Design, Synthesis and Biological Evaluation of Sulfamide and Triazole Benzodiazepines as Novel p53-MDM2 Inhibitors

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemistry


2.2. Disrupting the Binding of p53-MDM2

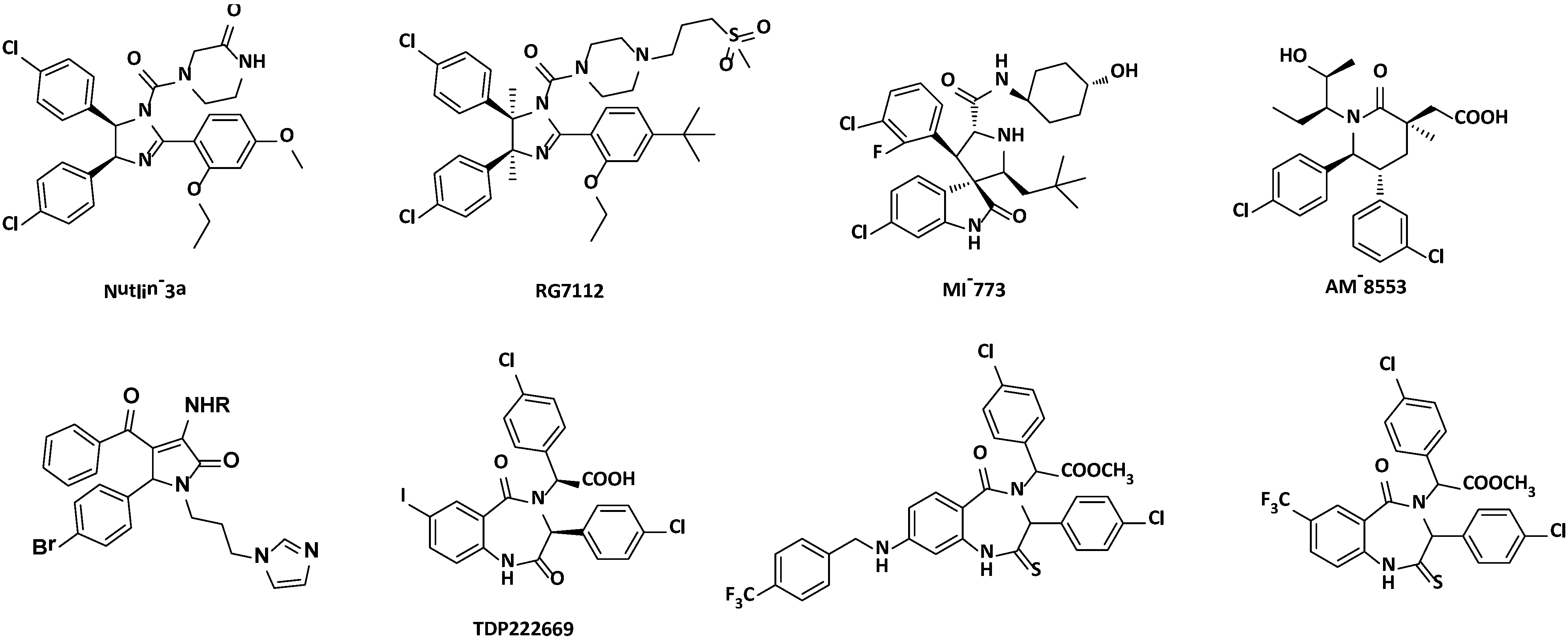{kind=link}
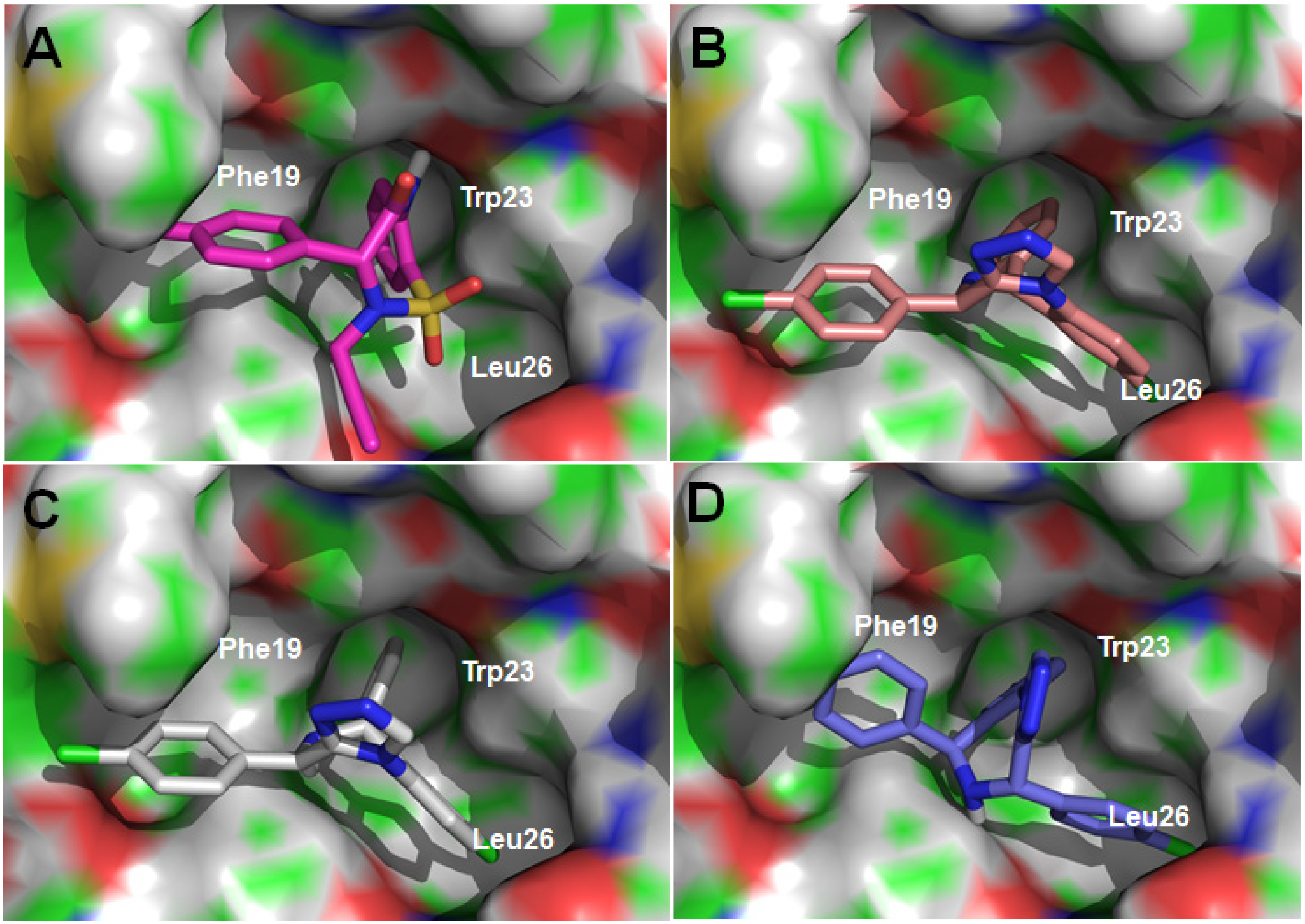{kind=link}
{kind=link}
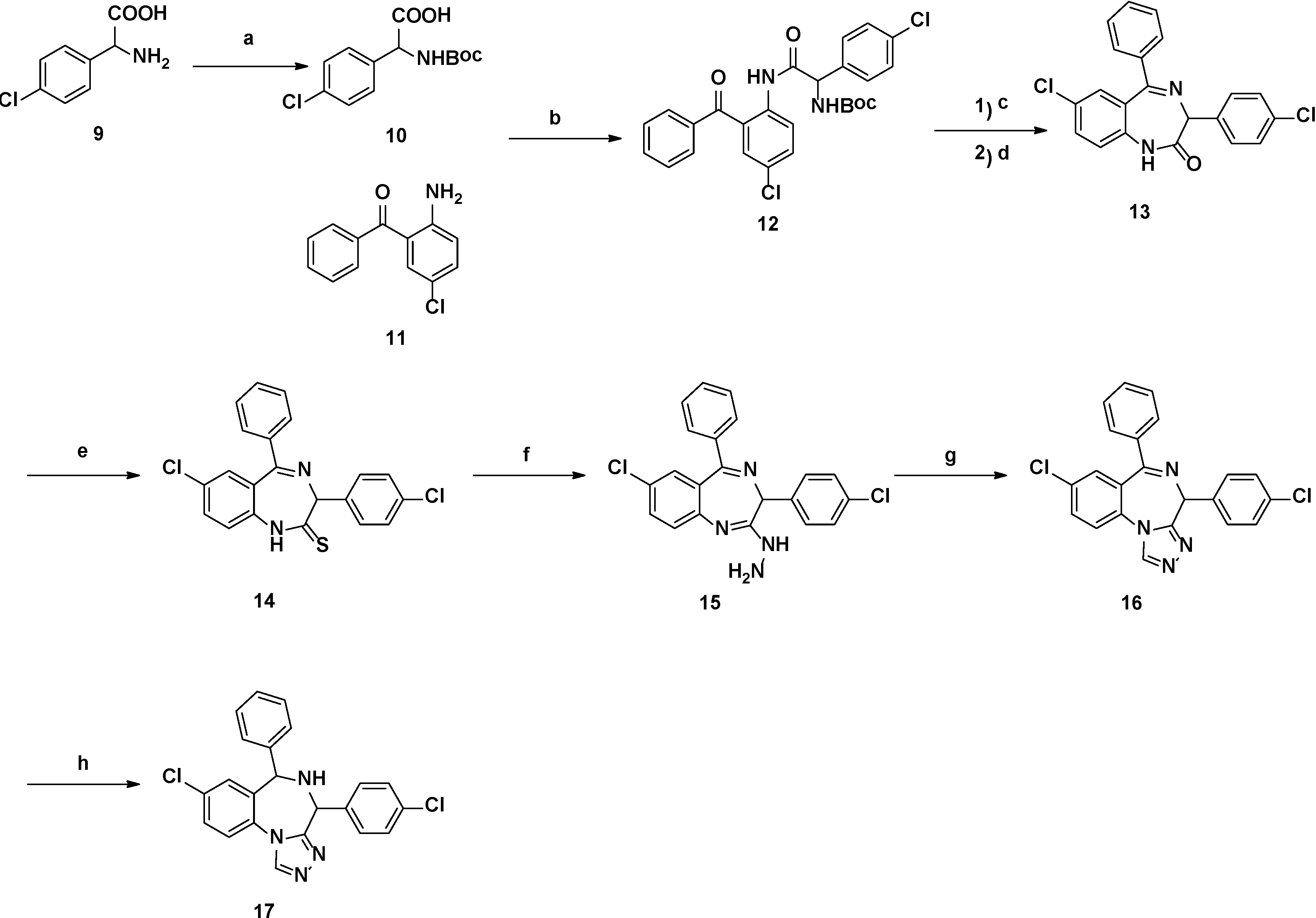{kind=link}
| Compounds | Ki (μM ) | IC50 (μM) | |||
|---|---|---|---|---|---|
| Saos-2 | U-2 OS | A549 | NCI-H1299 | ||
| (p53 null) | (wt-p53) | (wt-p53) | (p53 null) | ||
| 7a | 0.43 | >100 | >100 | >100 | >100 |
| 7b | 0.36 | >100 | >100 | >100 | >100 |
| 7c | 0.26 | >100 | >100 | >100 | >100 |
| 7d | >100 | >100 | >100 | 92.3 | >100 |
| 7e | >100 | >100 | 90.5 | >100 | >100 |
| 7f | 78.0 | >100 | >100 | >100 | >100 |
| 7g | 8.14 | >100 | >100 | >100 | >100 |
| 8 | 11.9 | >100 | >100 | >100 | >100 |
| 13 | 0.20 | 3.01 | 3.12 | 9.16 | 6.11 |
| 14 | 8.76 | 3.67 | 5.31 | 24.05 | 15.56 |
| 16 | 1.22 | 3.55 | 4.17 | 7.62 | 5.50 |
| 17 | 2.80 | 24.81 | 33.98 | 26.78 | 34.59 |
| Nutlin-3 | 0.09 | 20.8 | 16.3 | 12.7 | 4.15 |

2.3. In Vitro Antitumor Activity
3. Experimental Section
3.1. Chemistry
3.2. General Procedure
3.2.1. Synthesis of 7a and 7b
3.2.2. Synthesis of 7c–7g
3.3. Computational Protocol
3.4. p53-MDM2 Binding Assay
3.5. In Vitro Antitumor Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Cochran, A.G. Antagonists of protein-protein interactions. Chem. Biol. 2000, 7, R85–R94. [Google Scholar] [CrossRef]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? BioEssays 1993, 15, 689–690. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Fry, D.C. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84, 535–552. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.J.; Jiang, N.; Liu, J.J.; Zhao, C.L.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Wang, S.; Yu, S.; Sun, W.; Kumar, S.; Sun, D.; Zou, P.; McEachern, D.; Zhao, Y. Spiro-oxindole mdm2 antagonists. U.S. Patent 8518984 B2, 27 August 2013. [Google Scholar]
- Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 protein-protein interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Wu, Y.; Guo, Z.; Li, J.; Yao, J.; Xing, C.; Sheng, C.; Zhang, W. Double-edged Swords as Cancer Therapeutics: Novel Orally Active Small Molecules Simultaneously Inhibit p53-MDM2 Interaction and the NF-κB Pathway. J. Med. Chem. 2014, 57, 567–577. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef]
- Marugan, J.J.; Leonard, K.; Raboisson, P.; Gushue, J.M.; Calvo, R.; Koblish, H.K.; Lattanze, J.; Zhao, S.; Cummings, M.D.; Player, M.R.; et al. Enantiomerically pure 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3115–3120. [Google Scholar] [CrossRef]
- Parks, D.J.; LaFrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Marugan, J.J.; Raboisson, P.; Schubert, C.; Koblish, H.K.; Zhao, S.; Franks, C.F.; et al. Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorg. Med. Chem. Lett. 2006, 16, 3310–3314. [Google Scholar] [CrossRef]
- Pevarello, P.; Fancelli, D.; Vulpetti, A.; Amici, R.; Villa, M.; Pittala, V.; Vianello, P.; Cameron, A.; Ciomei, M.; Mercurio, C.; et al. 3-Amino-1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: A new class of CDK2 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1084–1090. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Zhang, Y.; Guo, Z.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; et al. Synthesis and biological evaluation of thio-benzodiazepines as novel small molecule inhibitors of the p53-MDM2 protein-protein interaction. Eur. J. Med. Chem. 2011, 46, 5654–5661. [Google Scholar] [CrossRef]
- Guo, Z.; Zhuang, C.; Zhu, L.; Zhang, Y.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; Sheng, C.; et al. Structure-activity relationship and antitumor activity of thio-benzodiazepines as p53-MDM2 protein-protein interaction inhibitors. Eur. J. Med. Chem. 2012, 56, 10–16. [Google Scholar] [CrossRef]
- Li, J.; Wu, Y.; Guo, Z.; Zhuang, C.; Yao, J.; Dong, G.; Yu, Z.; Min, X.; Wang, S.; Liu, Y.; et al. Discovery of 1-arylpyrrolidone derivatives as potent p53-MDM2 inhibitors based on molecule fusing strategy. Bioorg. Med. Chem. Lett. 2014, 24, 2648–2650. [Google Scholar] [CrossRef]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef]
- Miao, Z.; Zhu, L.; Dong, G.; Zhuang, C.; Wu, Y.; Wang, S.; Guo, Z.; Liu, Y.; Wu, S.; Zhu, S.; et al. A new strategy to improve the metabolic stability of lactone: Discovery of (20S,21S)-21-fluorocamptothecins as novel, hydrolytically stable topoisomerase I inhibitors. J. Med. Chem. 2013, 56, 7902–7910. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, Z.; Zhuang, C.; Wu, Y.; Guo, Z.; Li, J.; Dong, G.; Yao, J.; Sheng, C.; Miao, Z.; Zhang, W. Design, Synthesis and Biological Evaluation of Sulfamide and Triazole Benzodiazepines as Novel p53-MDM2 Inhibitors. Int. J. Mol. Sci. 2014, 15, 15741-15753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms150915741
Yu Z, Zhuang C, Wu Y, Guo Z, Li J, Dong G, Yao J, Sheng C, Miao Z, Zhang W. Design, Synthesis and Biological Evaluation of Sulfamide and Triazole Benzodiazepines as Novel p53-MDM2 Inhibitors. International Journal of Molecular Sciences. 2014; 15(9):15741-15753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms150915741
Chicago/Turabian StyleYu, Zhiliang, Chunlin Zhuang, Yuelin Wu, Zizhao Guo, Jin Li, Guoqiang Dong, Jianzhong Yao, Chunquan Sheng, Zhenyuan Miao, and Wannian Zhang. 2014. "Design, Synthesis and Biological Evaluation of Sulfamide and Triazole Benzodiazepines as Novel p53-MDM2 Inhibitors" International Journal of Molecular Sciences 15, no. 9: 15741-15753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms150915741