
Applications of Engineered DNA-Binding Molecules Such as TAL Proteins and the CRISPR/Cas System in Biology Research

Abstract
:
{kind=link}
{kind=link}
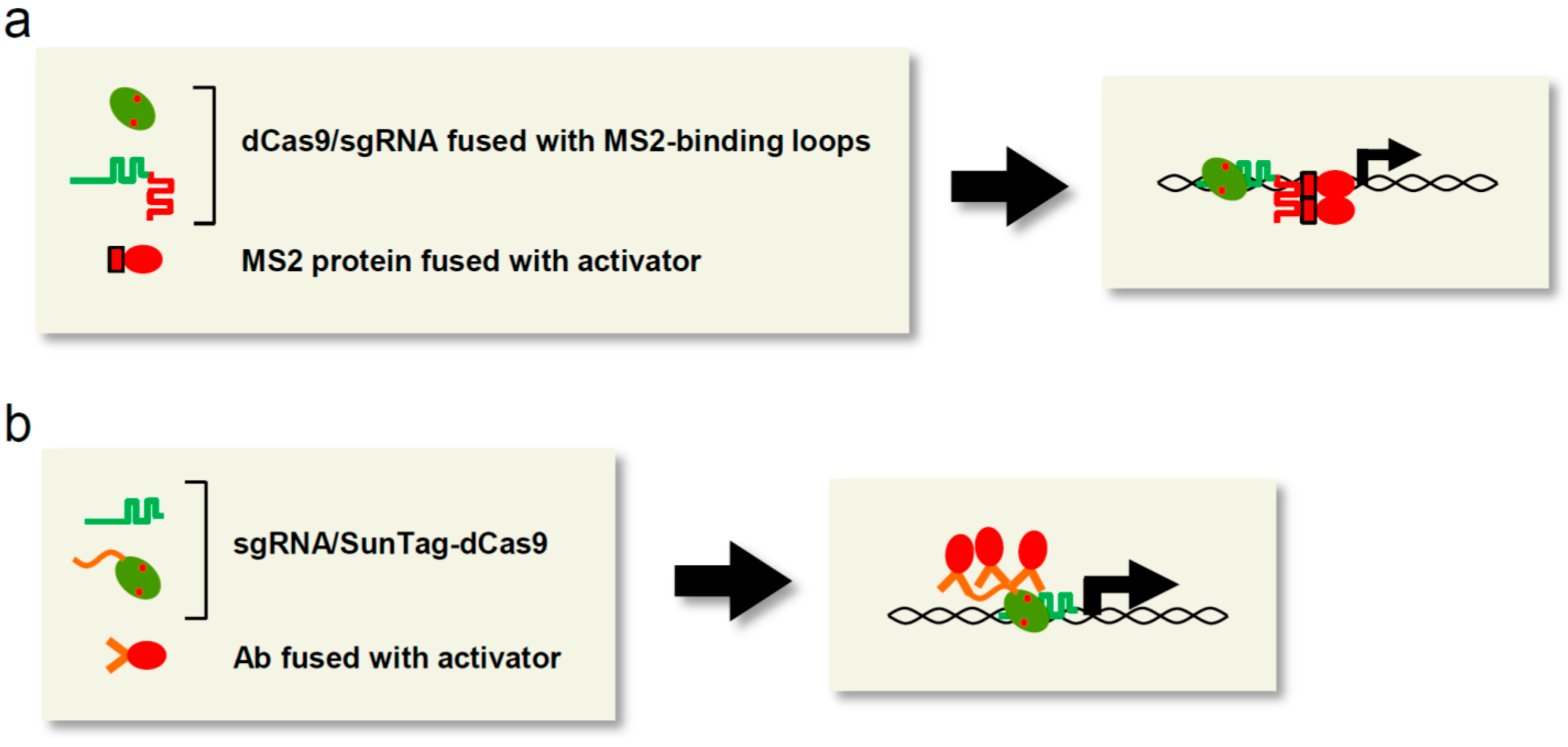{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. General Information on TAL and CRISPR/Cas
3. Applications of TAL and CRISPR/Cas to Locus-Specific Transcriptional Regulation
3.1. Transcriptional Activation

3.2. Transcriptional Activation by Scaffold RNA System

3.3. Transcriptional Activation by Synergistic Regulation Systems
3.4. Transcriptional Repression
3.5. Epigenetic Modification
3.6. Off-Target Activity
3.7. Inducible Systems for Locus-Specific Transcriptional Regulation
3.8. Influence of Chromatin Structures of Target Sites
3.9. Genome-Wide Approaches for Locus-Specific Transcriptional Regulation
3.10. Manipulation of Outputs of Biological Processes
4. Locus Visualization for Live Imaging

5. Biochemical Analysis of RNAs

6. Isolation of Genomic Regions of Interest for Analysis of Genome Functions

7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Wijshake, T.; Baker, D.J.; van de Sluis, B. Endonucleases: New tools to edit the mouse genome. Biochim. Biophys. Acta 2014, 1842, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, K.; Podevin, N.; Breyer, D.; Carroll, D.; Herman, P. Engineering nucleases for gene targeting: Safety and regulatory considerations. New Biotechnol. 2014, 31, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 6213. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.M.; Jenkins, B.V.; O’Connor-Giles, K.M.; Wildonger, J. A CRISPR view of development. Genes Dev. 2014, 28, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Herr, W. The herpes simplex virus VP16-induced complex: The makings of a regulatory switch. Trends Biochem. Sci. 2003, 28, 294–304. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent Oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, J.; Tsang, J.C.H.; Ooi, J.; Wu, D.; Liu, P. Reprogramming to pluripotency using designer TALE transcription factors targeting enhancers. Stem Cell Rep. 2013, 1, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Dove, S.L.; Hochschild, A. Conversion of the omega subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 1998, 12, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Shechner, D.M.; Hacisuleyman, E.; Younger, S.T.; Rinn, J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 2015, 12, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Ousterout, D.G.; Brunger, J.M.; Farin, A.M.; Glass, K.A.; Guilak, F.; Crawford, G.E.; Hartemink, A.J.; Gersbach, C.A. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat. Methods 2013, 10, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Reyon, D.; Angstman, J.F.; Fu, Y.; Sander, J.D.; Joung, J.K. Robust, synergistic regulation of human gene expression using TALE activators. Nat. Methods 2013, 10, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Zhou, R.; Kuo, Y.; Cunniff, M.; Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012, 3, 968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiang, D.; Heriyanto, F.; Gao, Y.; Qian, Z.; Wu, W.S. Dissecting the roles of miR-302/367 cluster in cellular reprogramming using TALE-based repressor and TALEN. Stem Cell Rep. 2013, 1, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, E.; Qian, Z.; Wu, W.S. A multicolor panel of TALE-KRAB based transcriptional repressor vectors enabling knockdown of multiple gene targets. Sci. Rep. 2014, 4, 7338. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dai, Z.; Liang, Y.; Yin, M.; Ma, K.; He, M.; Ouyang, H.; Teng, C.B. Sequence-specific inhibition of microRNA via CRISPR/CRISPRi system. Sci. Rep. 2014, 4, 3943. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tsang, J.C.H.; Gaba, F.; Wu, D.; Lu, L.; Liu, P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 2014, 42, e155. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Bochar, D.A.; Hakimi, M.A.; Cooch, N.; Shiekhattar, R. Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 2006, 26, 6395–6402. [Google Scholar] [CrossRef] [PubMed]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Ippolito, A.M.D.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Pang, J.; Cheng, H.; Liu, W.; Di, J.; Xiao, H. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget 2015, 6, 10030–10044. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Cencic, R.; Miura, H.; Malina, A.; Robert, F.; Ethier, S.; Schmeing, T.M.; Dostie, J.; Pelletier, J. Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage. PLoS ONE 2014, 9, e109213. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussolino, C.; Morbitzer, R.; Lütge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-based photoactivatable transcription system. Chem. Biol. 2015, 22, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Kawano, F.; Suzuki, H.; Furuya, A.; Sato, M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun. 2015, 6, 6256. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Q.; Rebar, E.J.; Zhang, L.; Liu, Q.; Jamieson, A.C.; Liang, Y.; Qi, H.; Li, P.X.; Chen, B.; Mendel, M.C.; et al. Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions: Activation of vascular endothelial growth factor A. J. Biol. Chem. 2001, 276, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Valton, J.; Dupuy, A.; Daboussi, F.; Thomas, S.; Maréchal, A.; Macmaster, R.; Melliand, K.; Juillerat, A.; Duchateau, P. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J. Biol. Chem. 2012, 287, 38427–38432. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Yin, P.; Yan, C.; Pan, X.; Gong, X.; Qi, S.; Xie, T.; Mahfouz, M.; Zhu, J.-K.; Yan, N.; et al. Recognition of methylated DNA by TAL effectors. Cell Res. 2012, 22, 1502–1504. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lei, Y.; Wong, W.K.; Liu, S.; Lee, K.C.; He, X.; You, W.; Zhou, R.; Guo, J.T.; Chen, X.; et al. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res. 2014, 42, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T. Violacein and related tryptophan metabolites produced by Chromobacterium violaceum: Biosynthetic mechanism and pathway for construction of violacein core. Appl. Microbiol. Biotechnol. 2011, 91, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, N.A.; Genga, R.M.J.; Enuameh, M.S.; Garber, M.; Wolfe, S.A.; Maehr, R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 2014, 141, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.M.; Shipman, S.L.; Byrne, S.M.; Sanjana, N.E.; Murn, J.; Li, Y.; Li, S. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Trask, B.J. Fluorescence in situ hybridization. Trends Genet. 1991, 7, 149–154. [Google Scholar] [CrossRef]
- Robinett, C.; Straight, A.; Li, G.; Willhelm, C.; Sudlow, G.; Murray, A.; Belmont, A.S. In vivo loclization of DNA sequences and visualization of large-scale chromatin organizacion using lac operator/represor recognition. J. Cell Biol. 1996, 135, 1685–1700. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Lam, E. Detection of chromosomes tagged with green fluorescent protein in live Arabidopsis thaliana plants. Genome Biol. 2001, 2, research0045.1–research0045.10. [Google Scholar] [CrossRef] [PubMed]
- Matzke, A.J.; Huettel, B.; van der Winden, J.; Matzke, M. Use of two-color fluorescence-tagged transgenes to study interphase chromosomes in living plants. Plant Physiol. 2005, 139, 1586–1596. [Google Scholar] [CrossRef]
- Lindhout, B.I.; Fransz, P.; Tessadori, F.; Meckel, T.; Hooykaas, P.J.J.; van der Zaal, B.J. Live cell imaging of repetitive DNA sequences via GFP-tagged polydactyl zinc finger proteins. Nucleic Acids Res. 2007, 35, e107. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Ziegler-Birling, C.; Torres-Padilla, M.E. Live visualization of chromatin dynamics with fluorescent TALEs. Nat. Struct. Mol. Biol. 2013, 20, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.R.; Zhao, P.; Olson, S.; Duff, M.O.; Graveley, B.R.; Wells, L.; Terns, R.M.; Terns, M.P. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 2009, 139, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Benda, C.; Ebert, J.; Scheltema, R.A.; Schiller, H.B.; Baumgärtner, M.; Bonneau, F.; Mann, M.; Conti, E. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol. Cell 2014, 56, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ramia, N.F.; Spilman, M.; Tang, L.; Shao, Y.; Elmore, J.; Hale, C.; Cocozaki, A.; Bhattacharya, N.; Terns, R.M.; Terns, M.P.; et al. Essential structural and functional roles of the Cmr4 subunit in RNA cleavage by the Cmr CRISPR-Cas complex. Cell Rep. 2014, 9, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Fujii, H. Insertional chromatin immunoprecipitation: A method for isolating specific genomic regions. J. Biosci. Bioeng. 2009, 108, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujii, H. Direct identification of insulator components by insertional chromatin immunoprecipitation. PLoS ONE 2011, 6, e26109. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujii, H. Efficient isolation of specific genomic regions by insertional chromatin immunoprecipitation (iChIP) with a second-generation tagged LexA DNA-binding domain. Adv. Biosci. Biotechnol. 2012, 3, 626–629. [Google Scholar] [CrossRef]
- Fujita, T.; Fujii, H. Locus-specific biochemical epigenetics/chromatin biochemistry by insertional chromatin immunoprecipitation. ISRN Biochem. 2013, 2013, 913273. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujii, H. Efficient isolation of specific genomic regions retaining molecular interactions by the iChIP system using recombinant exogenous DNA-binding proteins. BMC Mol. Biol. 2014, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Kitaura, F.; Fujii, H. A critical role of the Thy28-MYH9 axis in B cell-specific expression of the Pax5 gene in chicken B cells. PLoS ONE 2015, 10, e0116579. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujii, H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem. Biophys. Res. Commun. 2013, 439, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Asano, Y.; Ohtsuka, J.; Takada, Y.; Saito, K.; Ohki, R.; Fujii, H. Identification of telomere-associated molecules by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP). Sci. Rep. 2013, 3, 3171. [Google Scholar] [CrossRef]
- Fujita, T.; Fujii, H. Identification of proteins associated with an IFNγ-responsive promoter by a retroviral expression system for enChIP using CRISPR. PLoS ONE 2014, 9, e103084. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Yuno, M.; Okuzaki, D.; Ohki, R.; Fujii, H. Identification of non-coding RNAs associated with telomeres using a combination of enChIP and RNA sequencing. PLoS ONE 2015, 10, e0123387. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujii, H. Isolation of specific genomic regions and identification of associated molecules by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Methods Mol. Biol. 2015, 1288, 43–52. [Google Scholar] [PubMed]
- Nusinzon, I.; Horvath, C.M. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 14742–14747. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Paulson, M.; Holko, M.; Rice, C.M.; Williams, B.R.G.; Marié, I.; Levy, D.E. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 9578–9583. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujita, T.; Fujii, H. Applications of Engineered DNA-Binding Molecules Such as TAL Proteins and the CRISPR/Cas System in Biology Research. Int. J. Mol. Sci. 2015, 16, 23143-23164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161023143
Fujita T, Fujii H. Applications of Engineered DNA-Binding Molecules Such as TAL Proteins and the CRISPR/Cas System in Biology Research. International Journal of Molecular Sciences. 2015; 16(10):23143-23164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161023143
Chicago/Turabian StyleFujita, Toshitsugu, and Hodaka Fujii. 2015. "Applications of Engineered DNA-Binding Molecules Such as TAL Proteins and the CRISPR/Cas System in Biology Research" International Journal of Molecular Sciences 16, no. 10: 23143-23164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161023143