
Monosodium Urate Crystal-Induced Chondrocyte Death via Autophagic Process

Abstract
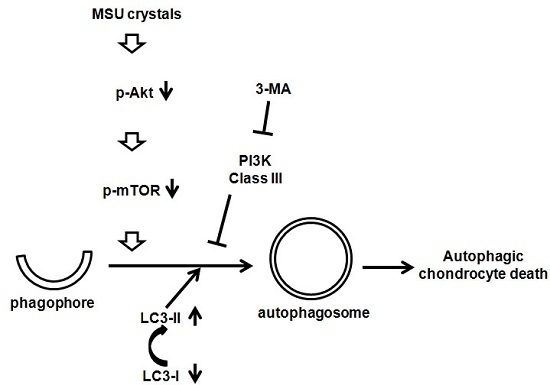
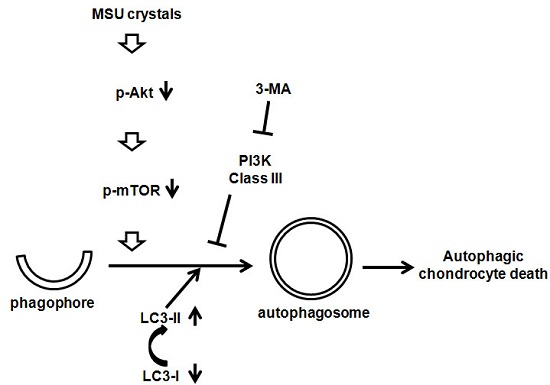
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
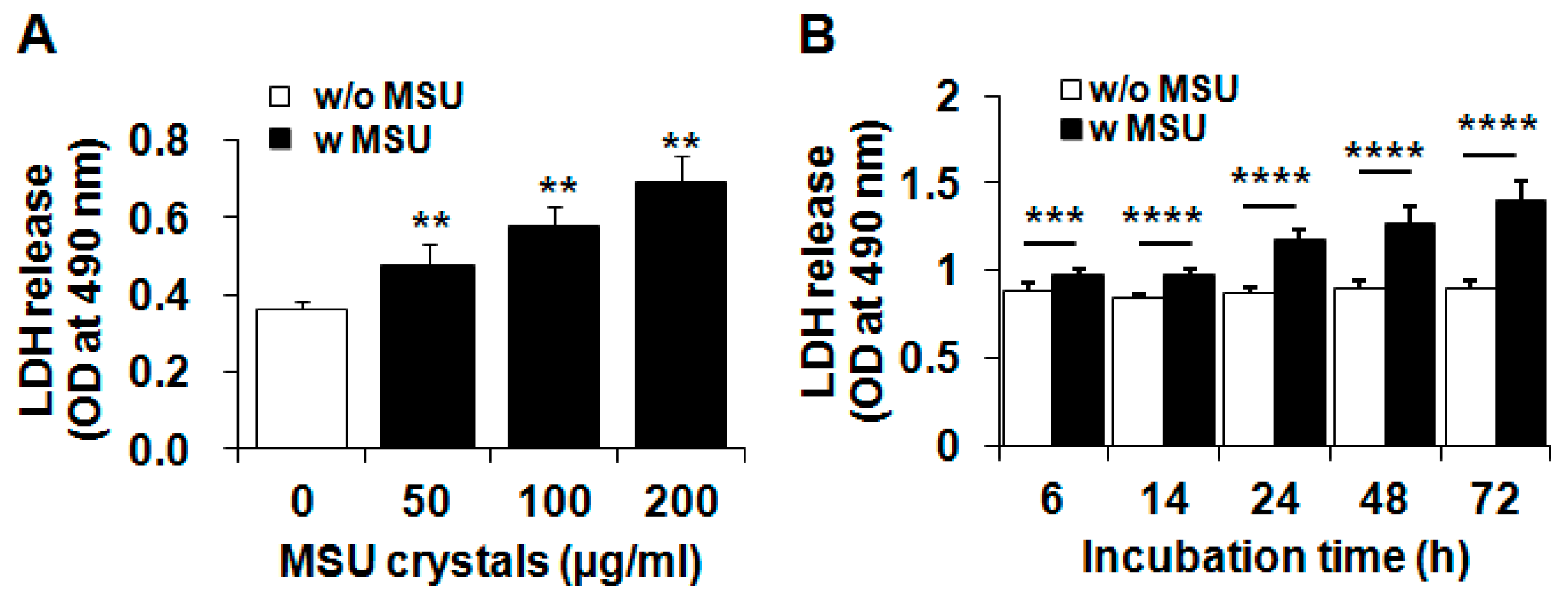
2.1. MSU (Monosodium Urate) Crystals Reduced the Viability of Articular Chondrocytes

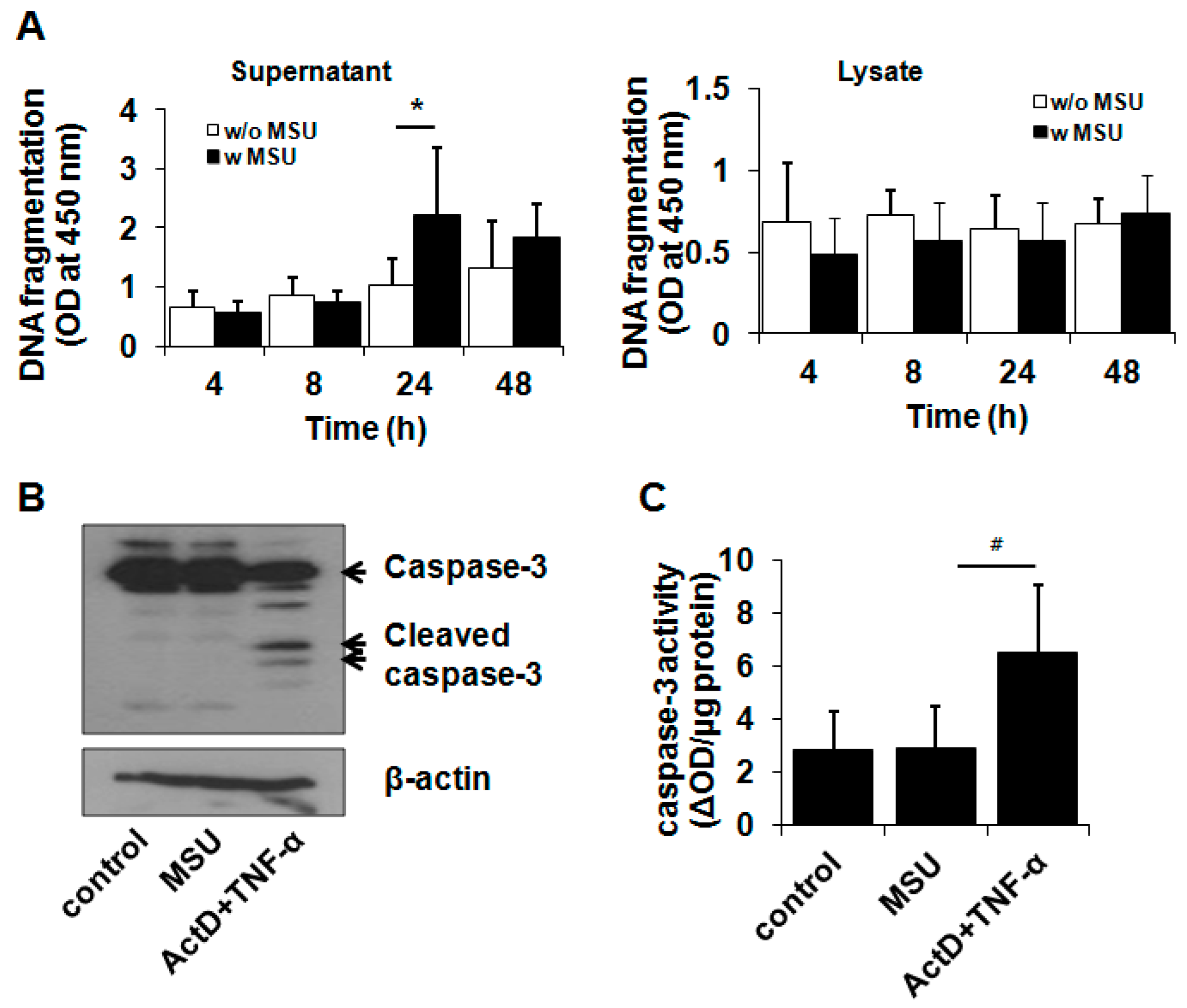
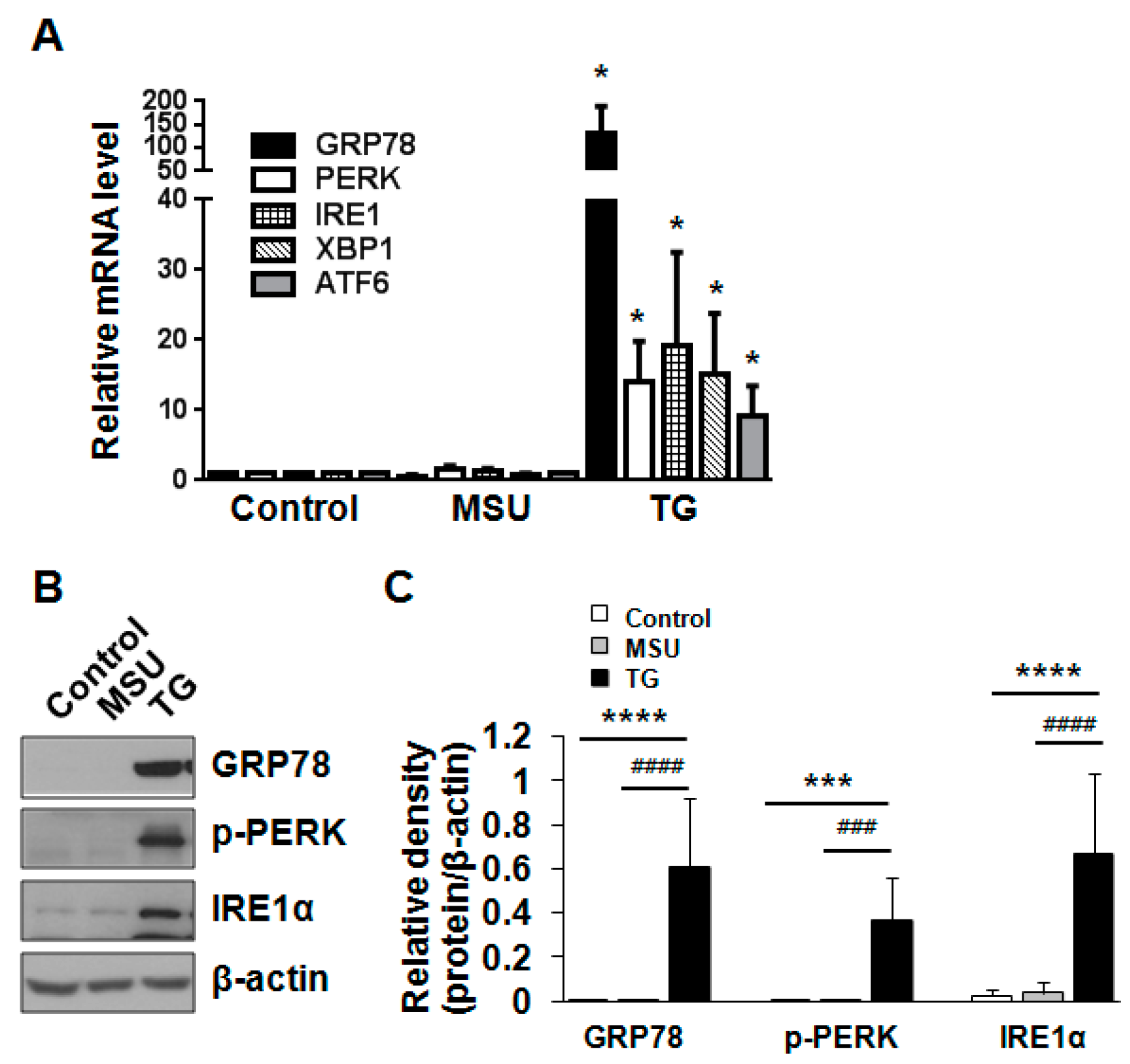
2.2. MSU Crystal-Induced Cell Death Is Independent of Apoptosis or ER Stress-Induced Death


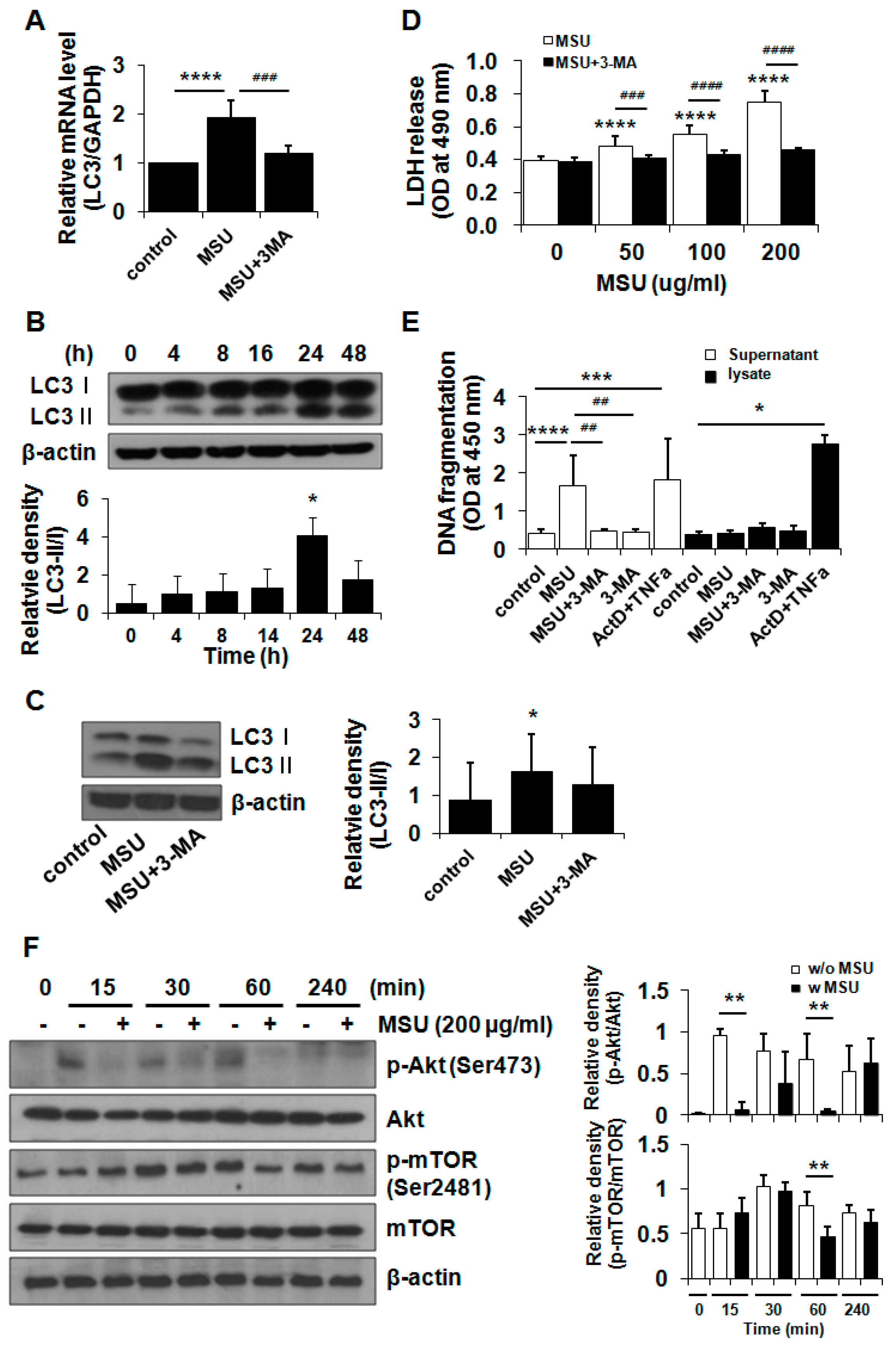
2.3. MSU Crystals Induced Articular Chondrocyte Death via Activation of the Autophagy Pathway

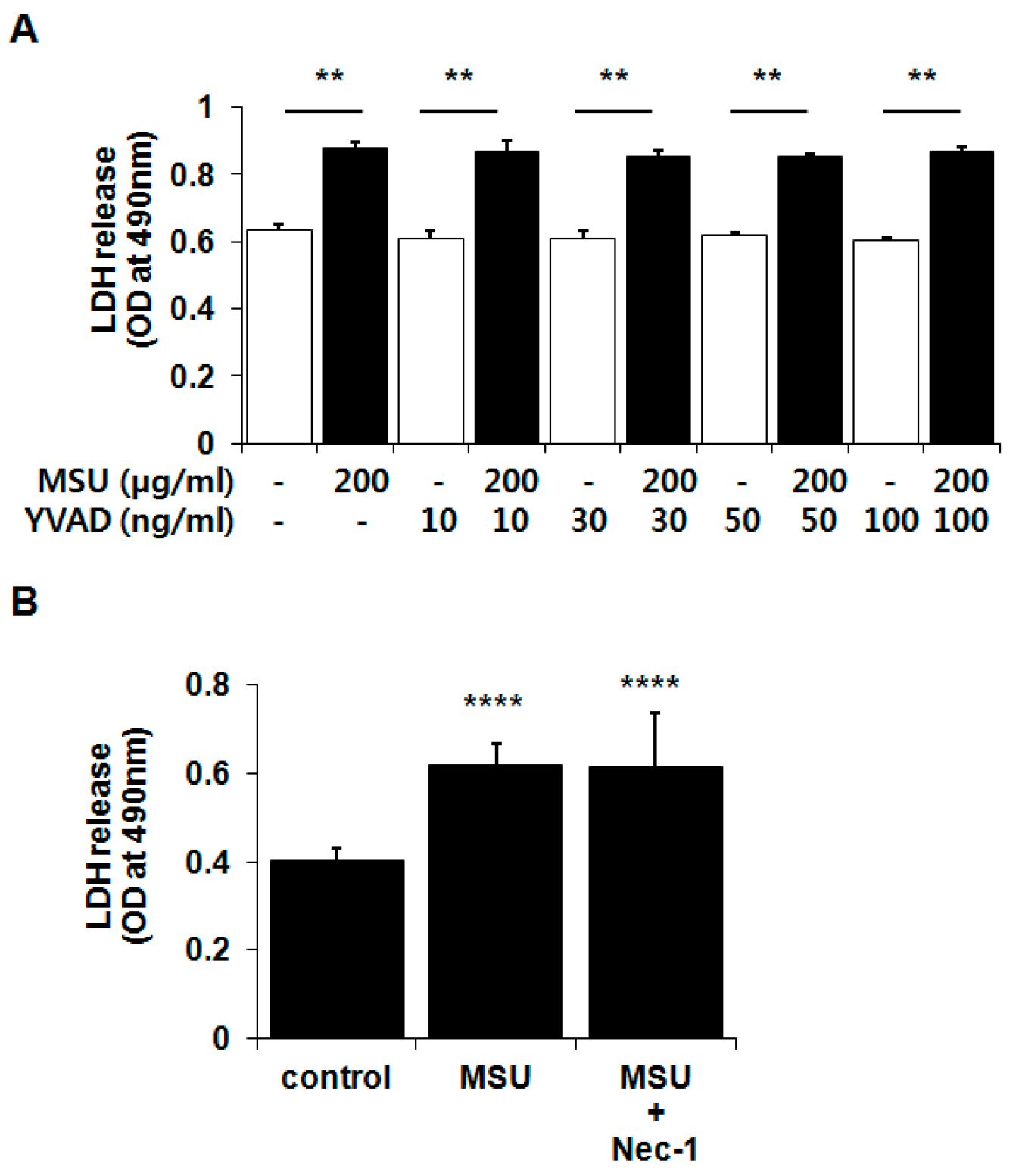
2.4. MSU Crystal-Induced Chondrocyte Death Was Independent of Pyroptosis and Necroptosis

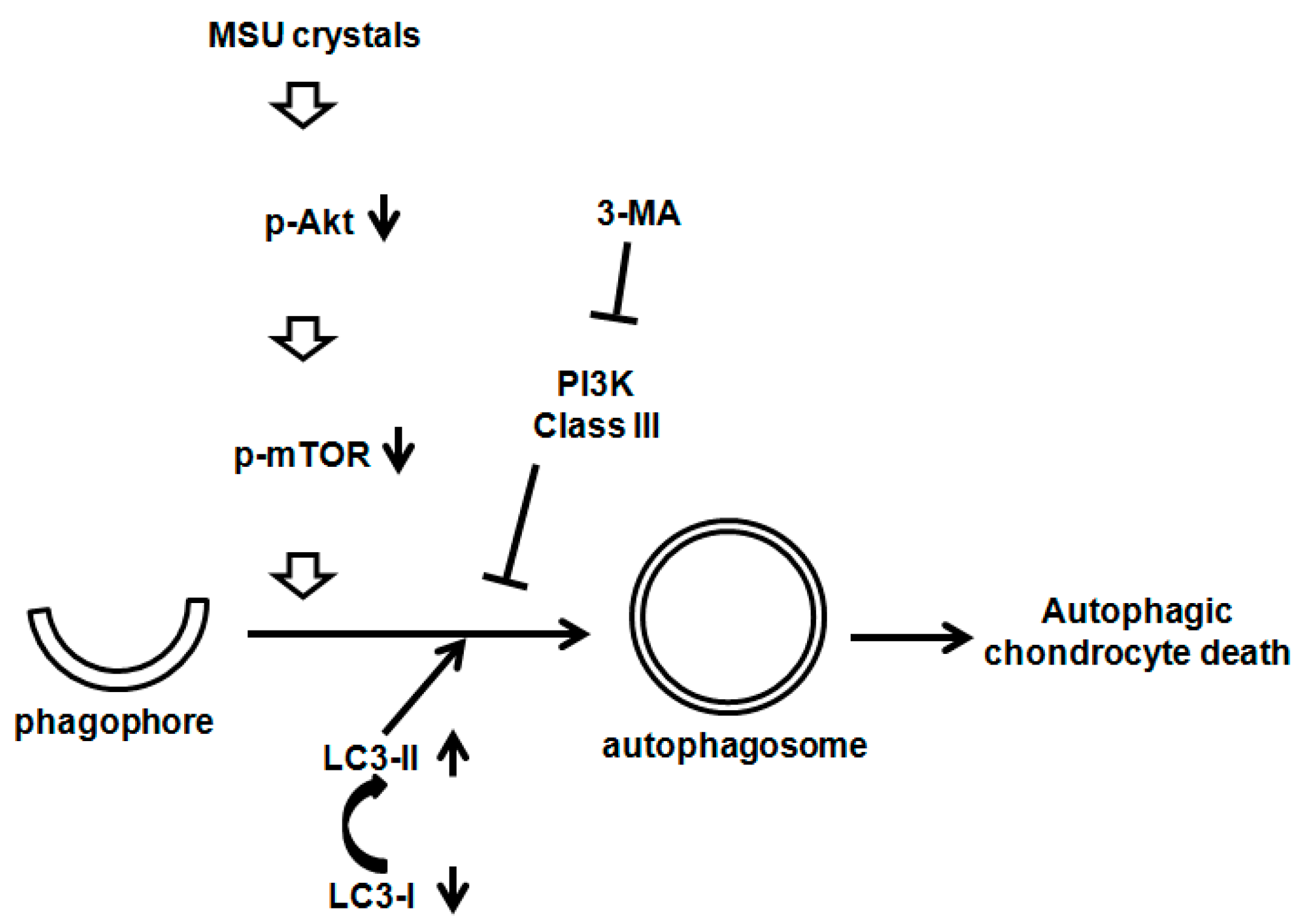
3. Discussion

4. Experimental Section
4.1. Materials
4.2. Sources of Tissues
4.3. Chondrocyte Isolation
4.4. MSU Preparation
4.5. Cell Viability Assay
4.6. Colorimetric TUNEL Assay
4.7. Caspase-3 Activity Measurement
4.8. RT-qPCR Analysis
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Oliviero, F.; Scanu, A.; Punzi, L. Metabolism of crystals within the joint. Reumatismo 2011, 63, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Terkeltaub, R. Update on gout: New therapeutic strategies and options. Nat. Rev. Rheumatol. 2010, 6, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Di Giovine, F.S.; Malawista, S.E.; Nuki, G.; Duff, G.W. Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1. J. Immunol. 1987, 138, 3213–3218. [Google Scholar] [PubMed]
- Di Giovine, F.S.; Malawista, S.E.; Thornton, E.; Duff, G.W. Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution. J. Clin. Investig. 1991, 87, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Guerne, P.A.; Terkeltaub, R.; Zuraw, B.; Lotz, M. Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes. Arthritis Rheumatol. 1989, 32, 1443–1452. [Google Scholar] [CrossRef]
- Chhana, A.; Callon, K.E.; Pool, B.; Naot, D.; Gamble, G.D.; Dray, M.; Pitto, R.; Bentley, J.; McQueen, F.M.; Cornish, J.; Dalbeth, N. The effects of monosodium urate monohydrate crystals on chondrocyte viability and function: Implications for development of cartilage damage in gout. J. Rheumatol. 2013, 40, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, C.J.; Mohr, W.; Haferkamp, O. The effect of soluble sodium urate on the proliferation and proteoglycan synthesis of lapine articular chondrocytes in monolayer culture. Rheumatol. Int. 1981, 1, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liote, F.; Rose, D.M.; Merz, D.; Terkeltaub, R. Proline-rich tyrosine kinase 2 and Src kinase signaling transduce monosodium urate crystal-induced nitric oxide production and matrix metalloproteinase 3 expression in chondrocytes. Arthritis Rheumatol. 2004, 50, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R.; Pritzker, K.; Firestein, G.S.; Terkeltaub, R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J. Immunol. 2005, 174, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, J.; Lu, L.; Qiu, Z.Y.; Zhang, X.; Yu, S.B.; Wu, Y.P.; Wang, M.Q. Enhancement of chondrocyte autophagy is an early response in the degenerative cartilage of the temporomandibular joint to biomechanical dental stimulation. Apoptosis 2013, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy signaling through reactive oxygen species. Antioxid. Redox Signal. 2011, 14, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheumatol. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Leshinsky, S.; Srinivas, V.; Shapiro, I.M. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 2010, 25, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Knowles, M.A.; Platt, F.M.; Ross, R.L.; Hurst, C.D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheumatol. 2009, 60, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheumatol. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, W.; Zhao, Z.; Zhang, T.; Song, Y. Autophagy in human articular chondrocytes is cytoprotective following glucocorticoid stimulation. Mol. Med. Rep. 2014, 9, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Van Raam, B.J.; Ehrnhoefer, D.E.; Hayden, M.R.; Salvesen, G.S. Intrinsic cleavage of receptor-interacting protein kinase-1 by caspase-6. Cell Death Differ. 2013, 20, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Clark, B.; McQueen, F.; Doyle, A.; Taylor, W. Validation of a radiographic damage index in chronic gout. Arthritis Rheumatol. 2007, 57, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Romero, C.; Carreira, V.; Rego, I.; Remeseiro, S.; Lopez-Armada, M.J.; Blanco, F.J. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 2008, 8, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.E.; Speicher, D.M.; Gradisar, I.; McBurney, D.L.; Baraga, A.; Doane, K.J.; Horton, W.E., Jr. Advanced osteoarthritis in humans is associated with altered collagen VI expression and upregulation of ER-stress markers Grp78 and bag-1. J. Histochem. Cytochem. 2009, 57, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Horton, W.E., Jr.; Bennion, P.; Yang, L. Cellular, molecular, and matrix changes in cartilage during aging and osteoarthritis. J. Musculoskelet. Neuronal Interact. 2006, 6, 379–381. [Google Scholar] [PubMed]
- Boot-Handford, R.P.; Briggs, M.D. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010, 339, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Carlson, S.G.; McBurney, D.; Horton, W.E., Jr. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J. Biol. Chem. 2005, 280, 31156–31165. [Google Scholar] [CrossRef] [PubMed]
- Husa, M.; Petursson, F.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res. Ther. 2013, 15, R218. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, K.; Goldring, M.B.; Yokota, H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch. Oral Biol. 2009, 54, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, J.; Zhang, P.; Song, F.; Jiang, R.; Li, M.; Xia, F.; Guo, F.J. IRE1alpha dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell. Signal. 2013, 25, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.J.; Liu, Y.; Zhou, J.; Luo, S.; Zhao, W.; Li, X.; Liu, C. XBP1S protects cells from ER stress-induced apoptosis through Erk1/2 signaling pathway involving CHOP. Histochem. Cell Biol. 2012, 138, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, F.; Cheng, S.; Wang, X.; Hao, Y. Uric acid-induced endoplasmic reticulum stress triggers phenotypic change in rat glomerular mesangial cells. Nephrology 2013, 18, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Shin, H.S.; Choi, H.S.; Park, J.W.; Jo, I.; Oh, E.S.; Lee, K.Y.; Lee, B.H.; Johnson, R.J.; Kang, D.H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Bohensky, J.; Shapiro, I.M. Autophagy: A new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs 2009, 189, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Bohensky, J.; Zahm, A.M.; Shapiro, I.M. Autophagy in mineralizing tissues: Microenvironmental perspectives. Cell Cycle 2009, 8, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Seino, D.; Blanco, F.J.; D’Lima, D.; Lotz, M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheumatol. 2012, 64, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [PubMed]
- Lee, H.S.; Lee, C.H.; Tsai, H.C.; Salter, D.M. Inhibition of cyclooxygenase 2 expression by diallyl sulfide on joint inflammation induced by urate crystal and IL-1beta. Osteoarthr. Cartil. 2009, 17, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Jung, H.Y.; Park, K.Y.; Kim, S.K. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology 2014, 53, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Schorn, C.; Janko, C.; Krenn, V.; Zhao, Y.; Munoz, L.E.; Schett, G.; Herrmann, M. Bonding the foe—NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front. Immunol. 2012, 3, 376. [Google Scholar] [CrossRef] [PubMed]
- Maueroder, C.; Kienhofer, D.; Hahn, J.; Schauer, C.; Manger, B.; Schett, G.; Herrmann, M.; Hoffmann, M.H. How neutrophil extracellular traps orchestrate the local immune response in gout. J. Mol. Med. 2015, 93, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, S.R.; Conaghan, P.G.; McDermott, M.F. The role of the NLRP3 inflammasome in gout. J. Inflamm. Res. 2011, 4, 39–49. [Google Scholar] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Asch, E.; Bloch, D.; Bole, G.; Borenstein, D.; Brandt, K.; Christy, W.; Cooke, T.D.; Greenwald, R.; Hochberg, M.; et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheumatol. 1986, 29, 1039–1049. [Google Scholar] [CrossRef]
- Kim, H.A.; Cho, M.L.; Choi, H.Y.; Yoon, C.S.; Jhun, J.Y.; Oh, H.J.; Kim, H.Y. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheumatol. 2006, 54, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, H.S.; Yang, C.M.; Park, S.J.; Kim, H.A. Monosodium Urate Crystal-Induced Chondrocyte Death via Autophagic Process. Int. J. Mol. Sci. 2015, 16, 29265-29277. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226164
Hwang HS, Yang CM, Park SJ, Kim HA. Monosodium Urate Crystal-Induced Chondrocyte Death via Autophagic Process. International Journal of Molecular Sciences. 2015; 16(12):29265-29277. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226164
Chicago/Turabian StyleHwang, Hyun Sook, Chung Mi Yang, Su Jin Park, and Hyun Ah Kim. 2015. "Monosodium Urate Crystal-Induced Chondrocyte Death via Autophagic Process" International Journal of Molecular Sciences 16, no. 12: 29265-29277. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226164