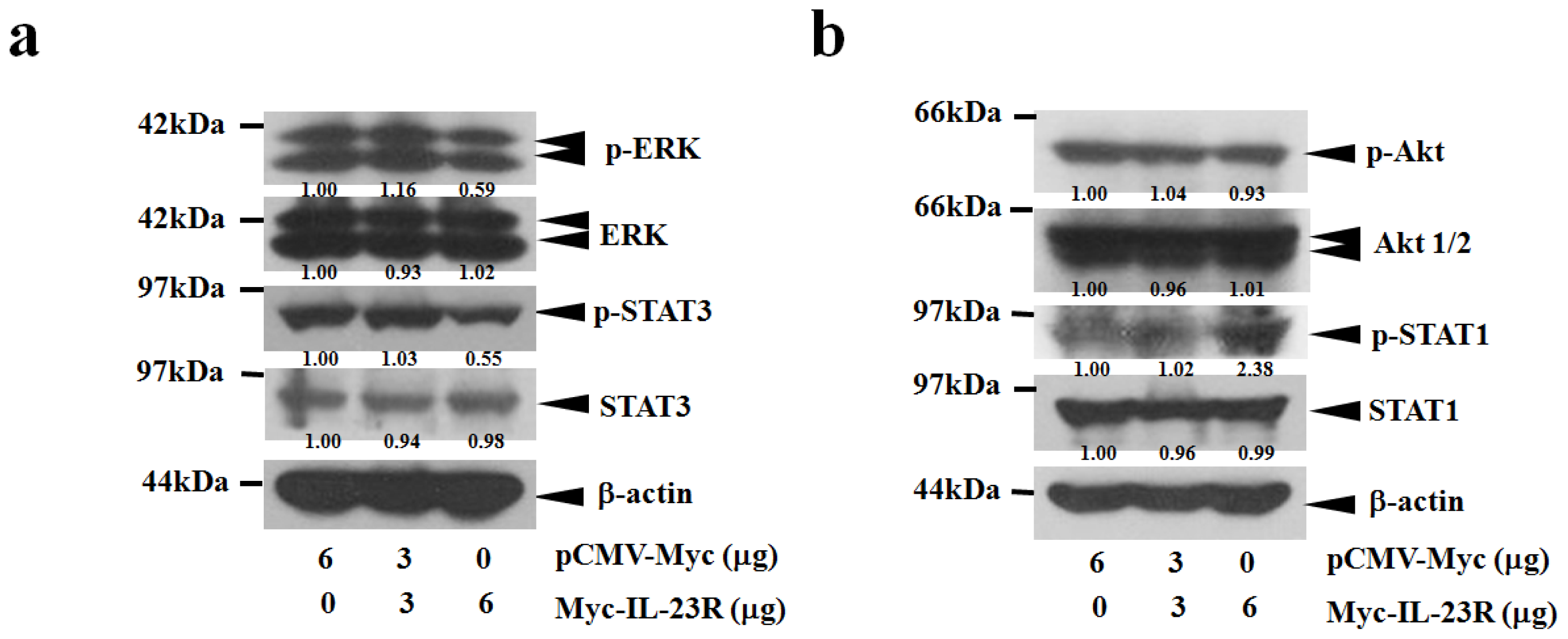
2.4. IL-23R Inhibits both RAS/MAPK and STAT3 Signaling Pathways in both 293ET and HeLa Cells
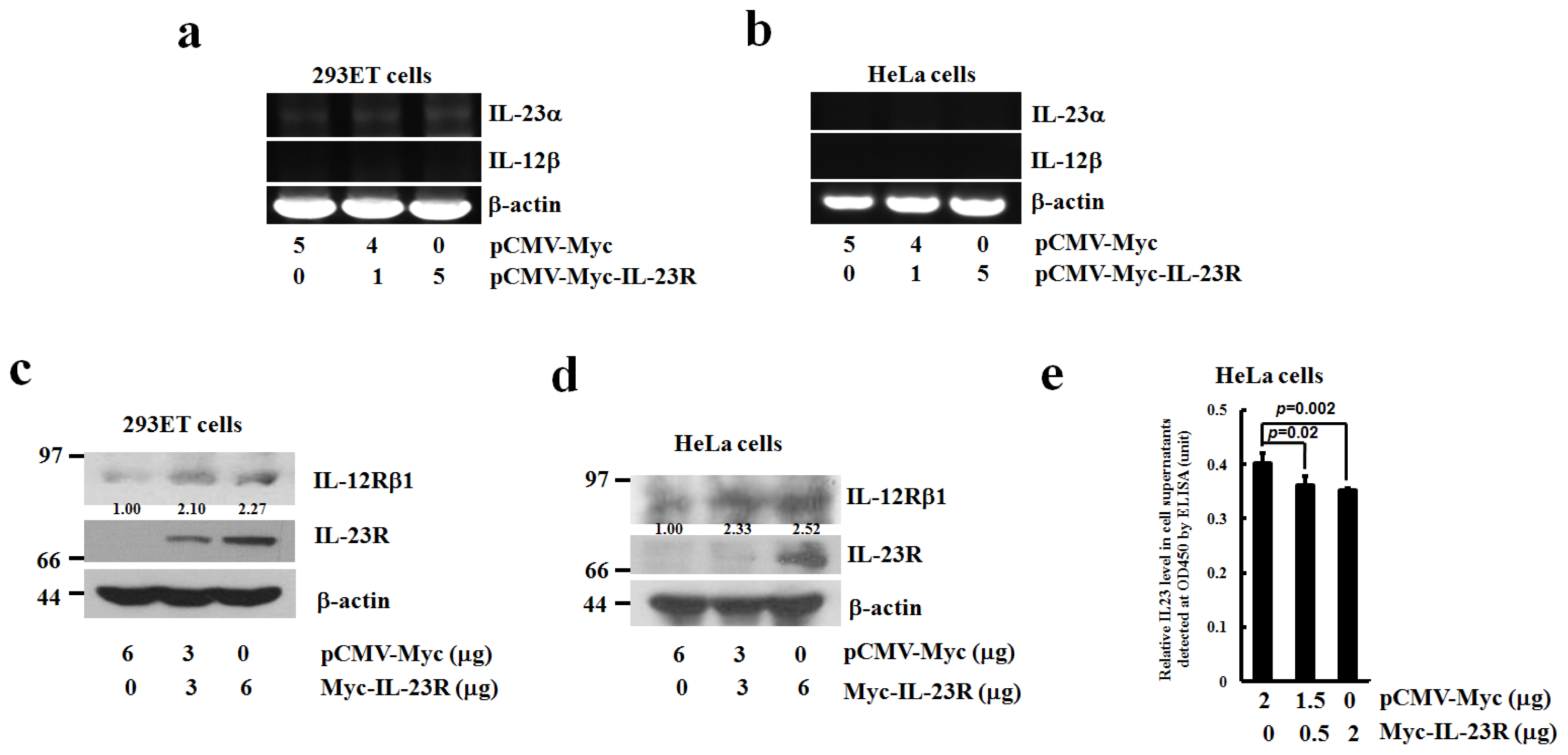
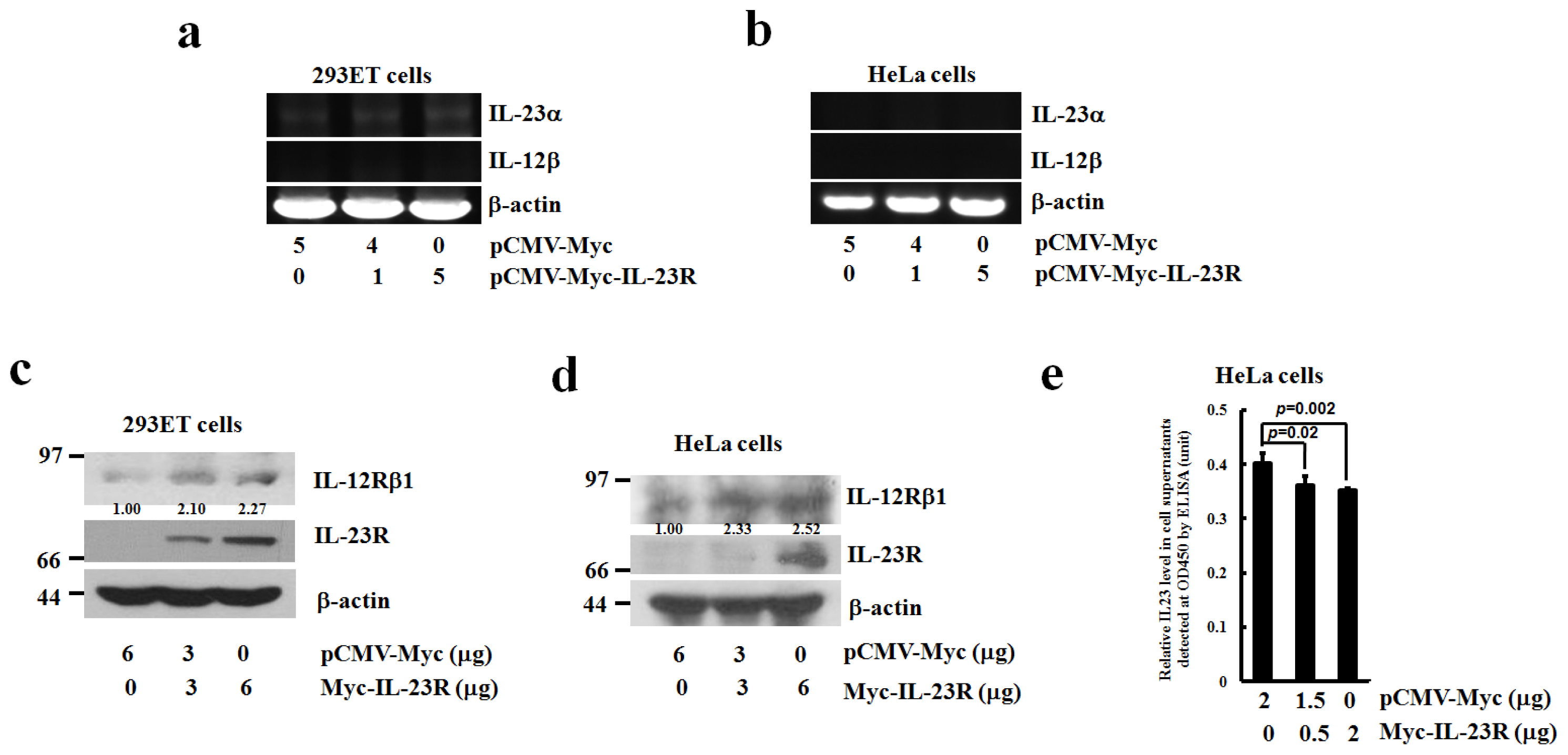
2.5. The Effects of IL-23R Overexpression on Endogenous Expressions of IL-23 Complex and IL-12Rβ1 in both 293ET and HeLa Cells
3. Experimental Section
3.1. Cell Lines, Chemicals and Reagents
3.2. Cell Culture and Transfection
3.3. Cell Proliferation Assay by Cell-Counting Kit-8 (CCK-8)
3.4. Flow Cytometric Analysis
3.5. Caspase 3/7 Assay
3.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
3.7. Western Blot Analysis
3.8. Enzyme Linked Immunosorbent Assay (ELISA)
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jones, L.L.; Vignali, D.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res 2011, 51, 5–14. [Google Scholar]
- van Wanrooij, R.L.; Zwiers, A.; Kraal, G.; Bouma, G. Genetic variations in interleukin-12 related genes in immune-mediated diseases. J. Autoimmun 2012, 39, 359–368. [Google Scholar]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol 2012, 13, 722–728. [Google Scholar]
- Chua, A.O.; Chizzonite, R.; Desai, B.B.; Truitt, T.P.; Nunes, P.; Minetti, L.J.; Warrier, R.R.; Presky, D.H.; Levine, J.F.; Gately, M.K.; et al. Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to gp130. J. Immunol 1994, 153, 128–136. [Google Scholar]
- Presky, D.H.; Yang, H.; Minetti, L.J.; Chua, A.O.; Nabavi, N.; Wu, C.Y.; Gately, M.K.; Gubler, U. A functional interleukin 12 receptor complex is composed of two β-type cytokine receptor subunits. Proc. Natl. Acad. Sci. USA 1996, 93, 14002–14007. [Google Scholar]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 prtein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol 2002, 168, 5699–5708. [Google Scholar]
- Pflanz, S.; Hibbert, L.; Mattson, J.; Rosales, R.; Vaisberg, E.; Bazan, J.F.; Phillips, J.H.; McClanahan, T.K.; de Waal Malefyt, R.; Kastelein, R.A. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol 2004, 172, 2225–2231. [Google Scholar]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol 2012, 13, 290–299. [Google Scholar]
- Zhang, X.Y.; Zhang, H.J.; Zhang, Y.; Fu, Y.J.; He, J.; Zhu, L.P.; Wang, S.H.; Liu, L. Identification and expression analysis of alternatively spliced isoforms of human interleukin-23 receptor gene in normal lymphoid cells and selected tumor cells. Immunogenetics 2006, 57, 934–943. [Google Scholar]
- Li, J.; Zhang, L.; Zhang, J.; Wei, Y.; Li, K.; Huang, L.; Zhang, S.; Gao, B.; Wang, X.; Lin, P. Interleukin 23 regulates proliferation of lung cancer cells in a concentration-dependent way in association with the interleukin-23 receptor. Carcinogenesis 2013, 34, 658–666. [Google Scholar]
- Tao, Y.P.; Wang, W.L.; Li, S.Y.; Zhang, J.; Shi, Q.Z.; Zhao, F.; Zhao, B.S. Associations between polymorphisms in IL-12A, IL-12B, IL-12Rβ1, IL-27 gene and serum levels of IL-12p40, IL-27p28 with esophageal cancer. J. Cancer Res. Clin. Oncol 2012, 138, 1891–1900. [Google Scholar]
- Ferretti, E.; Montagna, D.; Di Carlo, E.; Cocco, C.; Ribatti, D.; Ognio, E.; Sorrentino, C.; Lisini, D.; Bertaina, A.; Locatelli, F.; et al. Absence of IL-12Rβ2 in CD33(+)CD38(+) pediatric acute myeloid leukemia cells favours progression in NOD/SCID/IL2RgammaC-deficient mice. Leukemia 2012, 26, 225–235. [Google Scholar]
- Cardenes, M.; Angel-Moreno, A.; Fieschi, C.; Sologuren, I.; Colino, E.; Molines, A.; Garcia-Laorden, M.I.; Campos-Herrero, M.I.; Andujar-Sanchez, M.; Casanova, J.L.; et al. Oesophageal squamous cell carcinoma in a young adult with IL-12R beta 1 deficiency. J. Med. Genet 2010, 47, 635–637. [Google Scholar]
- Airoldi, I.; di Carlo, E.; Banelli, B.; Moserle, L.; Cocco, C.; Pezzolo, A.; Sorrentino, C.; Rossi, E.; Romani, M.; Amadori, A.; et al. The IL-12Rβ2 gene functions as a tumor suppressor in human B cell malignancies. J. Clin. Invest 2004, 113, 1651–1659. [Google Scholar]
- Ferretti, E.; di Carlo, E.; Cocco, C.; Ribatti, D.; Sorrentino, C.; Ognio, E.; Montagna, D.; Pistoia, V.; Airoldi, I. Direct inhibition of human acute myeloid leukemia cell growth by IL-12. Immunol. Lett 2010, 133, 99–105. [Google Scholar]
- Gorelik, E.; Edwards, R.P.; Feng, X.; Marrangoni, A.M.; Grandis, J.R.; Drenning, S.D.; Velikokhatnaya, L.; Kwon, J.A.; Lokshin, A.E. IL-12 receptor-mediated upregulation of FasL in human ovarian carcinoma cells. Int. J. Cancer 2004, 112, 620–627. [Google Scholar]
- Bubenik, J. Interleukin 12 in cancer treatment. Folia. Biol. (Praha) 2011, 57, 1–2. [Google Scholar]
- Engel, M.A.; Neurath, M.F. Anticancer properties of the IL-12 family—Focus on colorectal cancer. Curr. Med. Chem 2011, 17, 3303–3308. [Google Scholar]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res 2009, 15, 1126–1132. [Google Scholar]
- Sevilla, L.; Zaldumbide, A.; Pognonec, P.; Boulukos, K.E. Transcriptional regulation of the bcl-x gene encoding the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and AP1 transcription factor families. Histol. Histopathol 2001, 16, 595–601. [Google Scholar]
- Choi, H.J.; Smithgall, T.E. HIV-1 Nef promotes survival of TF-1 macrophages by inducing Bcl-XL expression in an extracellular signal-regulated kinase-dependent manner. J. Biol. Chem 2004, 279, 51688–51696. [Google Scholar]
- Jazirehi, A.R.; Vega, M.I.; Chatterjee, D.; Goodglick, L.; Bonavida, B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res 2004, 64, 7117–7126. [Google Scholar]
- Peng, C.L.; Guo, W.; Ji, T.; Ren, T.; Yang, Y.; Li, D.S.; Qu, H.Y.; Li, X.; Tang, S.; Yan, T.Q.; et al. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol. Ther 2009, 8, 1729–1736. [Google Scholar]
- Lu, X.; Tang, X.; Guo, W.; Ren, T.; Zhao, H. Sorafenib induces growth inhibition and apoptosis of human chondrosarcoma cells by blocking the RAF/ERK/MEK pathway. J. Surg. Oncol 2010, 102, 821–826. [Google Scholar]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Yokoyama, A. Inhibition of MEK signaling enhances the ability of cytarabine to induce growth arrest and apoptosis of acute myelogenous leukemia cells. Apoptosis 2009, 14, 1108–1120. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | GenBank ID | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Size of Product (bp) |
|---|---|---|---|---|
| β-actin | BC009275 | cacactgtgcccatctacga | ctgcttgctgatccacatct | 600 |
| IL-23a | NM_016584 | aagtggaagtgggcagagat | atccttgagctgctgccttt | 664 |
| IL-12β | NM_002187 | ttctggacgtttcacctgct | tttttgcggcagatgaccgt | 500 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shi, W.-Y.; Che, C.-Y.; Liu, L. Human Interleukin 23 Receptor Induces Cell Apoptosis in Mammalian Cells by Intrinsic Mitochondrial Pathway Associated with the Down-Regulation of RAS/Mitogen-Activated Protein Kinase and Signal Transducers and Activators of Transcription factor 3 Signaling Pathways. Int. J. Mol. Sci. 2013, 14, 24656-24669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms141224656
Shi W-Y, Che C-Y, Liu L. Human Interleukin 23 Receptor Induces Cell Apoptosis in Mammalian Cells by Intrinsic Mitochondrial Pathway Associated with the Down-Regulation of RAS/Mitogen-Activated Protein Kinase and Signal Transducers and Activators of Transcription factor 3 Signaling Pathways. International Journal of Molecular Sciences. 2013; 14(12):24656-24669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms141224656
Chicago/Turabian StyleShi, Wei-Ye, Chang-Yan Che, and Li Liu. 2013. "Human Interleukin 23 Receptor Induces Cell Apoptosis in Mammalian Cells by Intrinsic Mitochondrial Pathway Associated with the Down-Regulation of RAS/Mitogen-Activated Protein Kinase and Signal Transducers and Activators of Transcription factor 3 Signaling Pathways" International Journal of Molecular Sciences 14, no. 12: 24656-24669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms141224656