Novel Formulations for Antimicrobial Peptides


Abstract
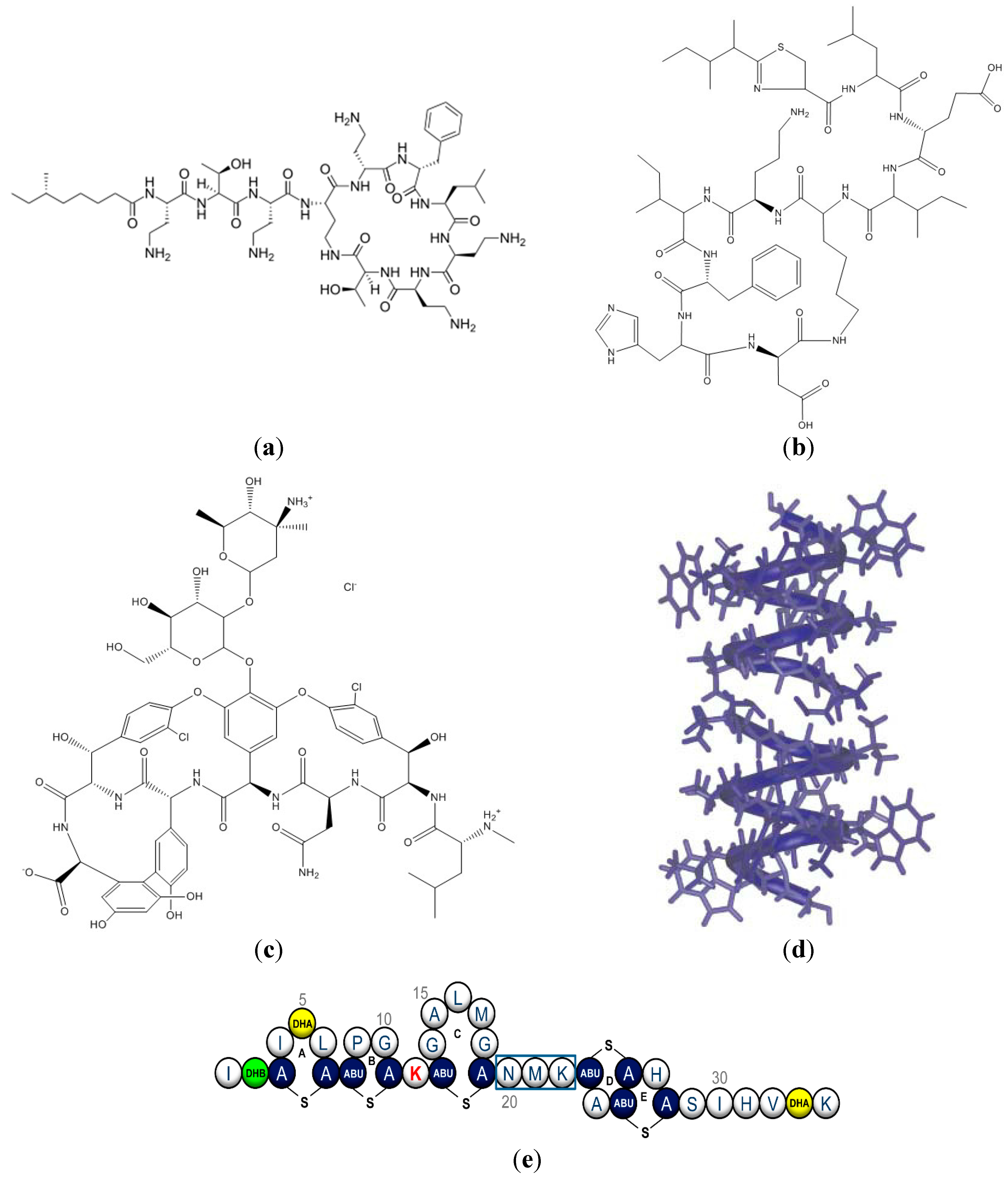
:1. Structure and Function of Antimicrobial Peptides



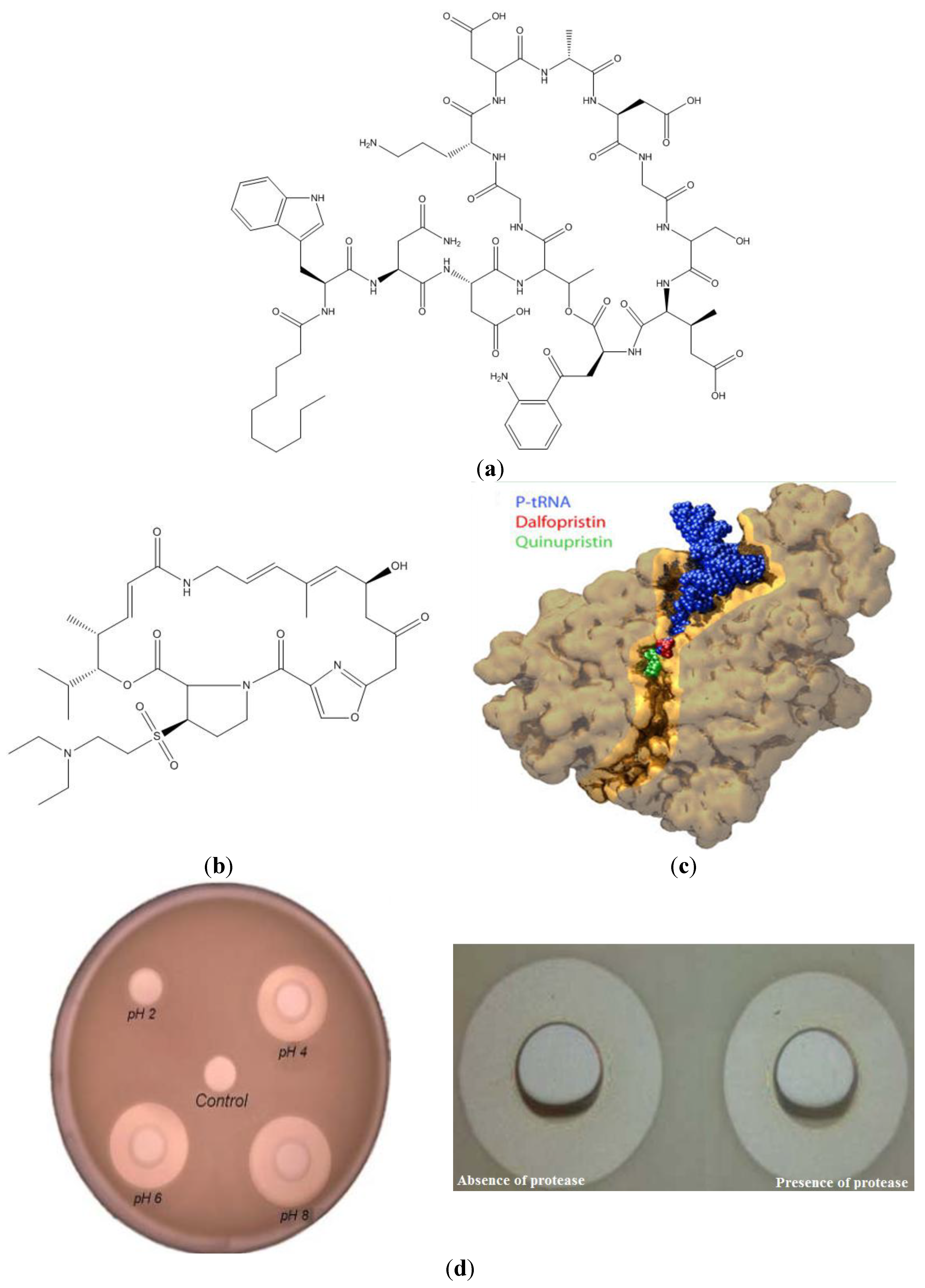
2. Novel Formulations for Peptides and the Cyclosporin Case

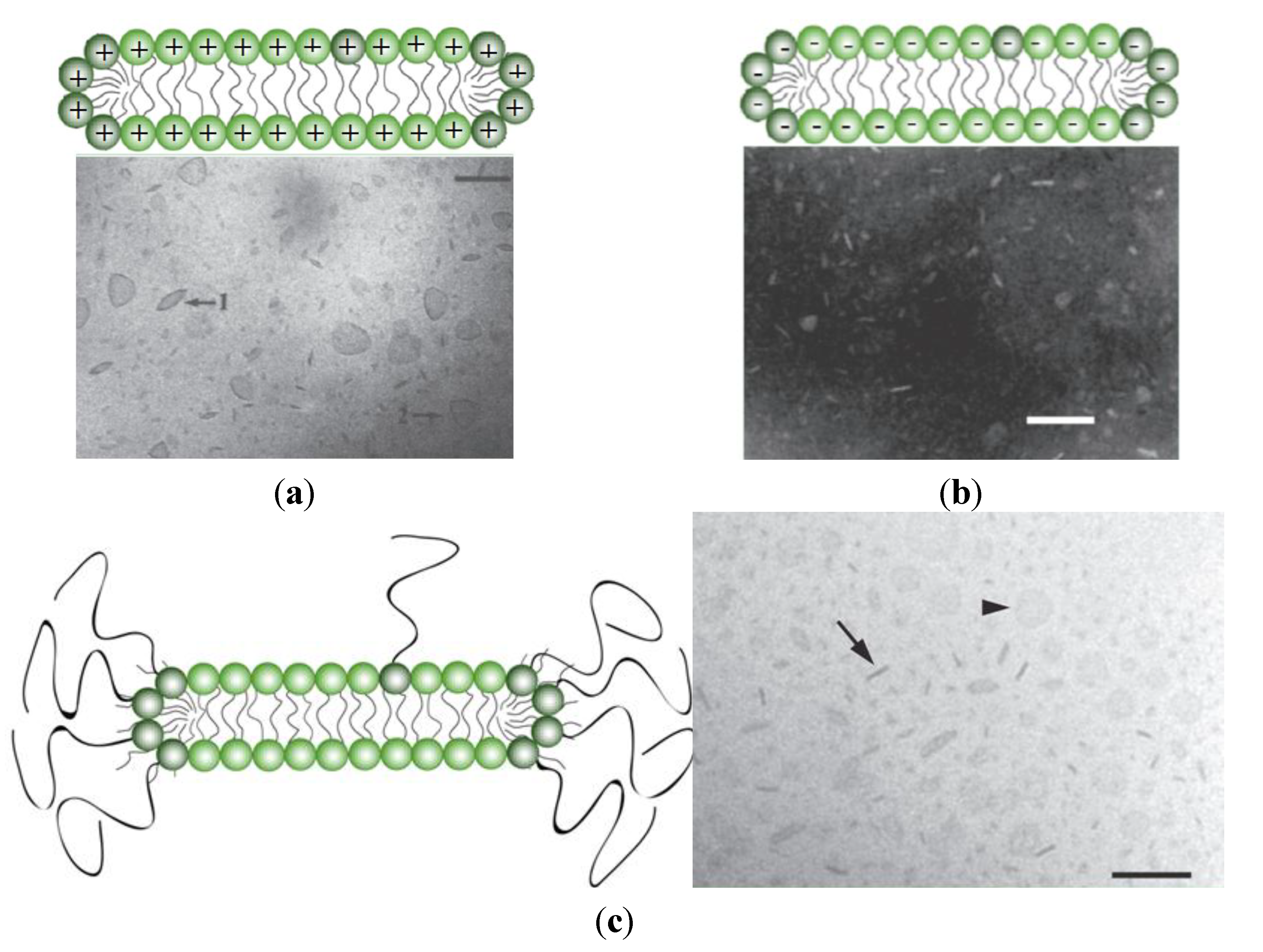
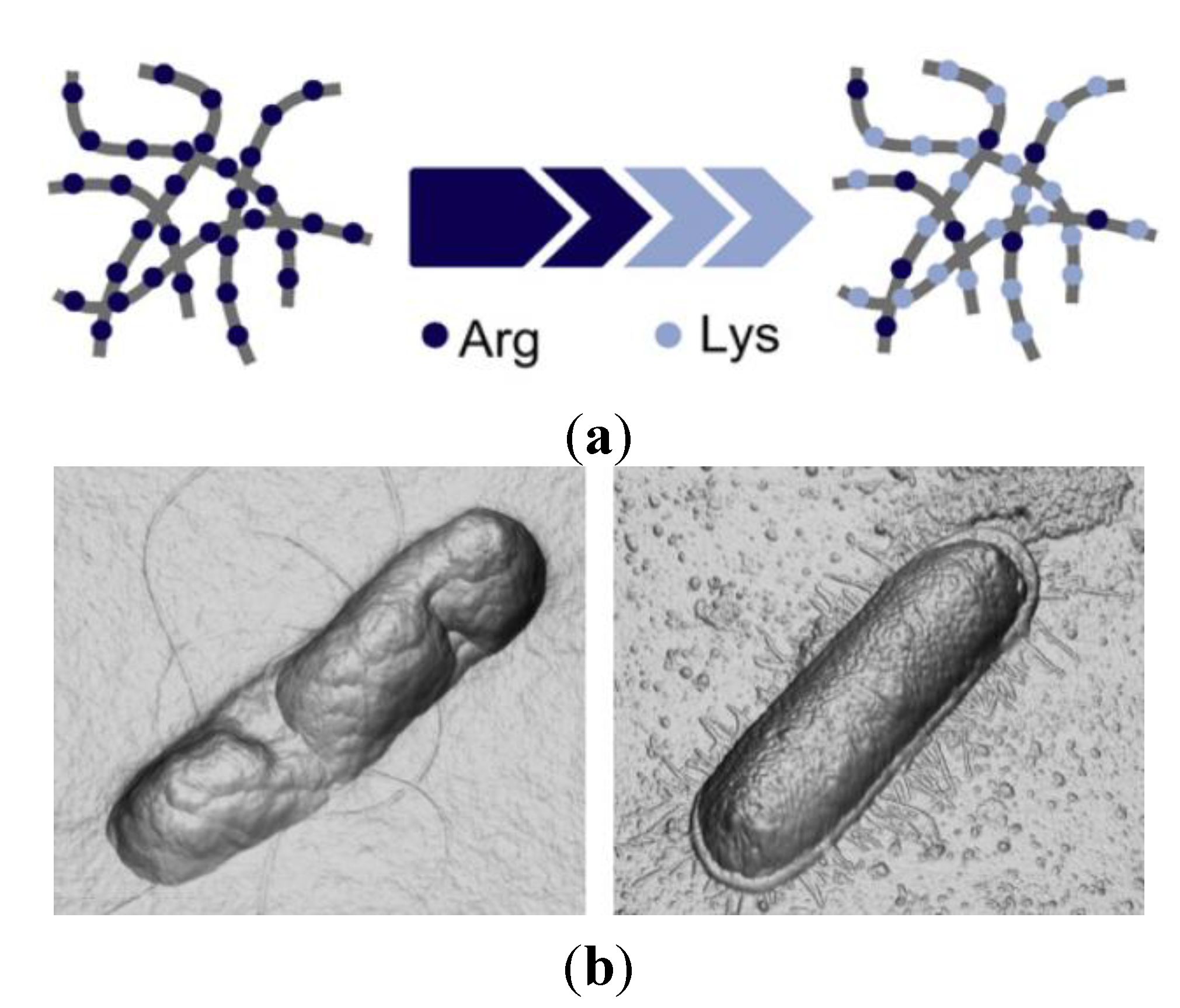
3. Novel Formulations for AMP




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | AMP | Spectrum of Activity | Indication | Ref. |
|---|---|---|---|---|
| Aerosol DMPC/DMPG liposomes | CM3 | P. aeruginosa | Pneumonia, lung infections | [164] |
| PEG-PLGA polymersomes | Lactoferrin | Not specified | Meningitis, brain-related infections | [219] |
| Liposomes | Nisin | Lactococcus lactis | Cheese manufacture | [180] |
| Liposomes | Nisin | L. monocytogenes | Cheese ripening | [178,179] |
| Fusogenic liposomes | Vancomycin | Gram-negative bacteria | Related infections | [191] |
| n-HA/CS/KGM scaffold for liposomes | Vancomycin | S. aureus | Osteomyelitis | [195] |
| DMGPC:Chol; DPPC:Chol liposomes | Polymyxin B | P. aeruginosa | Cystic fibrosis | [199] |
| DODAB liposome or bilayer disk | Gramicidin | E. coli, S. aureus | Related infections | [28] |
| POPC/cholesterol/ceramide-PEG5000 bilayer disk | Mellitin | E. coli | Related infections | [203] |
| Gelatin microspheres | AG-30 | P. aeruginosa, E. coli and S. aureus | Anti-ischaemia, angiogenic and antimicrobial | [223] |
| PGG nanoparticles | Nisin | L. monocytogenes | Food preservation | [227] |
| Hydroxypropyl cellulose gel | PXL150 | Gram-positive and Gram-negative bacteria, MRSA | Wound surgical site infections | [259] |
| Hydrogel + enzyme Dispersin B® | KSL-W | MRSA, S. epidermidis A. baumannii | Chronic wound infections with associated biofilm | [263] |
| Injectable peptidic hydrogel | PEP8R or derived with balanced arginine residues | MDR P. aeruginosa E. coli and S. aureus | Wound infections | [270] |
| Peptidic hydrogel + Ciprofloxacin | Tripeptide of Leu-Phe-Phe | S. aureus, E. coli and K. pneumoniae | Wound infections | [289] |
| Chewing gum | KSL | Oral bacterial pathogens | Dental plaque and caries | [294] |
| Chewing gum | KSL-W | Oral bacterial pathogens | Dental plaque and caries | [295] |
| Polystyrene fibers | Polymyxin B | Endotoxin of Gram-negative bacteria | Sepsis and septic shock | [298,299,300] |
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [PubMed]
- Fernández, L.; Hancock, R.E. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Fernández, L.; Hancock, R.E. Bacterial biofilm development as a multicellular adaptation: Antibiotic resistance and new therapeutic strategies. Curr. Opin. Microbiol. 2013, 16, 580–589. [Google Scholar]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta 2014, 1838, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [PubMed]
- Epand, R.F.; Mor, A.; Epand, R.M. Lipid complexes with cationic peptides and OAKs; Their role in antimicrobial action and in the delivery of antimicrobial agents. Cell Mol. Life Sci. 2011, 68, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M.; Lukic, M.L.; Flatt, P.R. Potential therapeutic applications of multifunctional host-defense peptides from frog skin as anti-cancer, anti-viral, immunomodulatory, and anti-diabetic agents. Peptides 2014, 57, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, A.F.; Vasconcelos, E.A.; Pelegrini, P.B.; Grossi de Sa, M.F. Antifungal defensins and their role in plant defense. Front. Microbiol. 2014, 5, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.; Gonçalves, S.; Santos, N.C. Defensins: Antifungal lessons from eukaryotes. Front. Microbiol. 2014, 5, 1–17. [Google Scholar] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Afacan, N.J.; Yeung, A.T.; Pena, O.M.; Hancock, R.E. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr. Pharm. Des. 2012, 18, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, J.; Marahiel, M.A. Chemoenzymatic and template-directed synthesis of bioactive macrocyclic peptides. Microbiol. Mol. Biol. Rev. 2006, 70, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Papagianni, M. Ribossomally synthesized peptides with antimicrobial properties: Biosynthesis, structure, function, and applications. Biotech. Adv. 2003, 21, 465–499. [Google Scholar] [CrossRef]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Economou, N.J.; Cocklin, S.; Loll, P.J. High-resolution crystal structure reveals molecular details of target recognition by bacitracin. Proc. Natl. Acad. Sci. USA 2013, 110, 14207–14212. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.M.; Anderson, J.M.; Marchant, R.E. Targeted delivery of vancomycin to Staphylococcus epidermidis biofilms using a fibrinogen-derived peptide. J. Biomed. Mater. Res. A 2012, 100, 2517–2525. [Google Scholar] [PubMed]
- Jordan, J.B.; Easton, P.L.; Hinton, J.F. Effects of phenylalanine substitutions in gramicidin A on the kinetics of channel formation in vesicles and channel structure in SDS micelles. Biophys. J. 2005, 88, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Molloy, E.M.; Field, D.; O’Connor, P.M.; Cotter, P.D.; Hill, C.; Ross, R.P. Saturation mutagenesis of lysine 12 leads to the identification of derivatives of nisin A with enhanced antimicrobial activity. PLoS One 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Biopolymers 1998, 47, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Cornut, I.; Thiaudière, E.; Dufourcq, J. The amphipathic helix in cytotoxic peptides. In The Amphipathic Helix; Epand, R.M., Ed.; CRC Press: Boca Raton, FL, USA, 1993; pp. 173–219. [Google Scholar]
- Marsh, D. Peptide models for membrane channels. Biochem. J. 1996, 315, 345–361. [Google Scholar] [PubMed]
- Pieta, P.; Mirza, J.; Lipkowski, J. Direct visualization of the alamethicin pore formed in a planar phospholipid matrix. Proc. Natl. Acad. Sci. USA 2012, 109, 21223–21227. [Google Scholar] [CrossRef] [PubMed]
- Ketchem, R.R.; Hu, W.; Cross, T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 1993, 261, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.A.; Olivares-Ortega, C.; Soto-Arriaza, M.A.; Carmona-Ribeiro, A.M. Interaction of gramicidin with DPPC/DODAB bilayer fragments. Biochim. Biophys. Acta 2012, 1818, 3064–3071. [Google Scholar] [CrossRef] [PubMed]
- Ragioto, D.A.M.T.; Carrasco, L.D.M.; Carmona-Ribeiro, A.M. Novel gramicidin formulations in cationic lipid as broad-spectrum microbicidal agents. Int. J. Nanomed. 2014, 9, 3183–3192. [Google Scholar]
- Wang, F.; Qin, L.; Pace, C.J.; Wong, P.; Malonis, R.; Gao, J. Solubilized gramicidin A as potential systemic antibiotics. Chembiochem 2012, 13, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.N.; Romo, T.D.; Grossfield, A. Simulating the mechanism of antimicrobial lipopeptides with all-atom molecular dynamics. Biochemistry 2013, 52, 5604–5610. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, P.; Biggin, P.C.; Tieleman, D.P.; Sansom, M.S. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta 1999, 1462, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.L.; Burton, M.; Grevis-James, A.; Hossain, M.A.; McArthur, S.; Palombo, E.A.; Wade, J.D.; Clayton, A.H. Imaging the action of antimicrobial peptides on living bacterial cells. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Shi, J.; Ceccarelli, A.; Kim, Y.H.; Park, A.; Ganz, T. Inhibition of neutrophil elastase prevents cathelicidin activation and impairs clearance of bacteria from wounds. Blood 2001, 97, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A.; Parra-Lopez, C.; Salcedo, M.; Lipps, C.J.; Heffron, F. Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc. Natl. Acad. Sci. USA 1992, 89, 11939–11943. [Google Scholar] [CrossRef] [PubMed]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef]
- Bals, R.; Weiner, D.; Meegalla, R.; Wilson, J. Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. J. Clin. Investig. 1999, 103, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef] [PubMed]
- De, Y.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Cherlov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schröder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R.E. Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Rudisill, J.A.; Lin, K.H.; Zhang, L.; Harris, S.M.; Falla, T.J.; Gallo, R.L. HB-107, a nonbacteriostatic fragment of the antimicrobial peptide cecropin B, accelerates murine wound repair. Wound Repair Regen. 2004, 12, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M. Host-defense peptides with therapeutic potential from skin secretions of frogs from the family pipidae. Pharmaceuticals 2014, 7, 58–77. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Gazdar, A.F.; Chen, H.C.; Johnson, B.E. Antitumor activity of magainin analogues against human lung cancer cell lines. Cancer Res. 1992, 52, 3534–3538. [Google Scholar] [PubMed]
- Lehmann, J.; Retz, M.; Sidhu, S.S.; Suttmann, H.; Sell, M.; Paulsen, F.; Harder, J.; Unteregger, G.; Stöckle, M. Antitumor activity of the antimicrobial peptide magainin II against bladder cancer cell lines. Eur. Urol. 2006, 50, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, R.A.; Barker, J.L.; Zasloff, M.; Chen, H.C.; Colamonici, O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc. Natl. Acad. Sci. USA. 1991, 88, 3792–3796. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M.; Prajeep, M.; Sonnevend, A.; Coquet, L.; Leprince, J.; Jouenne, T.; Vaudry, H.; King, J.D. Host-defense peptides in skin secretions of the tetraploid frog Silurana epitropicalis with potent activity against methicillin-resistant Staphylococcus aureus (MRSA). Peptides 2012, 37, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.C.; Lima, L.A.; Miranda, V.J.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013, 4, 321:1–321:56. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides—Challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Schnell, N.; Entian, K.D.; Schneider, U.; Gotz, F.; Zahner, H.; Kellner, R.; Jung, G. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 1988, 333, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Bierbaum, G.; Sahl, H.G. Lantibiotics: Mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 2009, 10, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.; Abriouel, H.; Lopez, R.L.; Ben Omar, N. Bacteriocin-based strategies for food biopreservation. Int. J. Food Microbiol. 2007, 120, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, A.; Mantella, A.; Goldstein, B.P.; Dei, R.; Benedetti, M.; Sbaragli, S.; Paradisi, F. In vitro activity of nisin against clinical isolates of Clostridium difficile. J. Chemother. 2004, 16, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Piper, C.; Draper, L.A.; Cotter, P.D.; Ross, R.P.; Hill, C. A comparison of the activities of lacticin3147 and nisin against drug-resistant Staphylococcus aureus and Enterococcus species. J. Antimicrob. Chemother. 2009, 64, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; McClean, S. Investigation of the cytotoxicity of eukaryotic and prokaryotic antimicrobial peptides in intestinal epithelial cells in vitro. Biochem. Pharmacol. 2006, 71, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Cresence, V.M.; Rejitha, J.S.; Lekshmi, M.U.; Dharsana, K.S.; Prasad, S.P.; Villa, H.M. Listeria—Review of epidemiology and pathogenesis. J. Microbiol. Immunol. Infect. 2007, 40, 4–13. [Google Scholar] [PubMed]
- Cintas, L.M.; Casaus, P.; Fernandez, M.F.; Hernandez, P.E. Comparative antimicrobial activity of enterocin L50, pediocin PA-1, nisin A and lactocin S against spoilage and foodborne pathogenic bacteria. Food Microbiol. 1998, 15, 289–298. [Google Scholar] [CrossRef]
- Millette, M.; Cornut, G.; Dupont, C.; Shareck, F.; Archambault, D.; Lacroix, M. Capacity of human nisin- and pediocin-producing lactic acid bacteria to reduce intestinal colonization by vancomycin-resistant enterococci. Appl. Environ. Microbiol. 2008, 74, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Cornut, G.; Fortin, C.; Soulières, D. Antineoplastic properties of bacteriocins: Revisiting potential active agents. Am. J. Clin. Oncol. 2008, 31, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Todorov, S.D.; Wachsman, M.; Tomé, E.; Dousset, X.; Destro, M.T.; Dicks, L.M.; Franco, B.D.; Vaz-Velho, M.; Drider, D. Characterisation of an antiviral pediocin-like bacteriocin produced by Enterococcus faecium. Food Microbiol. 2010, 27, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Lohans, C.T.; Vederas, J.C. Structural characterization of thioether-bridged bacteriocins. J. Antibiot. 2014, 67, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nissen-Meyer, J.; Rogne, P.; Oppegård, C.; Haugen, H.S.; Kristiansen, P.E. Structure-function relationships of the non-lanthionine-containing peptide (class II) bacteriocins produced by gram-positive bacteria. Curr. Pharm. Biotechnol. 2009, 10, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Drider, D.; Fimland, G.; Héchard, Y.; McMullen, L.M.; Prévost, H. The continuing story of class IIa bacteriocins. Microbiol. Mol. Biol. Rev. 2006, 70, 564–582. [Google Scholar] [CrossRef] [PubMed]
- Bionda, N.; Pitteloud, J.P.; Cudic, P. Cyclic lipodepsipeptides: A new class of antibacterial agents in the battle against resistant bacteria. Future Med. Chem. 2013, 5, 1311–1330. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.J.; Goult, C.M.; Donarski, J.A.; Micklefield, J.; Ramesh, V. NMR structure determination and calcium binding effects of lipopeptide antibiotic daptomycin. Org. Biomol. Chem. 2004, 2, 1872–1878. [Google Scholar] [CrossRef] [PubMed]
- Miao, V.; Coëffet-Legal, M.F.; Brian, P.; Brost, R.; Penn, J.; Whiting, A.; Martin, S.; Ford, R.; Parr, I.; Bouchard, M.; et al. Daptomycin biosynthesis in Streptomyces roseosporus: Cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 2005, 151, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.A.; Perlmutter, N.G.; Shapiro, H.M. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Woodford, N. Novel agents for the treatment of resistant Gram-positive infections. Expert Opin. Investig. Drugs 2003, 12, 117–137. [Google Scholar] [CrossRef]
- Kern, W.V. Daptomycin: First in a new class of antibiotics for complicated skin and soft-tissue infections. Int. J. Clin. Pract. 2006, 60, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, C.; Bettencourt, A. Daptomycin: A review of properties, clinical use, drug delivery and resistance. Mini Rev. Med. Chem. 2012, 12, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Arbeit, R.D.; Maki, D.; Tally, F.P.; Campanaro, E.; Eisenstein, B.I. The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin. Infect. Dis. 2004, 38, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Fowler, V.G.; Boucher, H.W.; Corey, G.R.; Abrutyn, E.; Karchmer, A.E.; Rupp, M.E.; Levine, D.P.; Chambers, H.F.; Tally, F.P.; Vigliani, G.A.; et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N. Engl. J. Med. 2006, 355, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, B.; Vijaysegaran, P.; Chaudhuri, A.; Crawford, S.; Ottley, M. Daptomycin resistance in prosthetic joint infections. Orthopedics 2012, 35, e603–e606. [Google Scholar] [CrossRef] [PubMed]
- Long, J.K.; Choueiri, T.K.; Hall, G.S.; Avery, R.K.; Sekeres, M.A. Daptomycin-resistant Enterococcus faecium in a patient with acute myeloid leukemia. Mayo Clin. Proc. 2005, 80, 1215–1216. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Bayer, A.S.; Tran, T.T.; Shamoo, Y.; Mileykovskaya, E.; Dowhan, W.; Guan, Z.; Arias, C.A. Daptomycin resistance in enterococci is associated with distinct alterations of cell membrane phospholipid content. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.B.; Jevitt, L.A.; Hageman, J.; McDonald, L.C.; Tenover, F.C. An association between reduced susceptibility to daptomycin and reduced susceptibility to vancomycin in Staphylococcus aureus. Clin. Infect. Dis. 2006, 42, 1652–1653. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Tominaga, E.; Neoh, H.M.; Hiramatsu, K. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Humphries, R.M.; Pollett, S.; Sakoulas, G. A current perspective on daptomycin for the clinical microbiologist. Clin. Microbiol. Rev. 2013, 26, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.W.; Greenough, A.; Burns, M.; Luteran, A.E.; McCafferty, D.G. Generation of ramoplanin resistant Staphylococcus aureus. FEMS Microbiol. Lett. 2010, 310, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Cocito, C.; di Giambattista, M.; Nyssen, E.; Vannuffel, P. Inhibition of protein synthesis by streptogramins and related antibiotics. J. Antimicrob. Chemother. 1997, 39, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.M.; Schlünzen, F.; Fucini, P.; Bartels, H.; Yonath, A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004, 2, 4:1–4:13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allignet, J.; Aubert, S.; Morvan, A.; el Solh, N. Distribution of genes encoding resistance to streptogramin A and related compounds among staphylococci resistant to these antibiotics. Antimicrob. Agents Chemother. 1996, 40, 2523–2528. [Google Scholar] [PubMed]
- Malbruny, B.; Canu, A.; Bozdogan, B.; Fantin, B.; Zarrouk, V.; Dutka-Malen, S.; Feger, C.; Leclercq, R. Resistance to quinupristin-dalfopristin due to mutation of L22 ribosomal protein in Staphylococcus aureus. Antimicrob. Agents Chemother. 2002, 46, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Chinali, G.; Moureau, P.; Cocito, C.G. The action of virginiamycin M on the acceptor, donor, and catalytic sites of peptidyltransferase. J. Biol. Chem. 1984, 259, 9563–9568. [Google Scholar] [PubMed]
- Arias, C.A.; Murray, B.E. The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 2012, 10, 266–278. [Google Scholar] [CrossRef]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; van Schepdael, A.; Orwa, J.A.; Roets, E.; Hoogmartens, J. Analysis of polymyxin B sulfate by capillary zone electrophoresis with cyclodextrin as additive. Method development and validation. J. Chromatogr. A 2000, 879, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Orwa, J.A.; Govaerts, C.; Busson, R.; Roets, E.; van Schepdael, A.; Hoogmartens, J. Isolation and structural characterization of polymyxin B components. J. Chromatogr. A 2001, 912, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J. Pharmacology of polymyxins: New insights into an “old” class of antibiotics. Future Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Clausell, A.; Garcia-Subirats, M.; Pujol, M.; Busquets, M.A.; Rabanal, F.; Cajal, Y. Gram-negative outer and inner membrane models: Insertion of cyclic cationic lipopeptides. J. Phys. Chem. B 2007, 111, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Jenssen, H.; Bains, M.; Wiegand, I.; Gooderham, W.J.; Hancock, R.E. The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob. Agents Chemother. 2012, 56, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Pueyo, M.T.; Mutafci, B.A.; Soto-Arriaza, M.A.; di Mascio, P.; Carmona-Ribeiro, A.M. The self-assembly of a cyclic lipopeptides mixture secreted by a B. megaterium strain and its implications on activity against a sensitive Bacillus species. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Pueyo, M.T.; Bloch, C., Jr.; Carmona-Ribeiro, A.M.; di Mascio, P. Lipopeptides produced by a soil Bacillus megaterium strain. Microb. Ecol. 2009, 57, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R. Road to clinical efficacy: Challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 2011, 6, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, F.; Gaggelli, E.; Molteni, E.; Porciatti, E.; Valensin, D.; Valensin, G. 1H- and 13C-NMR and molecular dynamics studies of cyclosporin a interacting with magnesium (II) or cerium (III) in acetonitrile. Conformational changes and cis-trans conversion of peptide bonds. Biophys. J. 2006, 90, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- Thell, K.; Hellinger, R.; Schabbauer, G.; Gruber, C.W. Immunosuppressive peptides and their therapeutic applications. Drug Discov. Today 2014, 19, 645–653. [Google Scholar] [CrossRef] [PubMed]
- De Mattos, A.M.; Olyaei, A.J.; Bennett, W.M. Nephrotoxicity of immunosuppressive drugs: Long-term consequences and challenges for the future. Am. J. Kidney Dis. 2000, 35, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.M.; Rhee, Y.S.; Wu, X. Nanomedicine in pulmonary delivery. Int. J. Nanomed. 2009, 4, 299–319. [Google Scholar] [CrossRef]
- Paranjpe, M.; Müller-Goymann, C.C. Nanoparticle-mediated pulmonary drug delivery: A review. Int. J. Mol. Sci. 2014, 15, 5852–5873. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Li, X.; Vogt, F.G.; Hayes, D., Jr.; Zwischenberger, J.B.; Park, E.S.; Mansour, H.M. Advanced spray-dried design, physicochemical characterization, and aerosol dispersion performance of vancomycin and clarithromycin multifunctional controlled release particles for targeted respiratory delivery as dry powder inhalation aerosols. Int. J. Pharm. 2013, 455, 374–392. [Google Scholar] [CrossRef]
- Mondon, K.; Zeisser-Labouèbe, M.; Gurny, R.; Möller, M. Novel cyclosporin A formulations using MPEG-hexyl-substituted polylactide micelles: A suitability study. Eur. J. Pharm. Biopharm. 2011, 77, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, Z.; Zhao, L.; Huang, L.; Yang, X.L.; Tang, J.; Feng, S.S. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, A. Oral cyclosporine A—The current picture of its liposomal and other delivery systems. Cell. Mol. Biol. Lett. 2009, 14, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Li, S. Advanced delivery of ciclosporin A: Present state and perspective. Expert Opin. Drug Deliv. 2007, 4, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P.R.; Chung, N.S.; Wasan, K.M. Cyclosporine A: A review of current oral and intravenous delivery systems. Drug Dev. Ind. Pharm. 2007, 33, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Degim, I.T.; Celebi, N. Controlled delivery of peptides and proteins. Curr. Pharm. Des. 2007, 13, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Italia, J.L.; Bhardwaj, V.; Kumar, M.N. Disease, destination, dose and delivery aspects of ciclosporin: The state of the art. Drug Discov. Today 2006, 11, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.J.; Sánchez, A. The potential of chitosan in ocular drug delivery. J. Pharm. Pharmacol. 2003, 55, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Delie, F.; Blanco-Príeto, M.J. Polymeric particulates to improve oral bioavailability of peptide drugs. Molecules 2005, 10, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, F.; Felt-Baeyens, O.; Besseghir, K.; Behar-Cohen, F.; Gurny, R. Cyclosporine A delivery to the eye: A pharmaceutical challenge. Eur. J. Pharm. Biopharm. 2003, 56, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, W.; Hayes, D., Jr.; Mansour, H.M. Physicochemical characterization and aerosol dispersion performance of organic solution advanced spray-dried cyclosporine A multifunctional particles for dry powder inhalation aerosol delivery. Int. J. Nanomed. 2013, 8, 1269–1283. [Google Scholar]
- Zhang, L.; Zhao, Z.L.; Wei, X.H.; Liu, J.H. Preparation and in vitro and in vivo characterization of cyclosporine A-loaded, PEGylated chitosan-modified, lipid-based nanoparticles. Int. J. Nanomed. 2013, 8, 601–610. [Google Scholar]
- Di Tommaso, C.; Torriglia, A.; Furrer, P.; Behar-Cohen, F.; Gurny, R.; Möller, M. Ocular biocompatibility of novel Cyclosporin A formulations based on methoxypoly (ethylene glycol)-hexylsubstituted poly (lactide) micelle carriers. Int. J. Pharm. 2011, 416, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Azzi, J.; Kwon, M.; Mounayar, M.; Tong, R.; Yin, Q.; Moore, R.; Skartsis, N.; Fan, T.M.; Abdi, R.; et al. Immunosuppressive activity of size-controlled PEG-PLGA nanoparticles containing encapsulated cyclosporine A. J. Transplant. 2012, 2012. [Google Scholar] [CrossRef]
- Gref, R.; Quellec, P.; Sanchez, A.; Calvo, P.; Dellacherie, E.; Alonso, M.J. Development and characterization of CyA-loaded poly (lactic acid)-poly (ethylene glycol) PEG micro- and nanoparticles. Comparison with conventional PLA particulate carriers. Eur. J. Pharm. Biopharm. 2001, 51, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, C.; Bourges, J.L.; Valamanesh, F.; Trubitsyn, G.; Torriglia, A.; Jeanny, J.C.; Behar-Cohen, F.; Gurny, R.; Möller, M. Novel micelle carriers for cyclosporin A topical ocular delivery: In vivo cornea penetration, ocular distribution and efficacy studies. Eur. J. Pharm. Biopharm. 2012, 81, 257–264. [Google Scholar] [CrossRef] [PubMed]
- El Tayar, N.; Mark, A.E.; Vallat, P.; Brunne, R.M.; Testa, B.; van Gunsteren, W.F. Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: Evidence from partition coefficients and molecular dynamics simulations. J. Med. Chem. 1993, 36, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Donnenfeld, E.; Pflugfelder, S.C. Topical ophthalmic cyclosporine: Pharmacology and clinical uses. Surv. Ophthalmol. 2009, 54, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Utine, C.A.; Stern, M.; Akpek, E.K. Clinical review: Topical ophthalmic use of cyclosporin A. Ocul. Immunol. Inflamm. 2010, 18, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, C.; Valamanesh, F.; Miller, F.; Furrer, P.; Rodriguez-Aller, M.; Behar-Cohen, F.; Gurny, R.; Möller, M. A novel cyclosporin a aqueous formulation for dry eye treatment: In vitro and in vivo evaluation. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2292–2299. [Google Scholar] [CrossRef]
- Daull, P.; Lallemand, F.; Philips, B.; Lambert, G.; Buggage, R.; Garrigue, J.S. Distribution of cyclosporine A in ocular tissues after topical administration of cyclosporine A cationic emulsions to pigmented rabbits. Cornea 2013, 32, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, F.; Daull, P.; Benita, S.; Buggage, R.; Garrigue, J.S. Successfully improving ocular drug delivery using the cationic nanoemulsion, novasorb. J. Drug Deliv. 2012. [Google Scholar] [CrossRef]
- Tamilvanan, S.; Benita, S. The potential of lipid emulsion for ocular delivery of lipophilic drugs. Eur. J. Pharm. Biopharm. 2004, 58, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Luschmann, C.; Herrmann, W.; Strauss, O.; Luschmann, K.; Goepferich, A. Ocular delivery systems for poorly soluble drugs: An in vivo evaluation. Int. J. Pharm. 2013, 455, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Klyashchitsky, B.A.; Owen, A.J. Drug delivery systems for cyclosporine: Achievements and complications. J. Drug Target. 1998, 5, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Stuhne-Sekalec, L.; Stanacev, N.Z. Liposomes as carriers of cyclosporin A. J. Microencapsul. 1991, 8, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Al-Angary, A.A.; Bayomi, M.A.; Khidr, S.H.; Al-Meshal, M.A.; Al-Dardiri, M. Characterization, stability and in vivo targeting of liposomal formulations containing cyclosporin. Int. J. Pharm. 1995, 114, 221–225. [Google Scholar] [CrossRef]
- Ouyang, C.; Choice, E.; Holland, J.; Meloche, M.; Madden, T.D. Liposomal cyclosporine. Characterization of drug incorporation and interbilayer exchange. Transplantation 1995, 60, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Al-Meshal, M.A.; Khidr, S.H.; Bayomi, M.A.; Al-Angary, A.A. Oral administration of liposomes containing cyclosporin: A pharmacokinetic study. Int. J. Pharm. 1998, 168, 163–168. [Google Scholar] [CrossRef]
- Lee, M.K.; Choi, L.; Kim, M.H.; Kim, C.K. Pharmacokinetics and organ distribution of cyclosporin A incorporated in liposomes and mixed micelles. Int. J. Pharm. 1999, 191, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ping, Q.; Chen, Y. Pharmacokinetic behavior of cyclosporin A in rabbits by oral administration of lecithin vesicle and Sandimmun Neoral. Int. J. Pharm. 2001, 216, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Lu, Y.; Qi, J.; Niu, M.; Lian, R.; Hu, F.; Wu, W. Enhanced oral bioavailability of cyclosporin A by liposomes containing a bile salt. Int. J. Nanomed. 2011, 6, 965–974. [Google Scholar]
- Karn, P.R.; Cho, W.; Park, H.J.; Park, J.S.; Hwang, S.J. Characterization and stability studies of a novel liposomal cyclosporin A prepared using the supercritical fluid method: Comparison with the modified conventional Bangham method. Int. J. Nanomed. 2013, 8, 365–377. [Google Scholar]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv. 2011. [Google Scholar] [CrossRef]
- Charkoftaki, G.; Kytariolos, J.; Macheras, P. Novel milk-based oral formulations: Proof of concept. Int. J. Pharm. 2010, 390, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yordanov, G. Nanocarriers for antibiotics. In Nanotechnology for Animal Health and Production; Garg, S.R., Ed.; Daya Publishing House: New Delhi, India, 2014; pp. 124–134. [Google Scholar]
- Drevets, D.; Leenen, P.; Greenfield, R. Invasion of the central nervous system by intracellular bacteria. Clin. Microbiol. Rev. 2004, 17, 323–347. [Google Scholar] [CrossRef] [PubMed]
- Garcia-del Portillo, F.; Finlay, B.B. The varied lifestyles of intracellular pathogens within eukaryotic vacuolar compartments. Trends Microbiol. 1995, 3, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M. Cationic nanostructures for vaccines. In Immune Response Activation; Duc, G.H.T., Ed.; InTech: Rijeka, Croatia, 2014; pp. 3–43. [Google Scholar]
- Aggarwal, P.; Hall, J.; McLeland, C.; Dobrovolskaia, M.; McNeil, S. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M. Interactions between bilayer vesicles, biomolecules, and interfaces. In Handbook of Surfaces and Interfaces of Materials; Nalwa, H.S., Ed.; Academic Press: Burlington, VT, USA, 2001; pp. 129–165. [Google Scholar]
- Sahay, G.; Alakhova, D.; Kabanov, A. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.A.; Carmona-Ribeiro, A.M. Interactions between cationic vesicles and serum proteins. Langmuir 1998, 14, 6077–6081. [Google Scholar] [CrossRef]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Briones, E.; Colino, C.; Lanao, J. Delivery systems to increase the selectivity of antibiotics in phagocytic cells. J. Control. Release 2008, 125, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, M.; Gatti, G.; Lazzarini, L.; Furlan, G.; Broccali, G.; Malena, M.; Franchini, C.; Concia, E. Penetration of vancomycin into human lung tissue. J. Antimicrob. Chemother. 1996, 38, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Pumerantz, A.; Muppidi, K.; Agnihotri, S.; Guerra, C.; Venketaraman, V.; Wang, J.; Betageri, G. Preparation of liposomal vancomycin and intracellular killing of meticillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2011, 37, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Malmsten, M.; Kasetty, G.; Pasupuleti, M.; Alenfall, J.; Schmidtchen, A. Highly selective end-tagged antimicrobial peptides derived from PRELP. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Kabanov, A.V. Brain delivery of proteins via their fatty acid and block copolymer modifications. J. Drug Target. 2013, 21, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.A.; Carmona-Ribeiro, A.M.; Petri, D.F. Catalytic behavior of lipase immobilized onto Congo red and PEG-decorated particles. Molecules 2014, 19, 8610–8628. [Google Scholar]
- Silva, R.A.; Carmona-Ribeiro, A.M.; Petri, D.F. Enzymatic activity of cholesterol oxidase immobilized onto polymer nanoparticles mediated by Congo red. Colloids Surf. B Biointerfaces 2013, 110, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Leão-Silva, A.C.; Naves, A.F.; Pereira, E.M.; Petri, D.F.; Carmona-Ribeiro, A.M. Assembly of horseradish peroxidase within supported cationic bilayers. Biotechnol. Prog. 2011, 27, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Naves, A.F.; Carmona-Ribeiro, A.M.; Petri, D.F. Immobilized horseradish peroxidase as a reusable catalyst for emulsion polymerization. Langmuir 2007, 23, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Haynie, S.L.; Crum, G.A.; Doele, B.A. Antimicrobial activities of amphiphilic peptides covalently bonded to a water-insoluble resin. Antimicrob. Agents Chemother. 1995, 39, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Beyermann, M.; Dathe, M. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrum. Antimicrob. Agents Chemother. 2009, 53, 1132–1141. [Google Scholar] [PubMed]
- Smola, M.; Vandamme, T.; Sokolowski, A. Nanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseases. Int. J. Nanomed. 2008, 3, 1–19. [Google Scholar] [CrossRef]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.F.; Hancock, R.E.W.; Samuel, J.; Finlay, W.H. In vitro aerosol delivery and regional airway surface liquid concentration of a liposomal cationic peptide. J. Pharm. Sci. 2001, 90, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.R.; Tyrrell, G.J.; Ng, T.; Finlay, W.H. In vitro evaluation of nebulization properties, antimicrobial activity, and regional airway surface liquid concentration of liposomal polymyxin B sulfate. Pharm. Res. 2003, 20, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Shim, W.S.; Cui, F.D.; Cheng, G.; Han, X.; Jin, Q.R.; Kim, D.D.; Chung, S.J.; Shim, C.K. Enhanced electrostatic interaction between chitosan-modified PLGA nanoparticle and tumor. Int. J. Pharm. 2009, 371, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Choi, J.S.; Kim, I.; Oh, K.T.; Lee, E.S.; Park, E.S.; Lee, K.C.; Youn, Y.S. Long-acting inhalable chitosan-coated poly (lactic-co-glycolic acid) nanoparticles containing hydrophobically modified exendin-4 for treating type 2 diabetes. Int. J. Nanomed. 2013, 8, 2975–2983. [Google Scholar]
- Li, Z.; Zhou, Z.; Huang, G.; Hu, F.; Xiang, Y.; He, L. Exendin-4 protects mitochondria from reactive oxygen species induced apoptosis in pancreatic Beta cells. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Makhlof, A.; Werle, M.; Tozuka, Y.; Takeuchi, H. Nanoparticles of glycol chitosan and its thiolated derivative significantly improved the pulmonary delivery of calcitonin. Int. J. Pharm. 2010, 397, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kuno, Y.; Sugimoto, S.; Takeuchi, H.; Kawashima, Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J. Control. Release 2005, 102, 373–381. [Google Scholar] [CrossRef]
- Zhang, Q.; Shen, Z.; Nagai, T. Prolonged hypoglycemic effect of insulin-loaded polybutylcyanoacrylate nanoparticles after pulmonary administration to normal rats. Int. J. Pharm. 2001, 218, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Dębek, C.; Olędzka, E.; Kozłowski, R. Polymeric systems of antimicrobial peptides—Strategies and potential applications. Molecules 2013, 18, 14122–14137. [Google Scholar] [CrossRef] [PubMed]
- Sobrino-López, A.; Martín-Belloso, O. Use of nisin and other bacteriocins for preservation of dairy products. Int. Dairy J. 2008, 18, 329–343. [Google Scholar] [CrossRef]
- Blanco-Padilla, A.; Soto, K.M.; Iturriaga, M.H.; Mendoza, S. Food antimicrobials nanocarriers. Sci. World J. 2014. [Google Scholar] [CrossRef]
- Da Silva Malheiros, P.; Daroit, D.J.; Brandelli, A. Food applications of liposome-encapsulated antimicrobial peptides. Trends Food Sci. Technol. 2010, 21, 284–292. [Google Scholar] [CrossRef]
- Benech, R.O.; Kheadr, E.E.; Lacroix, C.; Fliss, I. Antibacterial activities of nisin Z encapsulated in liposomes or produced in situ by mixed culture during cheddar cheese ripening. Appl. Environ. Microbiol. 2002, 68, 5607–5619. [Google Scholar] [CrossRef] [PubMed]
- Benech, R.O.; Kheadr, E.E.; Lacroix, C.; Fliss, I. Impactof nisin producing culture and liposome-encapsulated nisin on ripening of Lactobacillus added-cheddar cheese. J. Dairy Sci. 2003, 86, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Benech, R.O.; Kheadr, E.E.; Laridi, R.; Lacroix, C.; Fliss, I. Inhibition of Listeria innocua in cheddar cheese by addition of nisin Z in liposomes or by in situ production in mixed culture. Appl. Environ. Microbiol. 2002, 68, 3683–3690. [Google Scholar] [CrossRef] [PubMed]
- Laridi, R.; Kheadr, E.E.; Benech, R.O.; Vuillemard, J.C.; Lacroix, C.; Fliss, I. Liposome encapsulated nisin Z: Optimization, stability and release during milk fermentation. Int. Dairy J. 2003, 13, 325–336. [Google Scholar] [CrossRef]
- Prombutara, P.; Kulwatthanasal, Y.; Supaka, N.; Sramala, I.; Chareonpornwattana, S. Production of nisin-loaded solid lipid nanoparticles for sustained antimicrobial activity. Food Control 2012, 24, 184–190. [Google Scholar] [CrossRef]
- Chopra, M.; Kaur, P.; Bernela, M.; Thakur, R. Surfactant assisted nisin loaded chitosan-carageenan nanocapsule synthesis for controlling food pathogens. Food Control 2014, 37, 158–164. [Google Scholar] [CrossRef]
- Xiao, D.; Davidson, P.M.; Zhong, Q. Release and antilisterial properties of nisin from zein capsules spray-dried at different temperatures. LWT Food Sci. Technol. 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Colas, J.C.; Shi, W.; Rao, V.S.N.M.; Omri, A.; Mozafari, M.R.; Singh, H. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron 2007, 38, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Were, L.M.; Bruce, B.; Davidson, P.M.; Weiss, J. Encapsulation of nisin and lysozyme in liposomes enhances efficacy against Listeria monocytogenes. J. Food Prot. 2004, 67, 922–927. [Google Scholar] [PubMed]
- Malheiros, P.S.; Daroit, D.J.; Brandelli, A. Inhibition of Listeria monocytogenes in minas frescal cheese by free and nanovesicle-encapsulated nisin. Braz. J. Microbiol. 2012, 43, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Malheiros, P.S.; Sant’Anna, V.; Barbosa, M.S.; Brandelli, A.; Franco, B.D. Effect of liposome-encapsulated nisin and bacteriocin-like substance P34 on Listeria monocytogenes growth in Minas frescal cheese. Int. J. Food Microbiol. 2012, 156, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Martin-Visscher, L.A.; Yoganathan, S.; Sit, C.S.; Lohans, C.T.; Vederas, J.C. The activity of bacteriocins from Carnobacterium maltaromaticum UAL307 against gram-negative bacteria in combination with EDTA treatment. FEMS Microbiol. Lett. 2011, 317, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Prabha, V.; Rishi, P. Value addition in the efficacy of conventional antibiotics by nisin against Salmonella. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, Z.; Zhao, W.; Lu, T.; Wang, R.; Mei, Q.; Chen, T. Enhanced bactericidal potency of nanoliposomes by modification of the fusion activity between liposomes and bacterium. Int. J. Nanomed. 2013, 8, 2351–2360. [Google Scholar] [CrossRef]
- Nicolosi, D.; Scalia, M.; Nicolosi, V.M.; Pignatello, R. Encapsulation in fusogenic liposomes broadens the spectrum of action of vancomycin against Gram-negative bacteria. Int. J. Antimicrob. Agents 2010, 35, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Pumerantz, A.S. PEGylated liposomal vancomycin: A glimmer of hope for improving treatment outcomes in MRSA pneumonia. Recent Pat. Antiinfect. Drug Discov. 2012, 7, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Sande, L.; Sanchez, M.; Montes, J.; Wolf, A.J.; Morgan, M.A.; Omri, A.; Liu, G.Y. Liposomal encapsulation of vancomycin improves killing of methicillin-resistant Staphylococcus aureus in a murine infection model. J. Antimicrob. Chemother. 2012, 67, 2191–2194. [Google Scholar] [PubMed]
- Onyeji, C.O.; Nightingale, C.H.; Marangos, M.N. Enhanced killing of methicillin-resistant Staphylococcus aureus in human macrophages by liposome-entrapped vancomycin and teicoplanin. Infection 1994, 22, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Shang, B.C.; Tang, H.; Zhou, T.H.; Xu, G.L.; Li, H.L.; Chen, Q.H.; Xu, Y.Q. Nano-hydroxyapatite/chitosan/konjac glucomannan scaffolds loaded with cationic liposomal vancomycin: Preparation, in vitro release and activity against Staphylococcus aureus biofilms. J. Biomater. Sci. Polym. Ed. 2011, 22, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E. Liposomal antibiotic formulations for targeting the lungs in the treatment of Pseudomonas aeruginosa. Ther. Deliv. 2014, 5, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Halwani, M.; Omri, A.; Suntres, Z.E. Antimicrobial effectiveness of liposomal polymyxin B against resistant Gram-negative bacterial strains. Int. J. Pharm. 2008, 355, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Omri, A.; Suntres, Z.E.; Shek, P.N. Enhanced activity of liposomal polymyxin B against Pseudomonas aeruginosa in a rat model of lung infection. Biochem. Pharmacol. 2002, 64, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E.; Halwani, M.; Azghani, A.O.; Omri, A. Activity and interactions of liposomal antibiotics in presence of polyanions and sputum of patients with cystic fibrosis. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Abdelraouf, K.; Ledesma, K.R.; Chow, D.S.; Tam, V.H. Pharmacokinetics and efficacy of liposomal polymyxin B in a murine pneumonia model. Int. J. Antimicrob. Agents. 2013, 42, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M. Biomimetic nanoparticles: Preparation, characterization and biomedical applications. Int. J. Nanomed. 2010, 5, 249–259. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Lipid bilayer fragments and disks in drug delivery. Curr. Med. Chem. 2006, 13, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, M.M.; Reijmar, K.; Pränting, M.; Engström, A.; Andersson, D.I.; Edwards, K. PEG-stabilized lipid disks as carriers for amphiphilic antimicrobial peptides. J. Control. Release 2011, 156, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, A.; Wessman, P.; Rennie, A.R.; Edwards, K. Melittin-lipid interaction: A comparative study using liposomes, micelles and bilayer disks. Biochim. Biophys. Acta 2008, 1778, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Engvall, C.; Arfvidsson, M.; Lundahl, P.; Edwards, K. Development and initial evaluation of PEG-stabilized bilayer disks as novel model membranes. Biophys. Chem. 2005, 113, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Campanhã, M.T.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Interactions between cationic liposomes and bacteria: The physical-chemistry of the bactericidal action. J. Lipid Res. 1999, 40, 1495–1500. [Google Scholar] [PubMed]
- Martins, L.M.S.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Cationic vesicles as bactericides. Langmuir 1997, 13, 5583–5587. [Google Scholar] [CrossRef]
- Melo, L.D.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Antimicrobial particles from cationic lipid and polyelectrolytes. Langmuir 2010, 26, 12300–12306. [Google Scholar] [CrossRef] [PubMed]
- Campanhã, M.T.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Interactions between cationic vesicles and Candida albicans. J. Phys. Chem. B 2001, 105, 8230–8236. [Google Scholar] [CrossRef]
- Melo, L.D.; Carmona-Ribeiro, A.M. Fungicidal nanoparticles of low toxicity from cationic lipid and polyelectrolytes. NSTI Nanotech. 2012, 3, 350–353. [Google Scholar]
- Carmona-Ribeiro, A.M.; Carrasco, L.D.M. Fungicidal assemblies and their mode of action. OA Biotechnol. 2013, 2, 1–8. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Hammarstrom, L.; Edwards, K. Effect of bilayer phase transitions on vesicle structure, and its influence on the kinetics of viologen reduction. J. Phys. Chem. 1995, 99, 14531–14538. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Castuma, C.E.; Sesso, A.; Schreier, S. Bilayer structure and stability in dihexadecyl phosphate dispersions. J. Phys. Chem. 1991, 95, 5361–5366. [Google Scholar] [CrossRef]
- Wessman, P.; Morin, M.; Reijmar, K.; Edwards, K. Effect of alpha-helical peptides on liposome structure: A comparative study of melittin and alamethicin. J. Colloid Interface Sci. 2010, 346, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Rex, S.; Schwarz, G. Quantitative studies on the melittin-induced leakage mechanism of lipid vesicles. Biochemistry 1998, 37, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Effect of micellar charge on the conformation and dynamics of melittin. Eur. Biophys. J. 2004, 33, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Lauterwein, J.; Bosch, C.; Brown, L.R.; Wuthrich, K. Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim. Biophys. Acta 1979, 556, 244–264. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pang, Z.; Lu, W.; Yin, Q.; Gao, H.; Jiang, X. Self-assembled polymersomes conjugated with lactoferrin as novel drug carrier for brain delivery. Pharm. Res. 2012, 29, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Rice, K.C.; Liu, X.M.; Reinhardt, R.A.; Bayles, K.W.; Wang, D. Triclosan-loaded tooth-binding micelles for prevention and treatment of dental biofilm. Pharm. Res. 2010, 27, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, X.M.; Rice, K.C.; Li, X.; Yu, F.; Reinhardt, R.A.; Bayles, K.W.; Wang, D. Tooth-binding micelles for dental caries prevention. Antimicrob. Agents Chemother. 2009, 53, 4898–4902. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jia, Z.; Rice, K.C.; Reinhardt, R.A.; Bayles, K.W.; Wang, D. The development of dentotropic micelles with biodegradable tooth-binding moieties. Pharm. Res. 2013, 30, 2808–2817. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Nakagami, H.; Maeda, A.; Morishita, R.; Miyazaki, N.; Ogawa, T.; Tabata, Y.; Kikuchi, Y.; Hayashi, H.; Tatsu, Y.; et al. Development of a novel antimicrobial peptide, AG-30, with angiogenic properties. J. Cell. Mol. Med. 2009, 13, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Andreu, D.; Merrifield, R.B. Binding and action of cecropin and cecropin analogues: Antibacterial peptides from insects. Biochim. Biophys. Acta 1988, 939, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Veis, A. The physical chemistry of gelatin. Int. Rev. Connect. Tissue Res. 1965, 3, 113–200. [Google Scholar] [PubMed]
- Ikada, Y.; Tabata, Y. Protein release from gelatin matrices. Adv. Drug Deliv. Rev. 1998, 31, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Yang, L.; Narsimhan, G.; Bhunia, A.K.; Yao, Y. Designing carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide. J. Control. Release 2011, 150, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, S.L.; Wang, X.; Huang, L.; San-Martin Gonzalez, F.; Yao, Y. Phytoglycogenoctenyl succinate, an amphiphilic carbohydrate nanoparticle, and epsilon-polylysine to improve lipid oxidative stability of emulsions. J. Agric. Food Chem. 2010, 58, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, S.L.; Huang, L.; Bi, L.; Yao, Y. In vitro digestibility and emulsification properties of phytoglycogen octenyl succinate. J. Agric. Food Chem. 2010, 58, 5140–5146. [Google Scholar] [CrossRef] [PubMed]
- Wurzburg, O.B. Modified Starch. In Food Polysaccharides and Their Applications, 2nd ed.; Stephen, A.M., Phillips, G.O., Williams, P.A., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 67–98. [Google Scholar]
- Liu, L.; Xu, K.; Wang, H.; Tan, P.K.; Fan, W.; Venkatraman, S.S.; Li, L.; Yang, Y.Y. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 2009, 4, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Rentz, J.; Han, S.O.; Bull, D.A.; Kim, S.W. Water-soluble lipopolymer as an efficient carrier for gene delivery to myocardium. Gene Ther. 2003, 10, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, K.; Liu, L.; Tan, J.P.; Chen, Y.; Li, Y.; Fan, W.; Wei, Z.; Sheng, J.; Yang, Y.Y.; et al. The efficacy of self-assembled cationic antimicrobial peptide nanoparticles against Cryptococcus neoformans for the treatment of meningitis. Biomaterials 2010, 31, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Schröder, U.; Sabel, B.A. Nanoparticles, a drug carrier system to pass the blood-brain barrier, permit central analgesic effects of i.v. dalargin injections. Brain Res. 1996, 710, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Roney, C.; Kulkarni, P.; Arora, V.; Antich, P.; Bonte, F.; Wu, A.; Mallikarjuana, N.N.; Manohar, S.; Liang, H.F.; Kulkarni, A.R.; et al. Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer’s disease. J. Control. Release 2005, 108, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.N.; Zaro, J.L.; Shen, W.C. Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S. Arginine-rich peptides: Potential for intracellular delivery of macromolecules and the mystery of the translocation mechanisms. Int. J. Pharm. 2002, 245, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Urbán, P.; Valle-Delgado, J.J.; Moles, E.; Marques, J.; Díez, C.; Fernàndez-Busquets, X. Nanotools for the delivery of antimicrobial peptides. Curr. Drug Targets 2012, 13, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Akita, H.; Kogure, K.; Gräslund, A.; Langel, U.; Harashima, H.; Futaki, S. Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc. Chem. Res. 2012, 45, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Lorimer, C.P.; McCoy, C.P.; Gorman, S.P. Characterization of the physicochemical, antimicrobial, and drug release properties of thermoresponsive hydrogel copolymers designed for medical device applications. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 85, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Poon, Y.F.; Li, W.; Zhu, H.Y.; Yeap, S.H.; Cao, Y.; Qi, X.; Zhou, C.; Lamrani, M.; Beuerman, R.W.; et al. A polycationic antimicrobial and biocompatible hydrogel with microbe membrane suctioning ability. Nat. Mater. 2011, 10, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Petratos, P.B.; Chen, J.; Felsen, D.; Poppas, D.P. Local pharmaceutical release from a new hydrogel implant. J. Surg. Res. 2002, 103, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.E.; Patil, A.J.; Butler, M.F.; Mann, S. Guest-molecule-directed assembly of mesostructured nanocomposite polymer/organoclay hydrogels. Adv. Funct. Mater. 2011, 21, 674–681. [Google Scholar] [CrossRef]
- Vasconcelos, A.; Cavaco-Paulo, A. Wound dressings for a proteolytic-rich environment. Appl. Microbiol. Biotechnol. 2011, 90, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, R.; Miraftab, M.; Collyer, G. A critical review of modern and emerging absorbent dressings used to treat exuding wounds. Int. Wound J. 2012, 9, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Kundu, B.; Kundu, S.C. Silk sericin/polyacrylamide in situ forming hydrogels for dermal reconstruction. Biomaterials 2012, 33, 7456–7467. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Wood, K.M.; Blanchette, J.O. Hydrogels for oral delivery of therapeutic proteins. Expert Opin. Biol. Ther. 2004, 4, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.V. Colloidal microgels in drug delivery applications. Curr. Pharm. Des. 2006, 12, 4703–4712. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Amiji, M.M. Preparation and characterization of freeze-dried chitosan-poly (ethylene oxide) hydrogels for site-specific antibiotic delivery in the stomach. Pharm. Res. 1996, 13, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Wang, N.; Wu, X.S. Poly (vinyl alcohol) nanoparticles prepared by freezing-thawing process for protein/peptide drug delivery. J. Control. Release 1998, 56, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, S.; Semenzato, A.; Bersani, S.; Matricardi, P.; Rossi, F.; Caliceti, P. Cyclodextrin/PEG based hydrogels for multi-drug delivery. Int. J. Pharm. 2007, 345, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.V.; Shivakumar, H.G. Preparation and characterization of superporous hydrogels as pH-sensitive drug delivery system for Pantoprazole sodium. Curr. Drug Deliv. 2009, 6, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Haakansson, J.; Bjoern, C.; Lindgren, K.; Sjoestroem, E.; Sjoestrand, V.; Mahlapuu, M. Efficacy of the novel topical antimicrobial agent PXL150 in a mouse model of surgical site infections. Antimicrob. Agents Chemother. 2014, 58, 2982–2984. [Google Scholar] [CrossRef] [PubMed]
- Myhrman, E.; Hakansson, J.; Lindgren, K.; Bjorn, C.; Sjostrand, V.; Mahlapuu, M. The novel antimicrobial peptide PXL150 in the local treatment of skin and soft tissue infections. Appl. Microbiol. Biotechnol. 2013, 97, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Chen, S.; Etzler, F. Rheological characterization of hydroxypropylcellulose gels. Drug Dev. Ind. Pharm. 1999, 25, 153–161. [Google Scholar] [CrossRef] [PubMed]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Gawande, P.V.; Leung, K.P.; Madhyastha, S. Antibiofilm and antimicrobial efficacy of DispersinB®-KSL-W peptide-based wound gel against chronic wound infection associated bacteria. Curr. Microbiol. 2014, 68, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B.; Ragunath, C.; Ramasubbu, N.; Fine, D.H. Detachment of Actinobacillus actinomycetemcomitans biofilm cells by an endogenous beta-hexosaminidase activity. J. Bacteriol. 2003, 185, 4693–4698. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B. Therapeutic potential of biofilm-dispersing enzymes. Int. J. Artif. Organs 2009, 32, 545–554. [Google Scholar] [PubMed]
- Hong, S.Y.; Oh, J.E.; Kwon, M.Y.; Choi, M.J.; Lee, J.H.; Lee, B.L.; Moon, H.M.; Lee, K.H. Identification and characterization of novel antimicrobial decapeptides generated by combinatorial chemistry. Antimicrob. Agents Chemother. 1998, 42, 2534–2541. [Google Scholar] [PubMed]
- Na, D.H.; Faraj, J.; Capan, Y.; Leung, K.P.; DeLuca, P.P. Stability of antimicrobial decapeptide (KSL) and its analogues for delivery in the oral cavity. Pharm. Res. 2007, 24, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.P.; Abercrombie, J.J.; Campbell, T.M.; Gilmore, K.D.; Bell, C.A.; Faraj, J.A.; DeLuca, P.P. Antimicrobial peptides for plaque control. Adv. Dent. Res. 2009, 21, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, G.; Grossiord, J.L.; Agnely, F.; Chaumeil, J.C. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm. Res. 2006, 23, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Veiga, A.S.; Sinthuvanich, C.; Gaspar, D.; Franquelim, H.G.; Castanho, M.A.; Schneider, J.P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33, 8907–8916. [Google Scholar] [CrossRef] [PubMed]
- Salick, D.A.; Kretsinger, J.K.; Pochan, D.J.; Schneider, J.P. Inherent antibacterial activity of a peptide-based beta-hairpin hydrogel. J. Am. Chem. Soc. 2007, 129, 14793–14799. [Google Scholar] [CrossRef] [PubMed]
- Salick, D.A.; Pochan, D.J.; Schneider, J.P. Design of an injectable beta-hairpin peptide hydrogel that kills methicillin-resistant Staphylococcus aureus. Adv. Mater. 2009, 21, 4120–4123. [Google Scholar] [CrossRef]
- Flores, C.Y.; Diaz, C.; Rubert, A.; Benitez, G.A.; Moreno, M.S.; Fernandez Lorenzo de Mele, M.A.; Salvarezza, R.C.; Schilardi, P.L.; Vericat, C. Spontaneous adsorption of silver nanoparticles on Ti/TiO2 surfaces. Antibacterial effect on Pseudomonas aeruginosa. J. Colloid Interface Sci. 2010, 350, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Lange, D.; Hilpert, K.; Kindrachuk, J.; Zou, Y.; Cheng, J.T.; Kazemzadeh-Narbat, M.; Yu, K.; Wang, R.; Straus, S.K.; et al. The biocompatibility and biofilm resistance of implant coatings based on hydrophilic polymer brushes conjugated with antimicrobial peptides. Biomaterials 2011, 32, 3899–3909. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.; Palombo, R.R.; Petri, D.F.; Bruns, M.; Pereira, E.M.; Carmona-Ribeiro, A.M. Structure-activity relationship for quaternary ammonium compounds hybridized with poly (methyl methacrylate). ACS Appl. Mater. Interfaces 2011, 3, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Kurt, P.; Wood, L.; Ohman, D.E.; Wynne, K.J. Highly effective contact antimicrobial surfaces via polymer surface modifiers. Langmuir 2007, 23, 4719–4723. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Koepsel, R.R.; Matyjaszewski, K.; Russell, A.J. Permanent, non-leaching antibacterial surface-2: How high density cationic surfaces kill bacterial cells. Biomaterials 2007, 28, 4870–4879. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.; Hollander, A.; Jaeger, W.; Trick, I.; Oehr, C. Ultrathin antibacterial polyammonium coatings on polymer surfaces. Surf. Coat. Technol. 2003, 174, 584–587. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, E.; Rajabi, M.; Zou, G.; Pazgier, M.; Lu, W. Selective arginines are important for the antibacterial activity and host cell interaction of humanalpha-defensin 5. FEBS Lett. 2009, 583, 2507–2512. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Mishra, A.; Lai, G.H.; Davis, M.; Sanders, L.K.; Tran, D.; Garcia, A.; McCray, P.B.; Ouellette, A.J.; Selsted, M.E.; et al. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 2011, 133, 6720–6727. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, H.; Qu, X.; Weeks, C.S.; Cummings, J.E.; Kolusheva, S.; Walsh, K.B.; Jelinek, R.; Vanderlick, T.K.; Selsted, M.E.; Ouellette, A.J. Structure-activity determinants in paneth cell alpha-defensins: Loss-of-function in mouse cryptdin-4 by charge-reversal at arginine residue positions. J. Biol. Chem. 2004, 279, 11976–11983. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; de Leeuw, E.; Li, C.; Pazgier, M.; Li, C.; Zeng, P.; Lu, W.Y.; Lubkowski, J.; Lu, W. Toward understanding the cationicity of defensins. Arg and Lys versus their noncoded analogs. J. Biol. Chem. 2007, 282, 19653–19665. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Park, T.G. Self-assembled and nanostructured hydrogels for drug delivery and tissue engineering. Nano Today 2009, 4, 429–437. [Google Scholar] [CrossRef]
- Kopecek, J.I.; Yang, J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009, 5, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Dipeptide and tripeptide conjugates as low-molecular-weight hydrogelators. Macromol. Biosci. 2011, 11, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Marchesan, S.; Easton, C.D.; Kushkaki, F.; Waddington, L.; Hartley, P.G. Tripeptide self-assembled hydrogels: Unexpected twists of chirality. Chem. Commun. 2012, 48, 2195–2197. [Google Scholar] [CrossRef]
- Marchesan, S.; Waddington, L.; Easton, C.D.; Winkler, D.A.; Goodall, L.; Forsythe, J.; Hartley, P.G. Unzipping the role of chirality in nanoscale self-asssembly of tripeptide hydrogels. Nanoscale 2012, 4, 6752–6760. [Google Scholar] [CrossRef] [PubMed]
- Marchesan, S.; Qu, Y.; Waddington, L.J.; Easton, C.D.; Glattauer, V.; Lithgow, T.J.; McLean, K.M.; Forsythe, J.S.; Hartley, P.G. Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel. Biomaterials 2013, 34, 3678–3687. [Google Scholar] [CrossRef] [PubMed]
- Rassing, M.R. Chewing gum as a drug delivery system. Adv. Drug Deliv. Rev. 1994, 13, 89–121. [Google Scholar] [CrossRef]
- Ainamo, J.; Etemadzadeh, H. Prevention of plaque growth with chewing gum containing chlorhexidine acetate. J. Clin. Periodontol. 1987, 14, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D.; Bradshaw, D.J. Dental plaque as a biofilm. J. Ind. Microbiol. 1995, 15, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D. Dental plaque as a biofilm and a microbial community—Implications for health and disease. BMC Oral Health 2006, 6. [Google Scholar] [CrossRef]
- Na, D.H.; Faraj, J.; Capan, Y.; Leung, K.P.; DeLuca, P.P. Chewing gum of antimicrobial decapeptide (KSL) as a sustained antiplaque agent: Preformulation study. J. Control. Release 2005, 107, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Faraj, J.A.; Dorati, R.; Schoubben, A.; Worthen, D.; Selmin, F.; Capan, Y.; Leung, K.; DeLuca, P.P. Development of a peptide-containing chewing gum as a sustained release antiplaque antimicrobial delivery system. AAPS PharmSciTech. 2007, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Concannon, S.P.; Crowe, T.D.; Abercrombie, J.J.; Molina, C.M.; Hou, P.; Sukumaran, D.K.; Raj, P.A.; Leung, K.P. Susceptibility of oral bacteria to an antimicrobial decapeptide. J. Med. Microbiol. 2003, 52, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Rassing, M.R. Specialized oral mucosal drug delivery systems: Chewing gums. In Oral Mucosal Drug Delivery; Rathbone, M.J., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 319–357. [Google Scholar]
- Cruz, D.N.; Perazella, M.A.; Bellomo, R.; de Cal, M.; Polanco, N.; Corradi, V.; Lentini, P.; Nalesso, F.; Ueno, T.; Ranieri, V.M.; et al. Effectiveness of polymyxin B-immobilized fiber column in sepsis: A systematic review. Crit. Care 2007, 11, R47:1–R47:18. [Google Scholar] [CrossRef]
- Iba, T.; Nagaoka, I.; Yamada, A.; Nagayama, M.; Miki, T. Effect of hemoperfusion using polymyxin B-immobilized fibers on acute lung injury in a rat sepsis model. Int. J. Med. Sci. 2014, 11, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Esteban, E.; Ferrer, R.; Alsina, L.; Artigas, A. Immunomodulation in sepsis: The role of endotoxin removal by polymyxin B-immobilized cartridge. Mediators Inflamm. 2013. [Google Scholar] [CrossRef]
- Davies, B.; Cohen, J. Endotoxin removal devices for the treatment of sepsis and septic shock. Lancet. Infect. Dis. 2011, 11, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Hanasawa, K.; Sato, K.; Umeki, M.; Koga, N.; Naganuma, T.; Sato, S.; Shimonishi, T.; Ikeda, T.; Matsuno, N.; et al. Direct hemoperfusion with polymyxin-B-immobilized fiber columns improves septic hypotension and reduces inflammatory mediators in septic patients with colorectal perforation. Langenbecks Arch. Surg. 2008, 394, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Antonelli, M.; Fumagalli, R.; Foltran, F.; Brienza, N.; Donati, A.; Malcangi, V.; Petrini, F.; Volta, G.; Bobbio Pallavicini, F.M.; et al. Early use of polymyxin B hemoperfusion in abdominal septic shock: The EUPHAS randomized controlled trial. JAMA 2009, 301, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Inoue, Y.; Nishiyama, A.; Sugimoto, C.; Matsumuro, A.; Hirose, M.; Kitaichi, M.; Akira, M.; Arai, T.; Hayashi, S.; et al. Polymyxin-B hemoperfusion for acute exacerbation of idiopathic pulmonary fibrosis: Serum IL-7 as a prognostic marker. Sarcoidosis Vasc. Diffuse Lung Dis. 2011, 28, 113–122. [Google Scholar] [PubMed]
- Zhou, F.; Peng, Z.; Murugan, R.; Kellum, J.A. Blood purification and mortality in sepsis: A meta-analysis of randomized trials. Crit. Care Med. 2013, 41, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Sato, K.; Kurita, A.; Noda, T.; Okajima, M. Efficacy of endotoxin adsorption therapy (polymyxin B hemoperfusion) for methicillin-resistant Staphylococcus aureus toxic shock syndrome. A case report about five patients. Minerva Anestesiol. 2013, 79, 758–761. [Google Scholar] [PubMed]
- Vesentini, S.; Soncini, M.; Zaupa, A.; Silvestri, V.; Fiore, G.B.; Redaelli, A. Multi-scale analysis of the toraymyxin adsorption cartridge. Part I: Molecular interaction of polymyxin B with endotoxins. Int. J. Artif. Organs 2006, 29, 239–250. [Google Scholar] [PubMed]
- Manocha, S.; Feinstein, D.; Kumar, A.; Kumar, A. Novel therapies for sepsis: Antiendotoxin therapies. Expert Opin. Investig. Drugs 2002, 11, 1795–1812. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, E.J.; Fisher, C.J.; Sprung, C.L.; Straube, R.C.; Sadoff, J.C.; Foulke, G.E.; Wortel, C.H.; Fink, M.P.; Dellinger, R.P.; Teng, N.N.; et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N. Engl. J. Med. 1991, 324, 429–436. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, R.V.; Straube, R.C.; Sanders, C.; Smith, S.M.; Smith, C.R. Treatment of septic shock with human monoclonal antibodyHA-1A. A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann. Intern. Med. 1994, 121, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Laterre, P.F.; Cohen, J.; Burchardi, H.; Bruining, H.; Lerma, F.A.; Wittebole, X.; de Backer, D.; Brett, S.; Marzo, D.; et al. A pilot-controlled study of a polymyxin B-immobilized hemoperfusion cartridge in patients with severe sepsis secondary to intra-abdominal infection. Shock 2005, 23, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, K.; Shiohara, M.; Saito, S.; Tanaka, M.; Yanagisawa, R.; Tsuruta, G.; Fukuyama, T.; Hidaka, Y.; Nakazawa, Y.; Shimizu, T.; et al. Polymyxin-direct hemoperfusion for sepsis-induced multiple organ failure. Pediatr. Blood Cancer 2010, 55, 202–205. [Google Scholar] [PubMed]
- Shoji, H. Extracorporeal endotoxin removal for the treatment of sepsis: Endotoxin adsorption cartridge (Toraymyxin). Ther. Apher. Dial. 2003, 7, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ushiyama, C.; Suzuki, Y.; Inoue, T.; Shoji, H.; Shimada, N.; Koide, H. Combination therapy with polymyxin B-immobilized fibre haemoperfusion and teicoplanin for sepsis due to methicillin-resistant Staphylococcus aureus. J. Hosp. Infect. 2003, 53, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kawagoe, Y.; Sukuzi, T.; Shoji, H.; Ueda, Y.; Kobayashi, N.; Koide, H. Changes in plasma interleukin-18 by direct hemoperfusion with polymyxin B-immobilized fiber in patients with septic shock. Blood Purif. 2005, 23, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Osajima, A.; Kamochi, M.; Watanabe, H.; Aibara, K.; Kaizu, K. Endotoxin removal by direct hemoperfusion with an adsorbent column using polymyxin B-immobilized fiber ameliorates systemic circulatory disturbance in patients with septic shock. Am. J. Kidney Dis. 2002, 39, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ma, T.; Tang, H.; Fan, X.; Xu, Y. Vancomycin cationic liposome combined with nano-hydroxyapatite/chitosan/konjacglucomannan scaffold for treatment of infected bone defects in rabbits. Chin. J. Repar. Reconstr. Surg. 2012, 26, 190–195. [Google Scholar]
- Rishi, P.; Singh, A.P.; Arora, S.; Garg, N.; Kaur, I.P. Revisiting eukaryotic anti-infective biotherapeutics. Crit. Rev. Microbiol. 2014, 40, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.Y.; Yeaman, M.R. Emerging themes and therapeutic prospects for anti-infective peptides. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. New approaches in peptide antibiotics. Curr. Opin. Pharmacol. 2009, 9, 571–576. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona-Ribeiro, A.M.; De Melo Carrasco, L.D. Novel Formulations for Antimicrobial Peptides. Int. J. Mol. Sci. 2014, 15, 18040-18083. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151018040
Carmona-Ribeiro AM, De Melo Carrasco LD. Novel Formulations for Antimicrobial Peptides. International Journal of Molecular Sciences. 2014; 15(10):18040-18083. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151018040
Chicago/Turabian StyleCarmona-Ribeiro, Ana Maria, and Letícia Dias De Melo Carrasco. 2014. "Novel Formulations for Antimicrobial Peptides" International Journal of Molecular Sciences 15, no. 10: 18040-18083. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151018040