
The Actin Depolymerizing Factor (ADF)/Cofilin Signaling Pathway and DNA Damage Responses in Cancer

Abstract
:1. Introduction
2. The Actin Depolymerizing Factor (ADF)/Cofilin Signaling Pathways and Actin Dynamics
2.1. Biophysics and Biochemistry of Cofilin-1 (CFL-1)
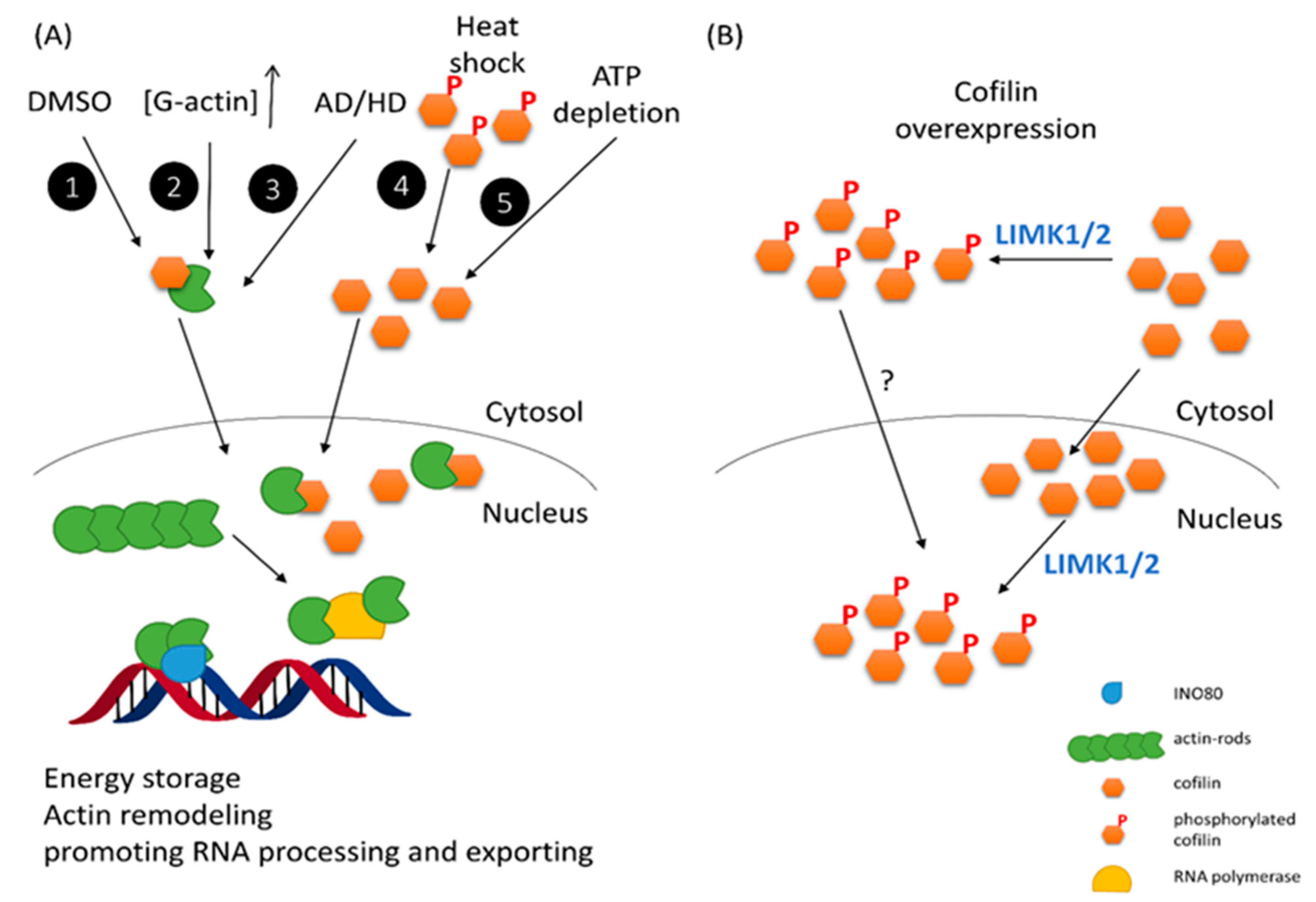
2.2. The Putative Role of CFL-1 in the Cell Nucleus

3. Genotoxicity and DNA Damage Response (DDR)
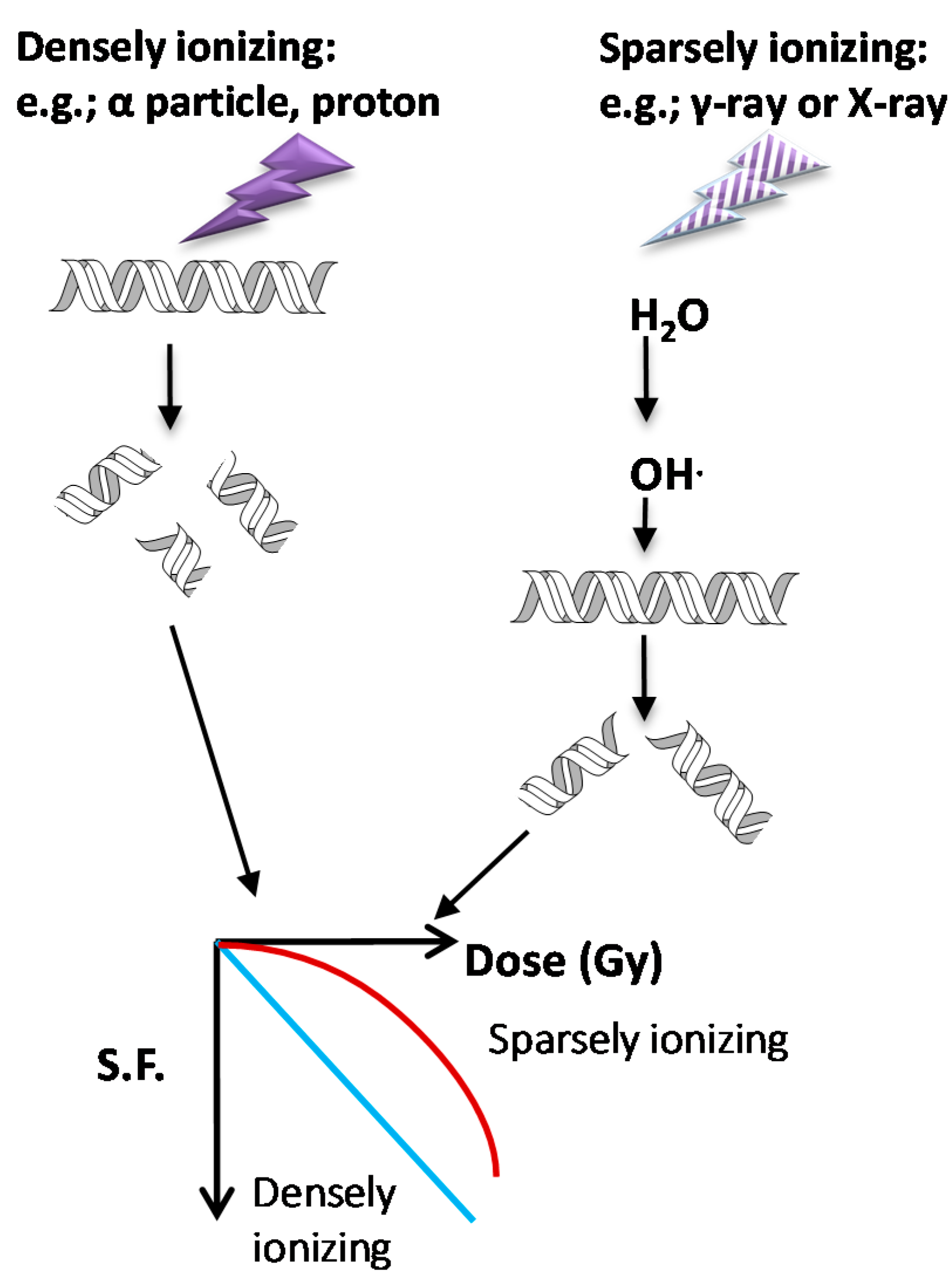
3.1. Types of Ionizing Radiation on Cell Survival and DDR

3.2. DNA Repair Mechanisms Following Ionizing Radiation (IR)
3.3. The Role of the Mre11-Rad50-NBS1 (MRN) Complex in Sensing IR-Induced DNA Damage
3.4. Ignition of DDR by Ataxia Telangiectasia Mutated (ATM) Kinase

4. Actin Dynamics, ADF/cofilin and DDR
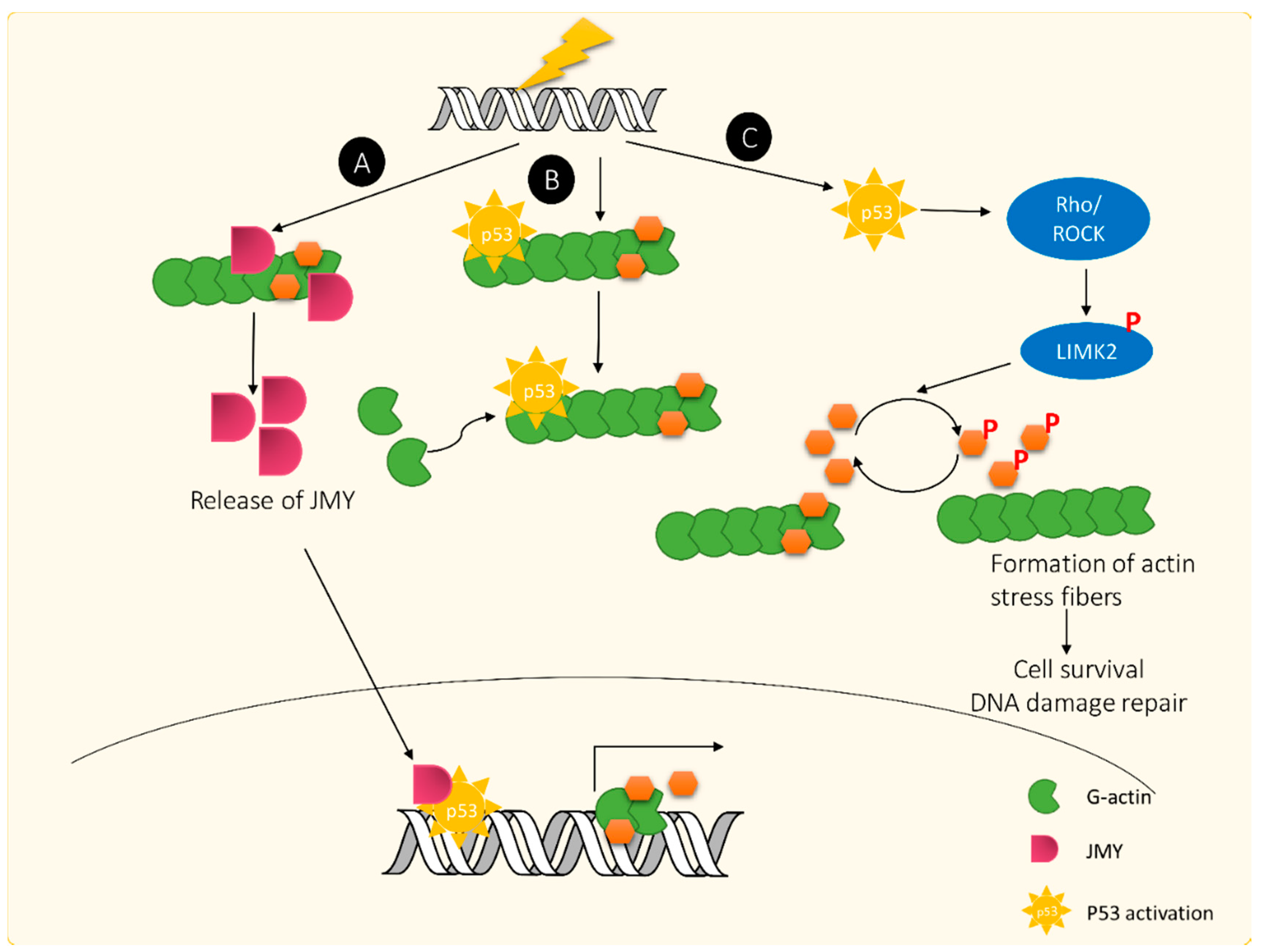
4.1. Actin Response Following DNA Damage

4.2. DDR Following Destabilization of the Actin Cytoskeleton

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Actin Inhibitors | Source/Host | Function |
|---|---|---|
| Cytochalasin B (CB) | Helminthosporium dematioideum/fungi | Blocking monomer add-on at the fast-growing end of actin filament |
| Cytochalasin D (CD) | Zygosporium mansonii/fungi | Blocking monomer add-on at the fast-growing end of actin filament, 10-fold more potent than CB |
| Latrunculin A | Latrunculia magnifica/Red Sea Sponge | Formation of a 1:1 complex with monomeric G-actin (Kd = 200 nM) |
| Misakinolid A (Bistheonellide A) | Theonella sp./marine sponge | Inhibits actin polymerization by forming a 1:2 complex with G-actin |
| Mycalolide B | Mycale sp./marine sponge | Severs F-actin and forms a 1:1 complex with G-actin to sequester it; it also suppresses actin-activated myosin Mg2+-ATPase activity |
| Swinholide A | Theonella swinhoei/marine sponge | Sequestering actin dimers with a binding stoichiometry of 1:1, and rapidly severing F-actin |
| Jasplakinolide | Jaspis johnstoni/marine sponge | A potent inducer of actin polymerization and stabilization in vitro, cell-permeable |
| Phalloidin | Amanita phalloides/fungi | A potent and specific F-actin binding agent; Inhibitor of F- to G-actin conversion, cell non-permeable |
4.3. AAPs and DDR
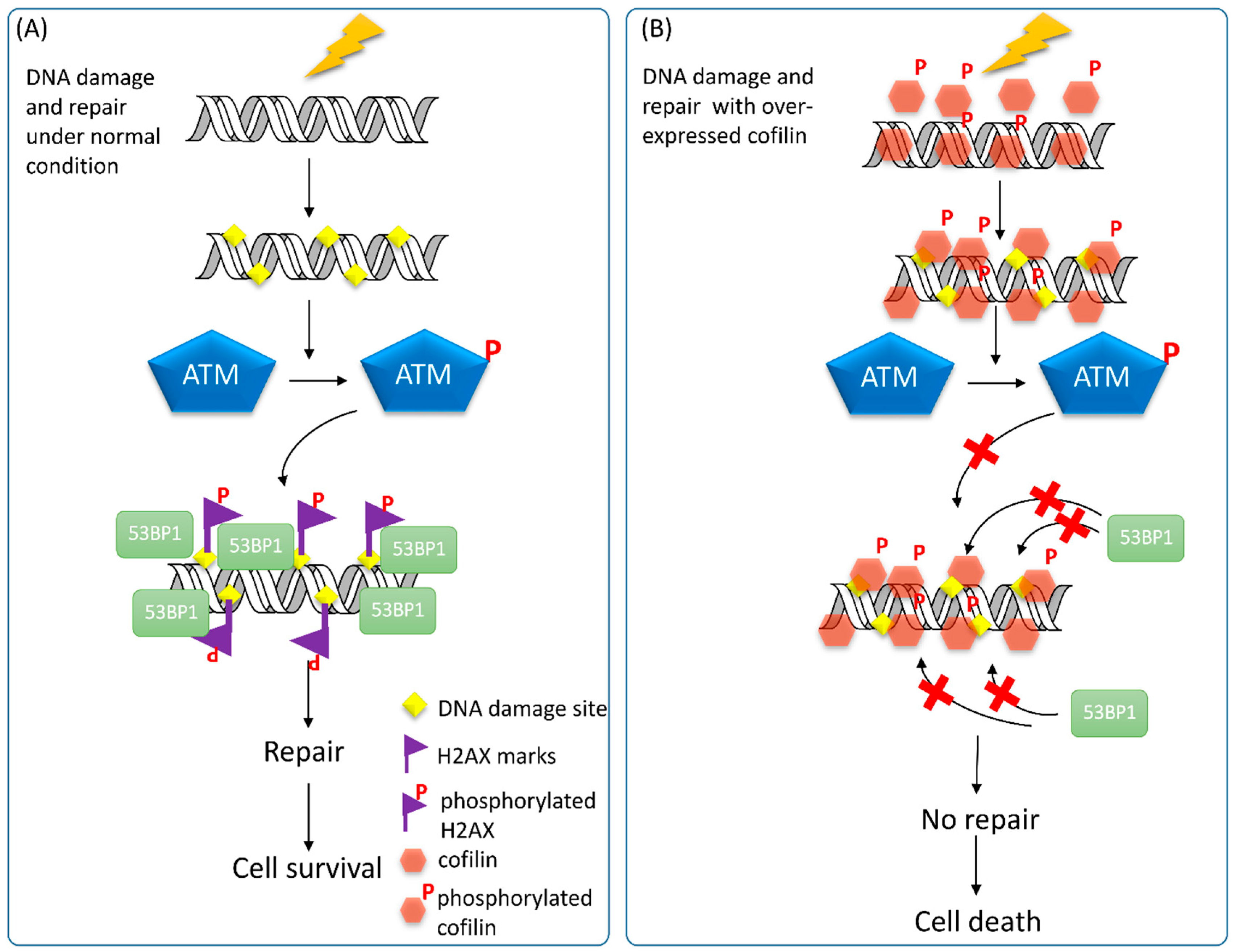
4.4. ADF/Cofilin and DDR

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rew, D.A. Cancer—A degenerative disorder? Eur. J. Surg. Oncol. 1998, 24, 362–366. [Google Scholar] [CrossRef]
- Fallowfield, L.J.; Hall, A.; Maguire, G.P.; Baum, M. Psychological outcomes of different treatment policies in women with early breast cancer outside a clinical trial. BMJ 1990, 301, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Smith, M.R.; Campbell, K.S. Immunotherapy of cancer. Eur. J. Pharmacol. 2009, 625, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.C. Chemotherapy of breast cancer. A general overview. Cancer 1983, 51, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Zhu, X. Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr. Opin. Cell Biol. 1997, 9, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ingber, D.E. The structural and mechanical complexity of cell-growth control. Nat. Cell Biol. 1999, 1, E131–E138. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikova, G.; Barkan, R.; Popov, B.; Nikolsky, N.; Chang, L.S. Disruption of the actin cytoskeleton leads to inhibition of mitogen-induced cyclin E expression, CDK2 phosphorylation, and nuclear accumulation of the retinoblastoma protein-related p107 protein. Exp. Cell Res. 2000, 259, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ingber, D.E. A discrete cell cycle checkpoint in late G1 that is cytoskeleton-dependent and map kinase (ERK)-independent. Exp. Cell Res. 2002, 275, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Iwig, M.; Czeslick, E.; Muller, A.; Gruner, M.; Spindler, M.; Glaesser, D. Growth regulation by cell shape alteration and organization of the cytoskeleton. Eur. J. Cell Biol. 1995, 67, 145–157. [Google Scholar] [PubMed]
- Bohmer, R.M.; Scharf, E.; Assoian, R.K. Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage-dependent expression of cyclin D1. Mol. Biol. Cell 1996, 7, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Lohez, O.D.; Reynaud, C.; Borel, F.; Andreassen, P.R.; Margolis, R.L. Arrest of mammalian fibroblasts in G1 in response to actin inhibition is dependent on retinoblastoma pocket proteins but not on p53. J. Cell Biol. 2003, 161, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Yi, M.; Zhang, X.; Xu, Y.; Jung, J.H.; Kim, D.K. Cytochalasin B induces apoptosis through the mitochondrial apoptotic pathway in HeLa human cervical carcinoma cells. Oncol. Rep. 2013, 30, 1929–1935. [Google Scholar] [PubMed]
- Kulms, D.; Dussmann, H.; Poppelmann, B.; Stander, S.; Schwarz, A.; Schwarz, T. Apoptosis induced by disruption of the actin cytoskeleton is mediated via activation of CD95 (Fas/Apo-1). Cell Death Differ. 2002, 9, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, S.N.; Kondratov, R.V.; Kopnin, P.B.; Chumakov, P.M.; Kopnin, B.P.; Vasiliev, J.M. Disruption of actin microfilaments by cytochalasin d leads to activation of p53. FEBS Lett. 1998, 430, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Trendowski, M.; Mitchell, J.M.; Corsette, C.M.; Acquafondata, C.; Fondy, T.P. Chemotherapy in vivo against m109 murine lung carcinoma with cytochalasin B by localized, systemic, and liposomal administration. Investig. New Drugs 2015. [Google Scholar] [CrossRef]
- Bousquet, P.F.; Paulsen, L.A.; Fondy, C.; Lipski, K.M.; Loucy, K.J.; Fondy, T.P. Effects of cytochalasin B in culture and in vivo on murine madison 109 lung carcinoma and on b16 melanoma. Cancer Res. 1990, 50, 1431–1439. [Google Scholar] [PubMed]
- Court, J.B.; Davies, G.; Davies, H.E.; Burn, C. Variation in radiosensitivity due to cell age and split-dose recovery in polykaryons induced by cytochalasin. Int. J. Radiat. Biol. 1995, 68, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R. Proteins of the ADF/cofilin family: Essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.F.; Ressad, F.; Pantaloni, D. Control of actin dynamics in cell motility. Role of ADF/cofilin. J. Biol. Chem. 1999, 274, 33827–33830. [Google Scholar] [CrossRef] [PubMed]
- Elam, W.A.; Kang, H.; de la Cruz, E.M. Biophysics of actin filament severing by cofilin. FEBS Lett. 2013, 587, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, J.; Song, X.; van Roosmalen, W.; Cammer, M.; Chen, X.; Desmarais, V.; Yip, S.C.; Backer, J.M.; Eddy, R.J.; Condeelis, J.S. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J. Cell Biol. 2007, 179, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Stope, M.B.; de Jesus, M.L.; Oude Weernink, P.A.; Urban, M.; Wieland, T.; Rosskopf, D.; Mizuno, K.; Jakobs, K.H.; Schmidt, M. Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 2007, 26, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Munsie, L.N.; Desmond, C.R.; Truant, R. Cofilin nuclear-cytoplasmic shuttling affects cofilin-actin rod formation during stress. J. Cell Sci. 2012, 125, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.D.; Chiu, Y.W.; Lo, C.C.; Chiang, P.H.; Chiu, S.J.; Tsai, C.H.; Hwang, J.J.; Chen, R.C.; Gorbunova, V.; Lee, Y.J. Enhanced cellular radiosensitivity induced by cofilin-1 over-expression is associated with reduced DNA repair capacity. Int. J. Radiat. Biol. 2013, 89, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Harris, H.E.; Weeds, A.G. Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett. 1980, 121, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, I. Purification from starfish eggs of a protein that depolymerizes actin. J. Biochem. 1981, 89, 1341–1344. [Google Scholar] [PubMed]
- Hosoya, H.; Mabuchi, I.; Sakai, H. Actin modulating proteins in the sea urchin egg. I. Analysis of G-actin-binding proteins by DNase I-affinity chromatography and purification of a 17,000 molecular weight component. J. Biochem. 1982, 92, 1853–1862. [Google Scholar] [PubMed]
- Berl, S.; Chou, M.; Mytilineou, C. Actin-stimulated myosin Mg2+-atpase inhibition by brain protein. J. Neurochem. 1983, 40, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Nishida, E.; Ohta, Y.; Sakai, H. Isolation of low molecular weight actin-binding proteins from porcine brain. J. Biochem. 1984, 95, 377–385. [Google Scholar] [PubMed]
- Nishida, E.; Muneyuki, E.; Maekawa, S.; Ohta, Y.; Sakai, H. An actin-depolymerizing protein (destrin) from porcine kidney. Its action on F-actin containing or lacking tropomyosin. Biochemistry 1985, 24, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of F-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Maekawa, S.; Sakai, H. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry 1984, 23, 5307–5313. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, F.; Matsumoto, S.; Yahara, I.; Yonezawa, N.; Nishida, E.; Sakai, H. Cloning and characterization of porcine brain cofilin cDNA. Cofilin contains the nuclear transport signal sequence. J. Biol. Chem. 1988, 263, 11564–11568. [Google Scholar] [PubMed]
- Hotulainen, P.; Paunola, E.; Vartiainen, M.K.; Lappalainen, P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell 2005, 16, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Gurniak, C.B.; Perlas, E.; Witke, W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev. Biol. 2005, 278, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; McGough, A.; Ono, S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999, 9, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Saunders, M.G.; Haddadian, E.J.; Freed, K.F.; De La Cruz, E.M.; Voth, G.A. Molecular origins of cofilin-linked changes in actin filament mechanics. J. Mol. Biol. 2013, 425, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, D.; Muhlrad, A.; Cooper, J.; Wear, M.; Reisler, E. Actin filament severing by cofilin. J. Mol. Biol. 2007, 365, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bernstein, B.W. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol. Rep. 2010, 2, 62. [Google Scholar] [PubMed]
- Okreglak, V.; Drubin, D.G. Loss of Aip1 reveals a role in maintaining the actin monomer pool and an in vivo oligomer assembly pathway. J. Cell Biol. 2010, 188, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Sakakibara, S.; Sokabe, M.; Tatsumi, H. Single-molecule imaging and kinetic analysis of cooperative cofilin-actin filament interactions. Proc. Natl. Acad. Sci. USA 2014, 111, 9810–9815. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Iida, K.; Yahara, I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1996, 1, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Gohla, A.; Bokoch, G.M. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr. Biol. 2002, 12, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin depolymerizing factor and cofilin phosphorylation dynamics: Response to signals that regulate neurite extension. Cell Motil. Cytoskelet. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Van Troys, M.; Huyck, L.; Leyman, S.; Dhaese, S.; Vandekerkhove, J.; Ampe, C. Ins and outs of ADF/cofilin activity and regulation. Eur. J. Cell Biol. 2008, 87, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Kane, S.; Medeiros, A.I.; Cornett, A.M.; Kim, S.H.; Marques, M.M.; Lee, S.P.; Lewis, C.; Bourdonnay, E.; Ballinger, M.N.; et al. PTEN directly activates the actin depolymerization factor cofilin-1 during PGE2-mediated inhibition of phagocytosis of fungi. Sci. Signal. 2012, 5, ra12. [Google Scholar] [PubMed]
- Pendleton, A.; Pope, B.; Weeds, A.; Koffer, A. Latrunculin B or ATP depletion induces cofilin-dependent translocation of actin into nuclei of mast cells. J. Biol. Chem. 2003, 278, 14394–14400. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Nagaoka, R.; Obinata, T. Cytoplasmic localization and nuclear transport of cofilin in cultured myotubes. Exp. Cell Res. 1993, 206, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Matsumoto, S.; Yahara, I. The KKRKK sequence is involved in heat shock-induced nuclear translocation of the 18-kDa actin-binding protein, cofilin. Cell Struct. Funct. 1992, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Iida, K.; Yonezawa, N.; Koyasu, S.; Yahara, I.; Sakai, H. Cofilin is a component of intranuclear and cytoplasmic actin rods induced in cultured cells. Proc. Natl. Acad. Sci. USA 1987, 84, 5262–5266. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Nishida, E.; Sakai, H.; Miyamoto, E. Dephosphorylation of cofilin accompanies heat shock-induced nuclear accumulation of cofilin. J. Biol. Chem. 1989, 264, 16143–16148. [Google Scholar] [PubMed]
- Nebl, G.; Meuer, S.C.; Samstag, Y. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. J. Biol. Chem. 1996, 271, 26276–26280. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.J.; Weeds, A.G.; Hussey, P.J. The maize actin-depolymerizing factor, ZmADF3, redistributes to the growing tip of elongating root hairs and can be induced to translocate into the nucleus with actin. Plant J. 1997, 12, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Yahara, I.; Aizawa, H.; Moriyama, K.; Iida, K.; Yonezawa, N.; Nishida, E.; Hatanaka, H.; Inagaki, F. A role of cofilin/destrin in reorganization of actin cytoskeleton in response to stresses and cell stimuli. Cell Struct. Funct. 1996, 21, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, R.; Abe, H.; Obinata, T. Site-directed mutagenesis of the phosphorylation site of cofilin: Its role in cofilin-actin interaction and cytoplasmic localization. Cell Motil. Cytoskelet. 1996, 35, 200–209. [Google Scholar] [CrossRef]
- Li, L.; Zhang, W.; Chai, X.; Zhang, Q.; Xie, J.; Chen, S.; Zhao, S. Neuronal maturation and laminar formation in the chicken optic tectum are accompanied by the transition of phosphorylated cofilin from cytoplasm to nucleus. Gene Expr. Patterns 2014, 16, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Mizuno, K. Nuclear export of LIM-kinase 1, mediated by two leucine-rich nuclear-export signals within the PDZ domain. Biochem. J. 1999, 338, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, C.A.; Bernard, O. LIM-kinase1. Int. J. Biochem. Cell Biol. 1999, 31, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Slee, J.B.; Lowe-Krentz, L.J. Actin realignment and cofilin regulation are essential for barrier integrity during shear stress. J. Cell. Biochem. 2013, 114, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, T.; Toyoshima, H.; Miura, M.; Wang, Y.; Iida, K.T.; Suzuki, H.; Sone, H.; Shimano, H.; Gotoda, T.; Nishimori, S.; et al. p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J. Biol. Chem. 2003, 278, 52919–52923. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Shen, X. Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol. 2014, 24, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Philimonenko, V.V.; Zhao, J.; Iben, S.; Dingova, H.; Kysela, K.; Kahle, M.; Zentgraf, H.; Hofmann, W.A.; de Lanerolle, P.; Hozak, P.; et al. Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 2004, 6, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Rivera, B.; Ruzicka, D.R.; Deal, R.B.; McKinney, E.C.; King-Reid, L.; Meagher, R.B. Actin depolymerizing factor 9 controls development and gene expression in arabidopsis. Plant Mol. Biol. 2008, 68, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.T.; Bamburg, J.R. Cofilin-mediated neurodegeneration in Alzheimerʼs disease and other amyloidopathies. Mol. Neurobiol. 2007, 35, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bernstein, B.W.; Davis, R.C.; Flynn, K.C.; Goldsbury, C.; Jensen, J.R.; Maloney, M.T.; Marsden, I.T.; Minamide, L.S.; Pak, C.W.; et al. ADF/cofilin-actin rods in neurodegenerative diseases. Curr. Alzheimer Res. 2010, 7, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Fertil, B.; Malaise, E.P. Intrinsic radiosensitivity of human cell lines is correlated with radioresponsiveness of human tumors: Analysis of 101 published survival curves. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Ye, R.; Veillette, C.J.; Lees-Miller, S.P. DNA-dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry 1999, 38, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, C.H. DNA-dependent protein kinase complex: A multifunctional protein in DNA repair and damage checkpoint. Mol. Cells 2002, 13, 159–166. [Google Scholar] [PubMed]
- Smith, G.C.; Divecha, N.; Lakin, N.D.; Jackson, S.P. DNA-dependent protein kinase and related proteins. Biochem. Soc. Symp. 1999, 64, 91–104. [Google Scholar] [PubMed]
- Jin, S.; Weaver, D.T. Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO J. 1997, 16, 6874–6885. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Schwarz, K.; Lieber, M.R. The artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair 2005, 4, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yuan, Y.; Maestas, A.; Shen, Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J. Biol. Chem. 2004, 279, 6098–6105. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994, 265, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast RAD52 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- New, J.H.; Kowalczykowski, S.C. RAD52 protein has a second stimulatory role in DNA strand exchange that complements replication protein-A function. J. Biol. Chem. 2002, 277, 26171–26176. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, A.; Sokol, K.; Burgman, P.; Li, L.; Li, G.C. Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: The effects of ionizing radiation on growth, survival, and development. Proc. Natl. Acad. Sci. USA 1997, 94, 13588–13593. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, C.; Sweeney, E.A.; Shirahama, T.; Igarashi, Y.; Hakomori, S.; Tsujimoto, H.; Imanishi, T.; Ohgaki, M.; Yamazaki, J.; Hagiwara, A.; et al. Overexpression of bax enhances the radiation sensitivity in human breast cancer cells. Surg. Today 1997, 27, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Taki, T.; Ohnishi, T.; Yamamoto, A.; Hiraga, S.; Arita, N.; Izumoto, S.; Hayakawa, T.; Morita, T. Antisense inhibition of the RAD51 enhances radiosensitivity. Biochem. Biophys. Res. Commun. 1996, 223, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S. Expression of human RAD52 confers resistance to ionizing radiation in mammalian cells. J. Biol. Chem. 1995, 270, 15467–15470. [Google Scholar] [CrossRef] [PubMed]
- Kraakman-van der Zwet, M.; Overkamp, W.J.; van Lange, R.E.; Essers, J.; van Duijn-Goedhart, A.; Wiggers, I.; Swaminathan, S.; van Buul, P.P.; Errami, A.; Tan, R.T.; et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol. Cell. Biol. 2002, 22, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; Tighe, A.; Scott, S.D.; Roberts, S.A.; Hendry, J.H.; Margison, G.P. Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res. 2001, 29, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Lopez, B.S. Inactivation of the RAD51 recombination pathway stimulates UV-induced mutagenesis in mammalian cells. Oncogene 2002, 21, 4065–4069. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors RAD50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shi, L.Z.; Truong, L.N.; Lu, C.S.; Razavian, N.; Li, Y.; Negrete, A.; Shiloach, J.; Berns, M.W.; Wu, X. RAD50 zinc hook is important for the Mre11 complex to bind chromosomal DNA double-stranded breaks and initiate various DNA damage responses. J. Biol. Chem. 2012, 287, 31747–31756. [Google Scholar] [CrossRef] [PubMed]
- Raymond, W.E.; Kleckner, N. RAD50 protein of s.Cerevisiae exhibits ATP-dependent DNA binding. Nucleic Acids Res. 1993, 21, 3851–3856. [Google Scholar] [CrossRef] [PubMed]
- Cerosaletti, K.; Wright, J.; Concannon, P. Active role for nibrin in the kinetics of ATM activation. Mol. Cell. Biol. 2006, 26, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Kim, S.T.; Xu, B.; Maser, R.S.; Lin, J.; Petrini, J.H.; Kastan, M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 2000, 404, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ranganathan, V.; Weisman, D.S.; Heine, W.F.; Ciccone, D.N.; OʼNeill, T.B.; Crick, K.E.; Pierce, K.A.; Lane, W.S.; Rathbun, G.; et al. Atm phosphorylation of nijmegen breakage syndrome protein is required in a DNA damage response. Nature 2000, 405, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Jakob, B.; Chen, P.; Kijas, A.W.; Becherel, O.J.; Gueven, N.; Birrell, G.; Lee, J.H.; Paull, T.T.; Lerenthal, Y.; et al. ATM protein-dependent phosphorylation of RAD50 protein regulates DNA repair and cell cycle control. J. Biol. Chem. 2011, 286, 31542–31556. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the Mrn complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Durocher, D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013, 14, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Adamietz, P.; Rudolph, A. ADP-ribosylation of nuclear proteins in vivo. Identification of histone H2B as a major acceptor for mono- and poly(ADP-ribose) in dimethyl sulfate-treated hepatoma Ah 7974 cells. J. Biol. Chem. 1984, 259, 6841–6846. [Google Scholar] [PubMed]
- Fontan-Lozano, A.; Suarez-Pereira, I.; Horrillo, A.; del-Pozo-Martin, Y.; Hmadcha, A.; Carrion, A.M. Histone H1 poly[ADP]-ribosylation regulates the chromatin alterations required for learning consolidation. J. Neurosci. 2010, 30, 13305–13313. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Horejsi, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; DʼAndrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [PubMed]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of Mre11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O'Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Acuna, L.; Di Tomaso, M.V.; Palitti, F.; Martinez-Lopez, W. Histone post-translational modifications in DNA damage response. Cytogenet. Genome Res. 2010, 128, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Rappold, I.; Iwabuchi, K.; Date, T.; Chen, J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 2001, 153, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.M.; Carpenter, P.B. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates atm kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Kamath, R.; Jin, S.; Balasubramani, M.; Pandita, T.K.; Rajasekaran, B. Tip60-mediated acetylation activates transcription independent apoptotic activity of Abl. Mol. Cancer 2011, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Takahashi, S.; Breitweiser, W.; Shiloh, Y.; Jones, N.; Ronai, Z. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell 2005, 18, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Shafman, T.; Khanna, K.K.; Kedar, P.; Spring, K.; Kozlov, S.; Yen, T.; Hobson, K.; Gatei, M.; Zhang, N.; Watters, D.; et al. Interaction between atm protein and c-Abl in response to DNA damage. Nature 1997, 387, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Weston, L.; Coutts, A.S.; La Thangue, N.B. Actin nucleators in the nucleus: An emerging theme. J. Cell Sci. 2012, 125, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, M.; Wang, S.; Qi, T.; Guo, L.; Li, J.; Qi, W.; Ampah, K.K.; Ba, X.; Zeng, X. Actin polymerization negatively regulates p53 function by impairing its nuclear import in response to DNA damage. PLoS One 2013, 8, e60179. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.R.; Crighton, D.; Samuel, M.S.; Lourenco, F.C.; Munro, J.; Wood, J.; Bensaad, K.; Vousden, K.H.; Sansom, O.J.; Ryan, K.M.; et al. p53-mediated transcriptional regulation and activation of the actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway promotes cell survival. Cell Res. 2011, 21, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Sayers, Z.; Koch, M.H.; Bordas, J.; Lindberg, U. Time-resolved X-ray scattering study of actin polymerization from profilactin. Eur. Biophys. J. 1985, 13, 99–108. [Google Scholar] [PubMed]
- Levee, M.G.; Dabrowska, M.I.; Lelli, J.L., Jr.; Hinshaw, D.B. Actin polymerization and depolymerization during apoptosis in HL-60 cells. Am. J. Physiol. 1996, 271, C1981–C1992. [Google Scholar] [PubMed]
- Guerra, L.; Carr, H.S.; Richter-Dahlfors, A.; Masucci, M.G.; Thelestam, M.; Frost, J.A.; Frisan, T. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 2008, 3, e2254. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.F.; Lin, T.Y.; Chen, J.Y.; Shieh, S.Y. p53-mediated transactivation of LIMK2b links actin dynamics to cell cycle checkpoint control. Oncogene 2010, 29, 2864–2876. [Google Scholar] [CrossRef] [PubMed]
- Andrin, C.; McDonald, D.; Attwood, K.M.; Rodrigue, A.; Ghosh, S.; Mirzayans, R.; Masson, J.Y.; Dellaire, G.; Hendzel, M.J. A requirement for polymerized actin in DNA double-strand break repair. Nucleus 2012, 3, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, S.; Weeds, A.; Okorokov, A.L.; Milner, J.; Cockman, M.; Pope, B. Wild-type p53 protein shows calcium-dependent binding to F-actin. Oncogene 1999, 18, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Belin, B.; Mullins, R.D. Actin binding to WH2 domains regulates nuclear import of the multifunctional actin regulator JMY. Mol. Biol. Cell 2012, 23, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Tsai, C.H.; Hwang, J.J.; Chiu, S.J.; Sheu, T.J.; Keng, P.C. Involvement of a p53-independent and post-transcriptional up-regulation for p21WAF/CIP1 following destabilization of the actin cytoskeleton. Int. J. Oncol. 2009, 34, 581–589. [Google Scholar] [PubMed]
- Coleman, M.L.; Densham, R.M.; Croft, D.R.; Olson, M.F. Stability of p21WAF1/CIP1 CDK inhibitor protein is responsive to RhoA-mediated regulation of the actin cytoskeleton. Oncogene 2006, 25, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; O'Neill, E.; Munro, J.; Konig, I.; Anderson, K.; Kolch, W.; Olson, M.F. MST kinases monitor actin cytoskeletal integrity and signal via c-Jun N-terminal kinase stress-activated kinase to regulate p21WAF1/CIP1 stability. Mol. Cell. Biol. 2009, 29, 6380–6390. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Dang, Y.; Shang, X.; Tsuruga, M.; Fujita, Y.; Tanaka, H.; Zhou, D.; Kawasaki, K.; Oka, S. Acceleration of DNA damage-induced apoptosis in leukemia cells by interfering with actin system. Exp. Hematol. 2000, 28, 1491. [Google Scholar] [CrossRef]
- Takeuchi, H.; Ara, G.; Sausville, E.A.; Teicher, B. Jasplakinolide: Interaction with radiation and hyperthermia in human prostate carcinoma and lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Jovceva, E.; Larsen, M.R.; Waterfield, M.D.; Baum, B.; Timms, J.F. Dynamic cofilin phosphorylation in the control of lamellipodial actin homeostasis. J. Cell Sci. 2007, 120, 1888–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitayama, K.; Kamo, M.; Oma, Y.; Matsuda, R.; Uchida, T.; Ikura, T.; Tashiro, S.; Ohyama, T.; Winsor, B.; Harata, M. The human actin-related protein hArp5: Nucleo-cytoplasmic shuttling and involvement in DNA repair. Exp. Cell Res. 2009, 315, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and γ-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; Boulahbel, H.; Graham, A.; la Thangue, N.B. Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 2007, 8, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Velkova, A.; Carvalho, M.A.; Johnson, J.O.; Tavtigian, S.V.; Monteiro, A.N. Identification of filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle 2010, 9, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Wang, Q.; Lu, H.; Brenneman, M.; Fan, F.; Shen, Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009, 69, 7978–7985. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shen, Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J. Biol. Chem. 2001, 276, 48318–48324. [Google Scholar] [PubMed]
- Sosne, G.; Qiu, P.; Goldstein, A.L.; Wheater, M. Biological activities of thymosin β4 defined by active sites in short peptide sequences. FASEB J. 2010, 24, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.E.; Adang, L.A.; Macara, I.G. Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell 2007, 130, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, K.; Xiao, H.; Zou, Y.J.; Zhang, W.B.; Liu, H.Y. Over-expression of cofilin-1 and phosphoglycerate kinase 1 in astrocytomas involved in pathogenesis of radioresistance. CNS Neurosci. Ther. 2012, 18, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Chiu, S.J.; Liu, C.C.; Sheu, T.J.; Hsieh, C.H.; Keng, P.C.; Lee, Y.J. Regulated expression of cofilin and the consequent regulation of p27(kip1) are essential for G1 phase progression. Cell Cycle 2009, 8, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Mordant, P.; Dorvault, N.; de la motte Rouge, T.; Bourhis, J.; Soria, J.C.; Deutsch, E. BMS-690514, a VEGFR and EGFR tyrosine kinase inhibitor, shows anti-tumoural activity on non-small-cell lung cancer xenografts and induces sequence-dependent synergistic effect with radiation. Br. J. Cancer 2010, 103, 347–353. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-Y.; Leu, J.-D.; Lee, Y.-J. The Actin Depolymerizing Factor (ADF)/Cofilin Signaling Pathway and DNA Damage Responses in Cancer. Int. J. Mol. Sci. 2015, 16, 4095-4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024095
Chang C-Y, Leu J-D, Lee Y-J. The Actin Depolymerizing Factor (ADF)/Cofilin Signaling Pathway and DNA Damage Responses in Cancer. International Journal of Molecular Sciences. 2015; 16(2):4095-4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024095
Chicago/Turabian StyleChang, Chun-Yuan, Jyh-Der Leu, and Yi-Jang Lee. 2015. "The Actin Depolymerizing Factor (ADF)/Cofilin Signaling Pathway and DNA Damage Responses in Cancer" International Journal of Molecular Sciences 16, no. 2: 4095-4120. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024095