2.1. Infectious Pancreatic Necrosis Virus (IPNV) Infection Induces Apoptosis and the Expression of Phosphatidylserine Receptor (PSR) in CHSE-214 Cells
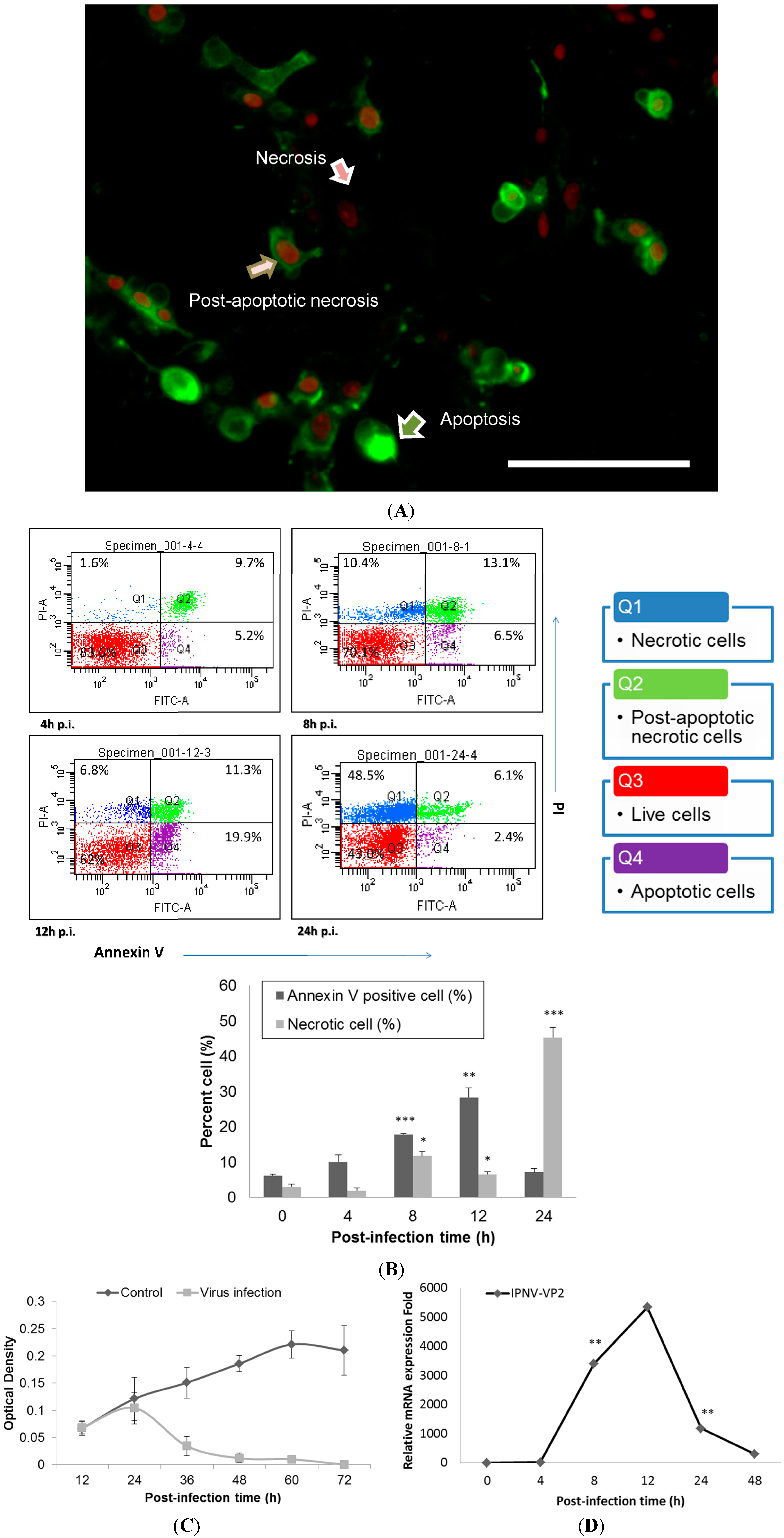
The cytopathic effect (CPE) of IPNV infection (MOI = 1) among CHSE-214 cells was observed at 8 h post-infection (h.p.i) and was found to increase dramatically as time increased; obvious cell death was observed between 12 and 48 h.p.i (Data not shown). IPNV infection induced apoptosis in CHSE-214 cells, and this was confirmed with double staining of annexin V and propidium iodide (PI) in the infected cells. Three types of the cells were identified at 8 h.p.i: Annexin V staining of exposed phospatidylserine (PS) indicated an apoptotic cell, PI in the nucleus indicated a necrotic cell, and dual staining indicated a post-apoptotic necrotic cell (
Figure 1A). In the flow cytometry analysis, PS-positive cells consistently increased in number, reaching 28.3% (
p < 0.01) of the total cells at 12 h.p.i., and then the majority of cells shifted to necrosis at 24 h.p.i. However, the CHSE-214 cell is very susceptible to the E1S strain IPNV, most of the cell showed CPE at 36 and 48 h.p.i. It is hard to collect the cells for fluorescence-activated cell sorting (FACS) assay. Hence, we only present the data from 0–24 h.p.i. only (
Figure 1B). To evaluate the viability of infected CHSE-214 cells, WST-1 assays were performed in triplicate, revealing that viability decreased gradually after 24 h.p.i., which corresponded to the peak in necrosis (
Figure 1B,C). Additionally, viral replication was analyzed by detecting the expression of viral capsid protein with real-time qPCR (
Figure 1D) and immunoblotting (Data not shown), allowing us to monitor the progression of IPNV replication in infected salmonid cells. Real-time qPCR for viral VP2, a major capsid protein of IPNV, indicated that replication peaked at 12 h.p.i. (
Figure 1D). Immunoblots confirmed the expression of PSR in apoptotic CHSE-214 cells during IPNV infection, and indicated that the PSR protein was induced by viral infection starting at 8 h.p.i. and remained consistently elevated until 48 h.p.i. (
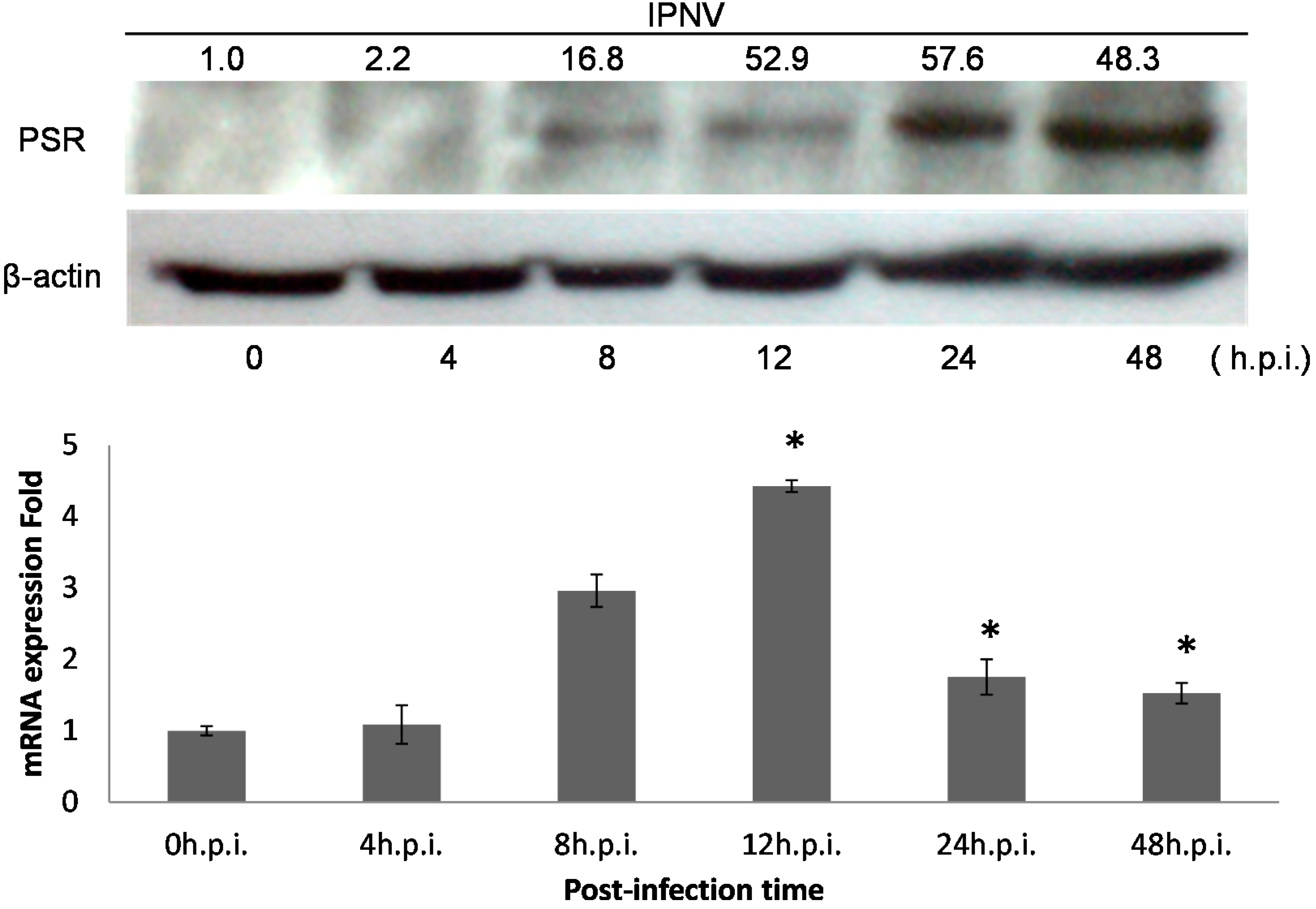
Figure 2).
Figure 1.
Apoptosis occurs in Infectious Pancreatic Necrosis Virus (IPNV)-infected CHSE-214 cells and viral replication reaches a peak at 12 h.p.i. (MOI = 1). (A) Annexin V and propidium iodide (PI) staining in infected CHSE-214 cells at 8 h.p.i.; three types of cells (pointed arrows) are delineated (scale bar = 100 μm). Annexin V-labeled cells indicated apoptosis, PI-labeled cells indicated necrosis, and double staining indicated post-apoptotic necrotic cells; (B) The infected cells were analyzed by flow cytometry for apoptosis, revealing a dynamic shift among the four types of cells. Double stained and PS-positive cells increased prior to 12 h.p.i., and then most cells became necrotic at 24 h.p.i. (* p < 0.05; ** p < 0.01; and *** p < 0.0001). All reported values have been corrected for background; (C) Cell viability was determined in triplicate by a WST-1 assay, which indicated that cell viability decreased after 24 h.p.i.; and (D) Real-time qPCR to determine the mRNA levels of viral VP2, a major capsid protein of IPNV. The results indicated high levels of replication among the infected cells that peaked and corresponded to the peak in apoptosis during IPNV infection. (** p < 0.01).
Figure 1.
Apoptosis occurs in Infectious Pancreatic Necrosis Virus (IPNV)-infected CHSE-214 cells and viral replication reaches a peak at 12 h.p.i. (MOI = 1). (A) Annexin V and propidium iodide (PI) staining in infected CHSE-214 cells at 8 h.p.i.; three types of cells (pointed arrows) are delineated (scale bar = 100 μm). Annexin V-labeled cells indicated apoptosis, PI-labeled cells indicated necrosis, and double staining indicated post-apoptotic necrotic cells; (B) The infected cells were analyzed by flow cytometry for apoptosis, revealing a dynamic shift among the four types of cells. Double stained and PS-positive cells increased prior to 12 h.p.i., and then most cells became necrotic at 24 h.p.i. (* p < 0.05; ** p < 0.01; and *** p < 0.0001). All reported values have been corrected for background; (C) Cell viability was determined in triplicate by a WST-1 assay, which indicated that cell viability decreased after 24 h.p.i.; and (D) Real-time qPCR to determine the mRNA levels of viral VP2, a major capsid protein of IPNV. The results indicated high levels of replication among the infected cells that peaked and corresponded to the peak in apoptosis during IPNV infection. (** p < 0.01).
![Ijms 15 19281 g001]()
Figure 2.
PSR expression is induced by IPNV infection in CHSE-214 cells. PSR expression was analyzed with polyclonal anti-PSR or anti-β-actin (loading control) antibodies by immunoblot (upper panel). Normalized density were calculated and listed above. Cells were infected with IPNV (MOI = 1), and proteins were isolated from cells at the times indicated. PSR (48 kDa) and β-actin (45 kDa) were analyzed at 0, 4, 8, 12, 24 and 48 h.p.i. in lanes 1–6, respectively (0 h.p.i. as control). PSR proteins were detected starting at 8 h.p.i. and continuously increased until 48 h.p.i. The mRNA expression level was measured by real-time PCR in triplicate and normalized by β-actin. (lower panel) The data was expressed as the mean ± SD (* p < 0.05)
Figure 2.
PSR expression is induced by IPNV infection in CHSE-214 cells. PSR expression was analyzed with polyclonal anti-PSR or anti-β-actin (loading control) antibodies by immunoblot (upper panel). Normalized density were calculated and listed above. Cells were infected with IPNV (MOI = 1), and proteins were isolated from cells at the times indicated. PSR (48 kDa) and β-actin (45 kDa) were analyzed at 0, 4, 8, 12, 24 and 48 h.p.i. in lanes 1–6, respectively (0 h.p.i. as control). PSR proteins were detected starting at 8 h.p.i. and continuously increased until 48 h.p.i. The mRNA expression level was measured by real-time PCR in triplicate and normalized by β-actin. (lower panel) The data was expressed as the mean ± SD (* p < 0.05)
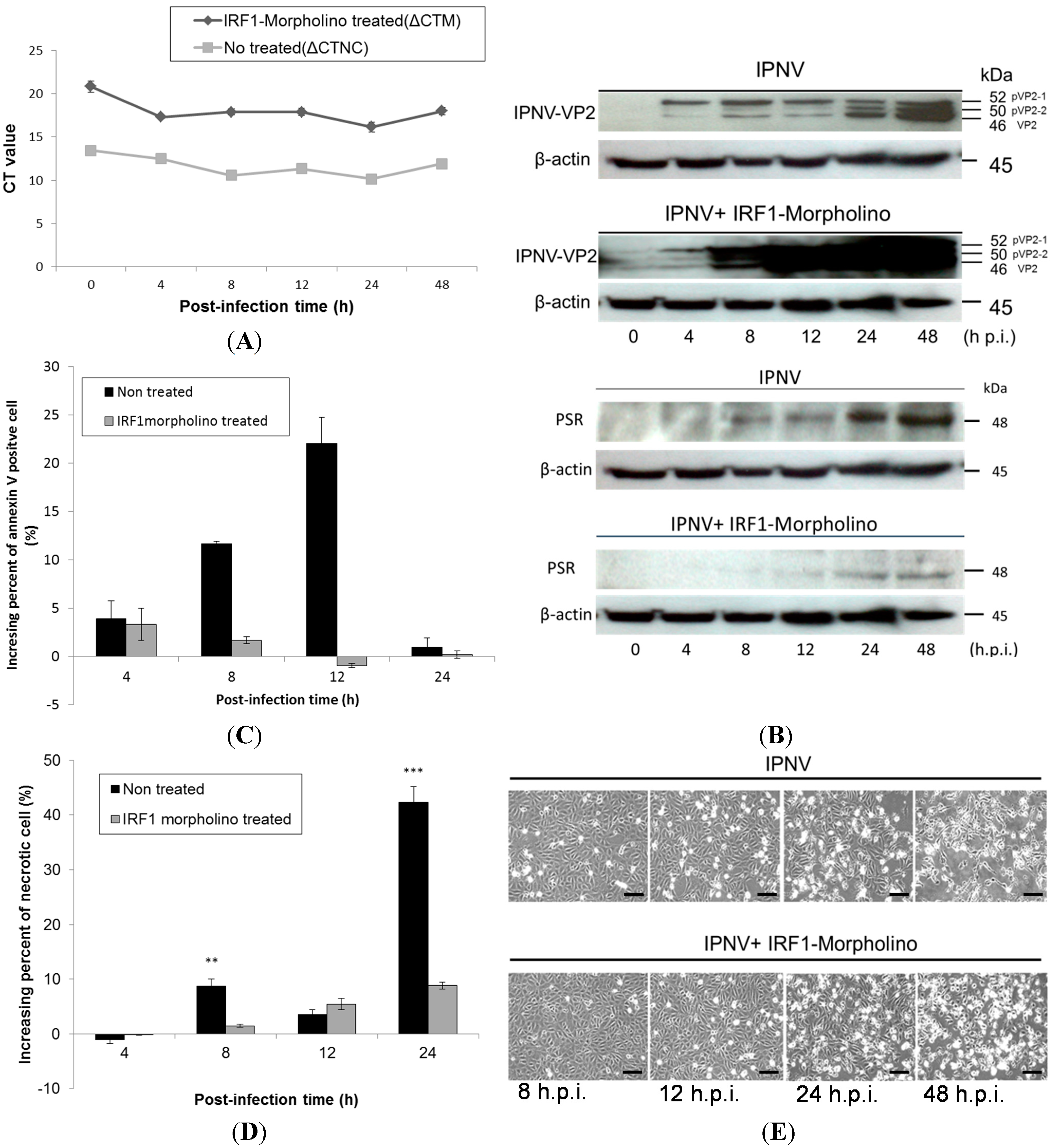
2.4. Effects of IRF-1 Knockdown on CHSE-214 Cells during IPNV Infection
To clarify the relationship between IRF-1 and PSR, IRF-1 was knocked down with morpholino oligos during IPNV infection. We delivered the salmon IRF-1 morpholino (10 μM) into CHSE-214 cells first by electroporation and then incubated the transfected cells for 18 h before IPNV infection (MOI = 1). The morpholino oligos were conjugated with fluorescein isothiocyanate (FITC) to enable easy observation of the delivery efficiency by fluorescence microscopy (Data not shown). After the IRF-1 morpholino was delivered into the cells, its inhibitory effects on IRF-1 were confirmed by real-time qPCR (
Figure 6A) and immunoblotting. Both tests showed the reduction of IRF-1 mRNA and protein expression in IRF-1 morpholino-treated CHSE-214 cells during IPNV infection.
Figure 6A shows IRF-1 mRNA expression with or without IRF-1 morpholino treatment following IPNV infection in terms of Δ
Ct value, which was normalized with the internal control (β-actin). The results indicated that IRF-1 mRNA expression was inhibited by approximately 38% with morpholino treatment. We next examined PSR expression in IRF-1 knockdown CHSE-214 cells during IPNV infection and found that PSR expression was delayed in IRF-1 knockdown cells during viral infection (
Figure 6B, β-actin as internal control). All samples were probed with anti-PSR antibodies, and morpholino-treated samples exhibited delayed PSR expression until 24 h.p.i., whereas non-morpholino-treated samples expressed PSR earlier at 8 h.p.i. We also wanted to investigate viral replication under IRF-1-inhibited conditions. To analyze viral replication in IRF-1 morpholino-treated or untreated infected CHSE-214 cells, the expression of the major capsid protein VP2 of IPNV was evaluated by immunoblot. The viral VP2 protein was detected as early as 4 h.p.i. in infected CHSE-214 cells, and the expression of VP2 protein gradually transitioned from the precursor form, pVP2-1 (52 kDa), and intermediate form, pVP2-2 (50 kDa), to the mature form VP2 (46 kDa) between 8 and 48 h.p.i. (
Figure 6B, lanes 3–6) in the infected cells. The morpholino-treated samples exhibited dramatically increasing expression of the VP2 protein between 8 and 48 h.p.i. in IRF-1 knockdown cells (
Figure 6B, lower panel). Additionally, expression of the viral protein increased constitutively from 4–48 h after viral infection in non-morpholino treated cells (
Figure 6B, upper panel).
Figure 5.
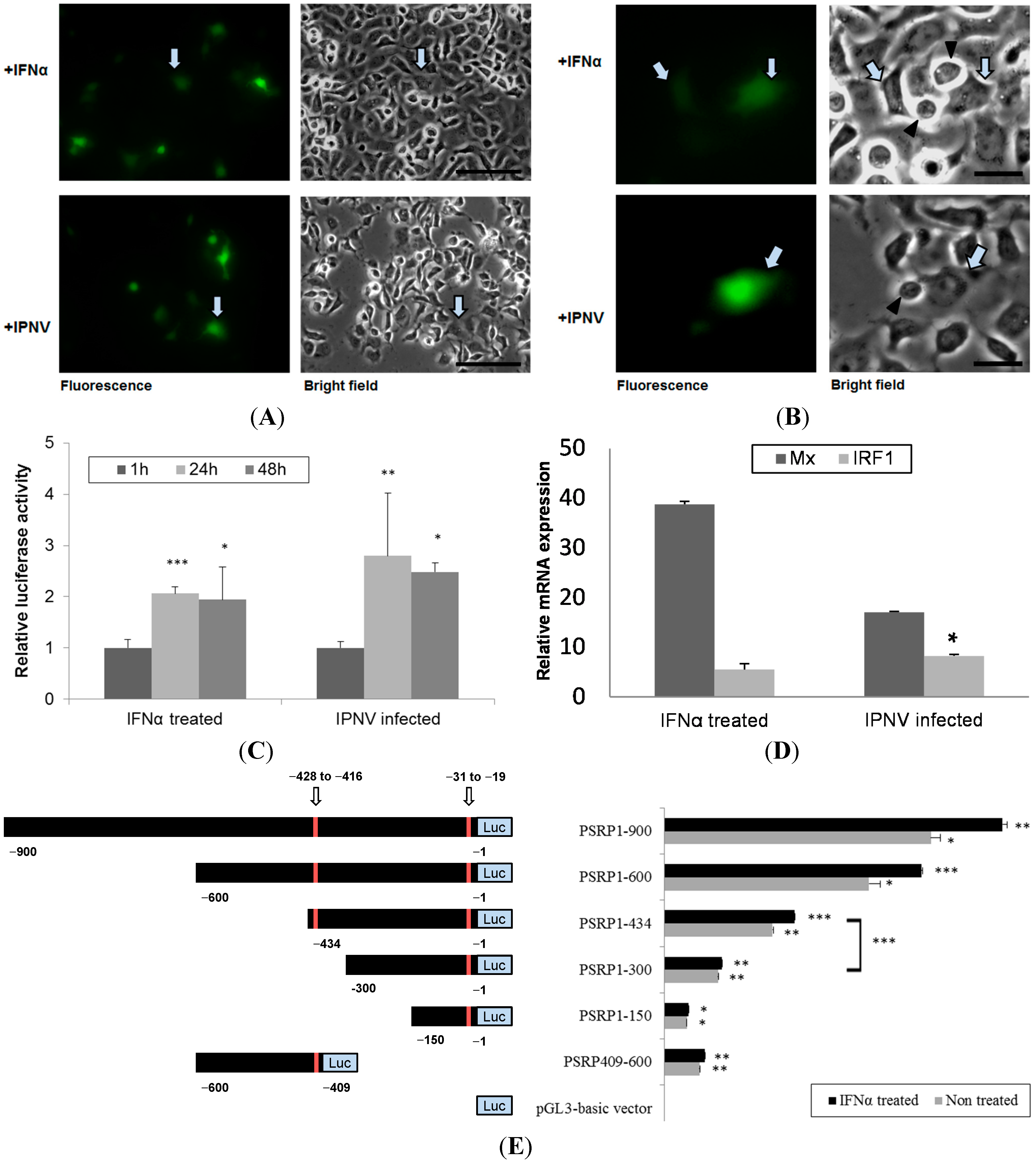
The PSR promoter can be induced by rIFN-α or IPNV in CHSE-214 cells. (A) CHSE-214 cells were transfected with the pPSRP:EGFP construct, stimulated with either rIFN-α (0.5 μg/mL) or IPNV (MOI = 1) for 24 h, and then visualized by fluorescent microscopy. The PSR promoter was activated by the stimuli (fluorescent green pointed by arrows; scale bar = 100 μm); (B) PSR promoter-activated cells (green) had the potential to engulf apoptotic cells (pointed triangle) in both rIFN-α or IPNV-stimulated cells (scale bar = 25 μm); (C) CHSE-214 cells were transfected with the pGL3-basic-PSRP construct, stimulated with either rIFN-α or IPNV for 24 and 48 h, and then evaluated with a luciferase assay. PSR promoter activity was expressed as fold induction of relative luciferase activity (RLU) compared to the promoterless vector and then compared with the values at 1 h. The data indicated that the PSR promoter was activated by the stimuli (* p < 0.05; ** p < 0.01; and *** p < 0.0001); (D) Mx and IRF-1 were induced in pPSRP:EGFP expressing cells as determinedby real-time qPCR. Relative expression is compared to beta-actin expression as internal control (* p < 0.05); and (E) Promoter activity of 900-bp PSR promoter and IRF-1-deleted PSR promoter. The left panel shows a schematic representation of the PSR gene promoter constructs. Full-length PSR promoter (−900 to −1) as well as various deleted fragments were inserted into the pGL3-basic plasmid with luciferase (Luc). Putative IRF-1 binding sites in the fragment are indicated by arrows and marked in red. PSR promoter activity in response to rIFN-α (0.5 μg/mL) was assessed 48 h after the addition of the cytokine and expressed as fold induction of relative luciferase activity (RLU) compared to the promoterless vector. Cells exposed to basal medium without the cytokine were used as a control. Luciferase assays (right panel) were performed in triplicate, and the data are expressed as the mean ± SD (* p < 0.05; ** p < 0.01; and *** p < 0.0001).
Figure 5.
The PSR promoter can be induced by rIFN-α or IPNV in CHSE-214 cells. (A) CHSE-214 cells were transfected with the pPSRP:EGFP construct, stimulated with either rIFN-α (0.5 μg/mL) or IPNV (MOI = 1) for 24 h, and then visualized by fluorescent microscopy. The PSR promoter was activated by the stimuli (fluorescent green pointed by arrows; scale bar = 100 μm); (B) PSR promoter-activated cells (green) had the potential to engulf apoptotic cells (pointed triangle) in both rIFN-α or IPNV-stimulated cells (scale bar = 25 μm); (C) CHSE-214 cells were transfected with the pGL3-basic-PSRP construct, stimulated with either rIFN-α or IPNV for 24 and 48 h, and then evaluated with a luciferase assay. PSR promoter activity was expressed as fold induction of relative luciferase activity (RLU) compared to the promoterless vector and then compared with the values at 1 h. The data indicated that the PSR promoter was activated by the stimuli (* p < 0.05; ** p < 0.01; and *** p < 0.0001); (D) Mx and IRF-1 were induced in pPSRP:EGFP expressing cells as determinedby real-time qPCR. Relative expression is compared to beta-actin expression as internal control (* p < 0.05); and (E) Promoter activity of 900-bp PSR promoter and IRF-1-deleted PSR promoter. The left panel shows a schematic representation of the PSR gene promoter constructs. Full-length PSR promoter (−900 to −1) as well as various deleted fragments were inserted into the pGL3-basic plasmid with luciferase (Luc). Putative IRF-1 binding sites in the fragment are indicated by arrows and marked in red. PSR promoter activity in response to rIFN-α (0.5 μg/mL) was assessed 48 h after the addition of the cytokine and expressed as fold induction of relative luciferase activity (RLU) compared to the promoterless vector. Cells exposed to basal medium without the cytokine were used as a control. Luciferase assays (right panel) were performed in triplicate, and the data are expressed as the mean ± SD (* p < 0.05; ** p < 0.01; and *** p < 0.0001).
![Ijms 15 19281 g005]()
The variation in the percentages of apoptotic and necrotic cells among IRF-1 morpholino-treated or untreated CHSE-214 cells was evaluated by flow cytometry during IPNV infection. The results showed that PS-positive cells (annexin V-stained) decreased in IRF-1 knockdown cells between 4 and 24 h.p.i. during IPNV infection (
Figure 6C), and necrotic cells were also decreased in IRF-1 knockdown CHSE-214 cells at 8 and 24 h.p.i. (
Figure 6D). The changes in morphology and mortality in IRF-1 morpholino-treated cells (
Figure 6E, lower panel) were less severe than in non-morpholino cells (
Figure 6E, upper panel) during IPNV infection, and these results hint that IRF-1 knockdown corresponds to reduced apoptotic cell death and necrotic cell death. Therefore, cell death was inhibited in IRF-1 knockdown CHSE-214 cells after IPNV infection, and IRF-1 might be required for the inhibition of viral replication. To evaluate viral titers in IRF-1 morpholino-treated CHSE-214 cells, we also determined the 50% tissue culture infective dose (TCID
50) but found that no increase in the number of whole viral particles was detected after infection of the IRF-1 morpholino-treated cells compared to the non-morpholino-treated samples (
Figure 7). This result implies that although viral replication is boosted in the IRF-1 morpholino-treated CHSE-214 cells, whole viral particles might not fully assemble to produce more viral particles.
Figure 6.
Effects of IRF-1 knockdown on CHSE-214 cells during IPNV infection. (A) Real-time qPCR was used to determine the mRNA levels of IRF-1 with or without IRF-1 morpholino treatment following IPNV infection (MOI = 1). Expression were normalized to the internal control (β-actin) and is shown as the ΔCt value. The results indicated that IRF-1 mRNA expression was inhibited by approximately 38% with the morpholino; (B) Immunoblots of PSR or IPNV-VP2 expressionin IPNV-infected CHSE-214 cells (MOI = 1); The upper panels of (B) show the expression without IRF-1 morpholino treatment during infection, and the lower panels show the expression with IRF-1 morpholino treatment (10 μM, 18 h incubation) during infection. Lanes 1–6 correspond to 0, 4, 8, 12, 24 and 48 h.p.i., respectively (0 h as positive control, β-actin as internal control). PSR expression exhibited a delay in IRF-1 knockdown CHSE-214 cells compared to the control group (non-morpholino treated), whereas the IPNV viral protein VP2 dramatically increased in the same cells during IPNV infection; (C,D) Flow cytometry to analyze apoptosis or necrosis in CHSE-214 cells during IPNV infection. The data indicated that PS-positive cells and necrotic cells decreased in IRF-1 knockdown CHSE-214 cells (** p < 0.01; and *** p < 0.0001); and (E) Cell death was inhibited in IRF-1 knockdown CHSE-214 cells (lower panel), compared with control (upper panel) after IPNV infection (scale bar = 100 μm).
Figure 6.
Effects of IRF-1 knockdown on CHSE-214 cells during IPNV infection. (A) Real-time qPCR was used to determine the mRNA levels of IRF-1 with or without IRF-1 morpholino treatment following IPNV infection (MOI = 1). Expression were normalized to the internal control (β-actin) and is shown as the ΔCt value. The results indicated that IRF-1 mRNA expression was inhibited by approximately 38% with the morpholino; (B) Immunoblots of PSR or IPNV-VP2 expressionin IPNV-infected CHSE-214 cells (MOI = 1); The upper panels of (B) show the expression without IRF-1 morpholino treatment during infection, and the lower panels show the expression with IRF-1 morpholino treatment (10 μM, 18 h incubation) during infection. Lanes 1–6 correspond to 0, 4, 8, 12, 24 and 48 h.p.i., respectively (0 h as positive control, β-actin as internal control). PSR expression exhibited a delay in IRF-1 knockdown CHSE-214 cells compared to the control group (non-morpholino treated), whereas the IPNV viral protein VP2 dramatically increased in the same cells during IPNV infection; (C,D) Flow cytometry to analyze apoptosis or necrosis in CHSE-214 cells during IPNV infection. The data indicated that PS-positive cells and necrotic cells decreased in IRF-1 knockdown CHSE-214 cells (** p < 0.01; and *** p < 0.0001); and (E) Cell death was inhibited in IRF-1 knockdown CHSE-214 cells (lower panel), compared with control (upper panel) after IPNV infection (scale bar = 100 μm).
![Ijms 15 19281 g006]()
Figure 7.
Viral titers in IRF-1 knockdown CHSE-214 cells during IPNV infection. After IPNV infection, supernatants and cell lysates were harvested and titrated for 50% tissue culture infective dose (TCID50).
Figure 7.
Viral titers in IRF-1 knockdown CHSE-214 cells during IPNV infection. After IPNV infection, supernatants and cell lysates were harvested and titrated for 50% tissue culture infective dose (TCID50).
Several defense mechanisms to combat viral infection have been elucidated among the innate immune responses in host cells. In general, a variety of mechanisms have evolved. One antiviral defense mechanism is the apoptosis of host cells to facilitate voluntary cell death and prevent viral replication and spreading. Additionally, the host cell can also induce protective antiviral immune responses via the release of interferon (IFN), a cytokine that can enhance the antiviral response and stimulate the immune response to limit the extent of viral infection. In this study, we used IPNV, an important fish pathogen belonging to the family
Birnaviridae, to infect salmonid cells and investigated cellular defenses in host cells upon viral infection. IPNV infection primarily causes symptoms of cellular necrosis in fishes, but a previous study showed that infection can induce atypical apoptosis preceding necrosis in a salmonid fish cell line [
33]. Specifically, this study demonstrated that infected cells first appeared apoptotic and subsequently switched to secondary necrosis following pre-necrosis in salmonid cells during IPNV infection. The present findings indicated that IPNV infection caused cell death in salmonid cells and that apoptosis preceded necrosis (
Figure 1A,B). We observed that the CPE of the cells severely increased as the infection progressed (data not shown), and the viability of the infected cells dramatically decreased after 24 h.p.i. (
Figure 1C), corresponding to the peak in necrosis in the infected cells (
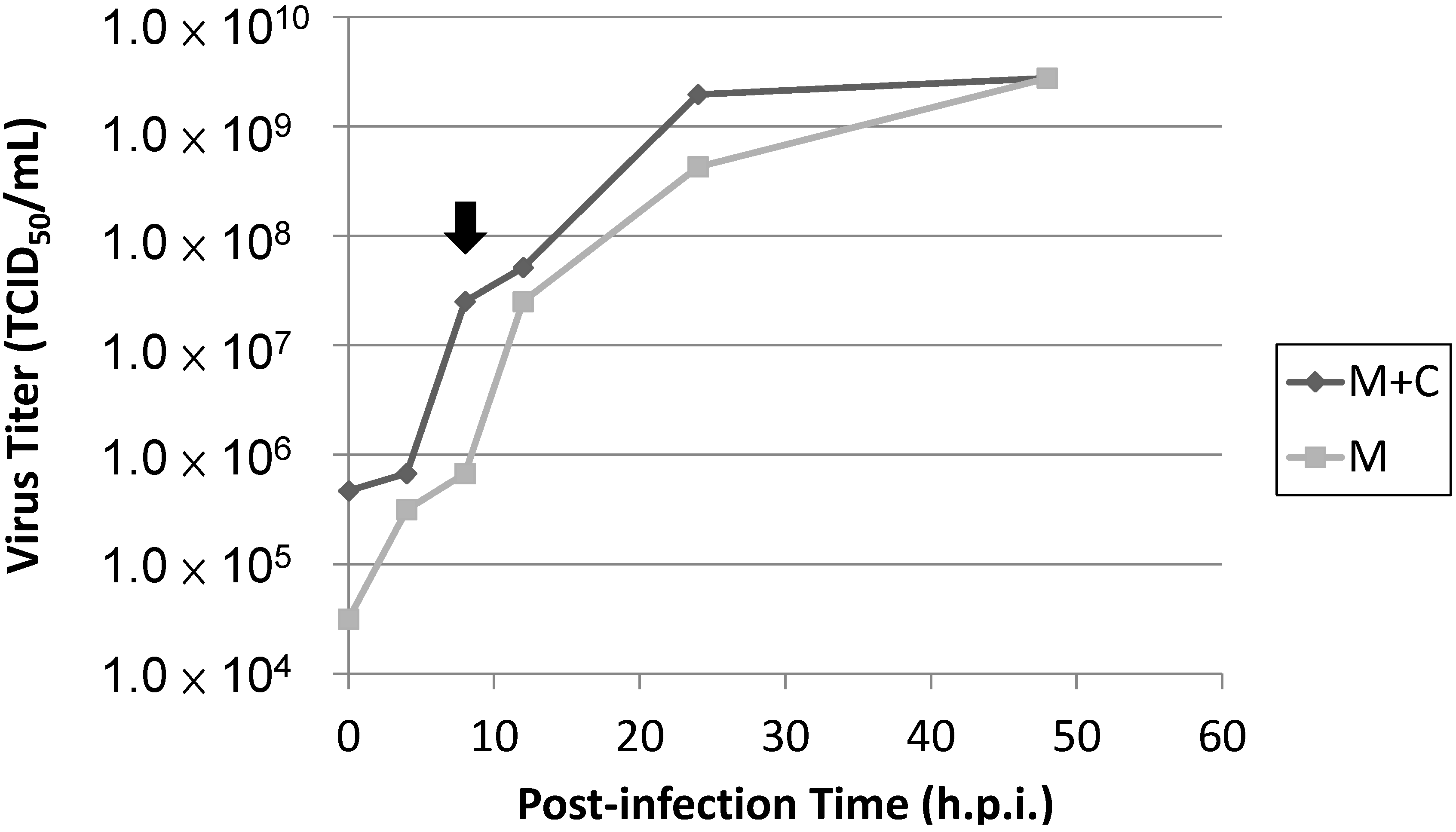
Figure 1B). Additionally, the presence of IPNV viral titers in the infected cells as detected by the TCID
50 assay indicated that the viral replication rate reached a peak at 8 h.p.i during infection (
Figure 8), and these data corresponded to previously published studies that indicate the total synthesis of virus-specific RNA reaches its maximum level between 8 and 10 h.p.i. [
34].
Figure 8.
TCID50 for viral titers in IPNV-infected cells. Totl viral particles were harvested from the medius as well as the cell pellets (M + C). The total number of viral particles was higher that the medius—Only viral particles (M) at each time point during the infection, while viral yields for both were equal at 48 h.p.i. The difference between (M + C) and (M) was due to the viral particles inside the nfected cells. Based on these results, viral replication rate reached a maximus inside the cells at 8 h.p.i. (point arrow). The time period between 8 and 10 h.p.i. represents the maximum total synthesis of virus-specific RNA.
Figure 8.
TCID50 for viral titers in IPNV-infected cells. Totl viral particles were harvested from the medius as well as the cell pellets (M + C). The total number of viral particles was higher that the medius—Only viral particles (M) at each time point during the infection, while viral yields for both were equal at 48 h.p.i. The difference between (M + C) and (M) was due to the viral particles inside the nfected cells. Based on these results, viral replication rate reached a maximus inside the cells at 8 h.p.i. (point arrow). The time period between 8 and 10 h.p.i. represents the maximum total synthesis of virus-specific RNA.
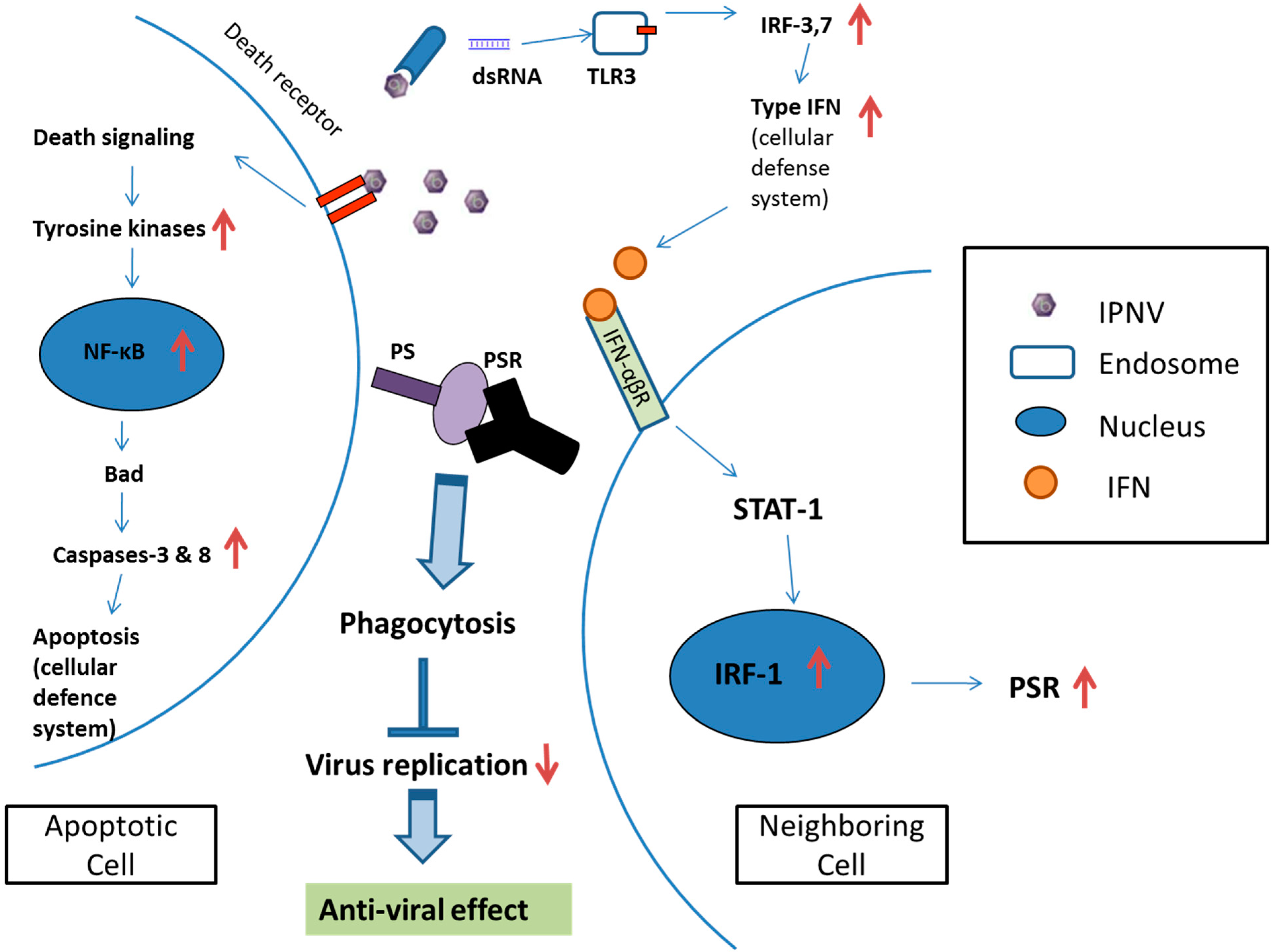
Elimination and clearance of virus-infected apoptotic cells is very important for host cell homeostasis and effective anti-viral action. Strikingly, previous results demonstrated that failure to engulf and remove these nearly dead cells may result in the survival of some cells, resembling the withdrawal from complete apoptosis. While such a reversal may not be of consequence in the context of the short lifespan of
C. elegans, a similar reversal in fate for a human cell could be dangerous, as it could prevent the quick clearance of apoptotic cells [
9]. Phagocytosis is a key process involved in eliminating virus-infected dying cells from an organism. Under normal physiological conditions, dying cells and pathogens are rapidly detected and removed by professional phagocytes such as macrophages and dendritic cells (DCs). In most cases, specific receptors on target cells are recognized and bound by phagocytes, triggering the intracellular signaling events required for phagocytosis [
10,
35]. Our previous study demonstrated that in addition to macrophages (professional phagocytes), which are capable of phagocytosing apoptotic cells, neighboring cells (non-professional phagocyte) were also capable of engulfing apoptotic salmonid cells during IPNV infection [
32]. Moreover, several research papers have determined that the phosphatidylserine receptor (PSR) plays a crucial role in the clearance of apoptotic cells. PSR serves as a membrane-associated receptor that regulates phagocytosis, and this has been confirmed in immature macrophages; additionally, this receptor has other functions when expressed in the cytosol and nuclei of mature macrophage-like cells [
7]. Therefore, we hypothesized that IPNV-induced apoptotic salmonid cells could be engulfed by neighboring cells via PSR binding with phosphatidylserine (PS). Although the mechanism of PSR induction is not yet clear, the type I IFN response is a general mechanism that is induced by viral infection, and thus we proposed a model suggesting a possible connection between them.
IFNs play an important role in innate immunity by inhibiting the replication and spread of pathogens, such as viruses, bacteria and parasites. Additionally, IFNs are key modulators of the immune response and exert anti-proliferative effects in some cell types. As a result, IFNs are used clinically to treat certain viral infections as well as some cancers and auto-immune diseases [
11]. IFNs are commonly grouped into two categories: Type I IFNs, also known as viral IFNs, including IFN-α and IFN-β, which are induced by viral infection, and type II IFN, also known as immune IFN or IFN-γ, which is induced by mitogenic or antigenic stimuli [
19,
32]. Robertsen B
et al. (2003) [
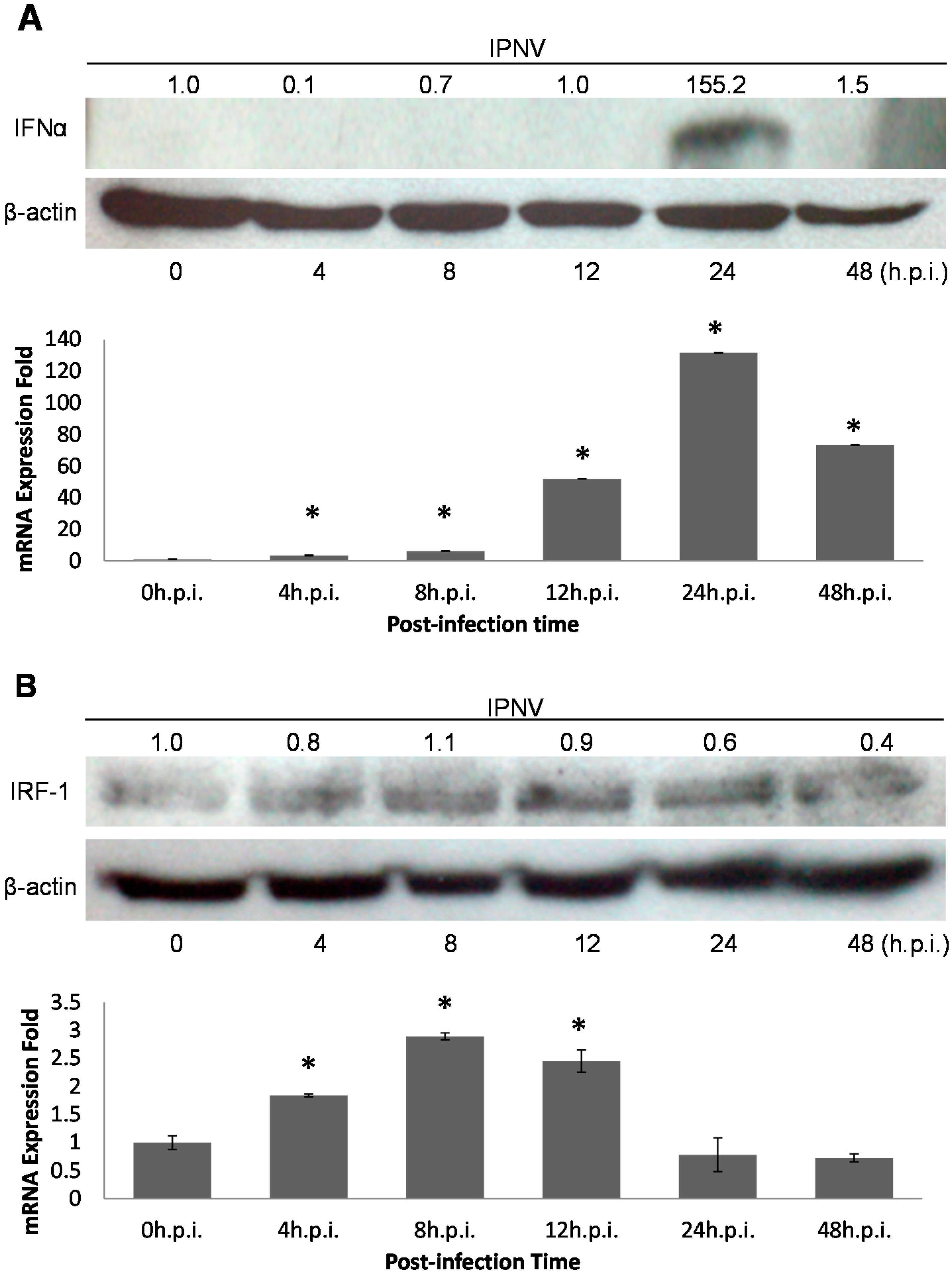
31] determined that IFN was induced by poly I:C treatment in the head kidneys of Atlantic salmon, and salmonid IFN-α1 promoted the induction of the antiviral protein Mx in CHSE-214 cells. This suggested that the type I IFN system existed in fishes as well as in mammals. We determined that the IFN pathway was induced by IPNV dsRNA, and PSR expression was also elevated upon IPNV infection in our system (
Figure 2 and
Figure 3A). Interestingly, rIFN-α proteins were induced early during IPNV infection but were not detected after 24 h.p.i. (
Figure 3A). This suggests that IPNV may be able to inhibit IFN signaling in fish cells and has evolved mechanisms to overcome IFN responses [
32,
36,
37]. Therefore, the virus may have its own strategies to combat cellular defenses, such as the secretion of proteins that can control cellular apoptotic processing by regulating the expression of related genes to promote a suitable environment for viral replication in host cells.
Cells secrete type I IFNs in response to viral infection following the elevated activity of various transcription factors (TFs) including the interferon regulatory factor (IRF) family [
28,
38], which regulates innate response gene transcription as transcriptional activators or repressors depending on the target gene and their binding partners [
39]. Nine members of the mammalian IRF family (IRFs 1–9) are known, and in particular, mammalian IRF-1, IRF-3 and IRF-7, are known to regulate the expression of type I IFNs, which are the cytokines involved in antiviral defense in response to virus-mediated signaling [
11,
26,
36]. During viral infection, IRF-3 plays an early role in inducing the transcription of IFN-β, which in turn induces the expression of IRF-7 and then promotes the transcription of members of the IFN-α gene family, thereby creating a positive-feedback loop that boosts the host antiviral response [
38]. IRF-1 is also induced by IFNs after IRF-3 and IRF-7 induction and hence it is located more downstream than IRF-3 and IRF-7 in the type I IFN signaling pathway [
19,
26,
36,
38,
40]. Based on this, we analyzed the promoter of PSR and found that there were IRF-1 binding elements located 900 bp upstream of the
PSR gene that might constitute the specific transcription factor binding region because the repetitive sequence that might be an enhancer region of the
PSR gene was located farther upstream. The putative IRF-1 binding sites in the PSR promoter suggested that IRF-1 can regulate
PSR gene expression via promoter binding, during viral infection (
Figure 2 and
Figure 4). Some variation between PSR protein levels and mRNA levels (data not shown) may be explained by the insufficient specificity of the primers used for
PSR gene detection, which were designed from the zebrafish sequence because the sequence of salmon PSR is unknown.
After locating the PSR promoter, which promotes
PSR gene expression during IPNV infection, we tried to elucidate the transcriptional mechanism of PSR by analyzing the TF binding sites in the promoter. Because the sequence of the salmonid PSR promoter is unknown, we selected the zebrafish sequence despite the differences between the two genes. Zebrafish IRF-1 displayed 40%–84% similarity to other fishes and 35%–39% similarity to other vertebrate IRF-1 sequences. We were still able to identify putative IRF-1 binding sites in the zebrafish PSR promoter. In addition, both PSR and IRF-1 were elevated after IPNV infection independent of the immunoblot results (
Figure 2 and
Figure 3) or the qPCR results. As the zebrafish PSR promoter contained putative IRF-1-binding sites, we tested whether rIFN-α or IPNV could induce the PSR promoter. We cloned the 900-bp promoter of zebrafish PSR and transfected it into salmonid cells following by IPNV infection or rIFN-α treatment to induce promoter activity. The data in
Figure 5A–C indicated that the PSR promoter was activated either by IPNV infection or rIFN-α treatment. Furthermore, we deleted the full-length PSR promoter and then observed that luciferase activity decreased with decreasing promoter length.
To clarify the relationship between PSR and IRF-1, we used an IRF-1-deleted PSR promoter construct and translation-blocking IRF-1 morpholinos to reduce the expression levels of IRF-1 in salmonid cells during viral infection and observed the effects after knockdown. When the IRF-1-deleted PSR gene promoter was analyzed, a markable decrease in the promoter activity was observed when the IRF-1 binding site from −434 to −300 was deleted, and pPSRP409-600 showed similar effects compared to the minimal promoter pPSRP1-150 but exhibited slightly higher promoter activity (
Figure 5E). These results suggested that the IRF-1 binding site in the PSR promoter is crucial for
PSR gene expression. Additionally, we determined that the expression of IRF-1 was suppressed during IPNV infection (
Figure 6A) after salmon IRF-1 morpholino oligos were transferred into the salmonid cells. Simultaneously, the PSR expression was delayed until 24 h.p.i., whereas the non-morpholino treated samples expressed PSR earlier at 8 h.p.i. (
Figure 6B), and viral protein replication dramatically increased IRF-1 knockdown CHSE-214 cells (
Figure 6C). Therefore, it can be assumed that PSR expression in salmonid cells during IPNV infection is induced through IRF-1 regulation. The variations in the percentages of virus-induced apoptosis and necrosis in IRF-1 knockdown CHSE-214 cells were quantitated with flow cytometry. The results indicated that the number of PS-positive cells decreased markedly in IRF-1 knockdown cells during IPNV infection (
Figure 6D), and the number of necrotic cells also decreased in IRF-1 knockdown CHSE-214 cells at 8 and 24 h.p.i. (
Figure 6E). However, the morphology and mortality of IRF-1 morpholino-treated cells changed, and the CPE was less severe compared to non-morpholino cells during IPNV infection (
Figure 6F).These results indicated that IRF-1 knockdown corresponds to reduced apoptotic cell death and necrotic cell death. Therefore, cell death is inhibited in IRF-1 knockdown CHSE-214 cells after IPNV infection, whereas viral replication is boosted, and it can be suggested that IRF-1 is required to inhibit viral replication. Conversely, the results obtained with the IRF-1 morpholino demonstrate that IRF-1 is required for virus-induced apoptosis, which corresponds with the data of MT Stang
et al. (2007) [
41], who showed that IRF-1 can induce ligand-independent FADD/caspase-8-mediated apoptosis in breast cancer cells despite the fact that they used IFNγ as an inducer. Thus, IRF-1 is required for apoptosis induction. In general, IRF-1 activity does not lead to cell death, whereas it promotes apoptosis under certain physiological and pathological conditions (e.g., DNA damage or viral stimuli) [
26,
40]. Overall, IRF-1 knockdown in infected cells will inhibit the IPNV-induced cytopathic effect as well as IPNV-induced apoptotic and necrotic events, thus increasing IPNV viral protein yields. Strikingly, viral titers measured by TCID
50 in IRF-1 knockdown cells did not reveal increased numbers of whole replicated viral particles in the culture medium (
Figure 7). However, we could not provide a full answer to this phenomenon in this study. This interesting question requires further experimentation in future research.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








