
Potential Role of Dipeptidyl Peptidase IV in the Pathophysiology of Heart Failure

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Dipeptidyl Peptidase IV (DPPIV) and Cardiac Dysfunction

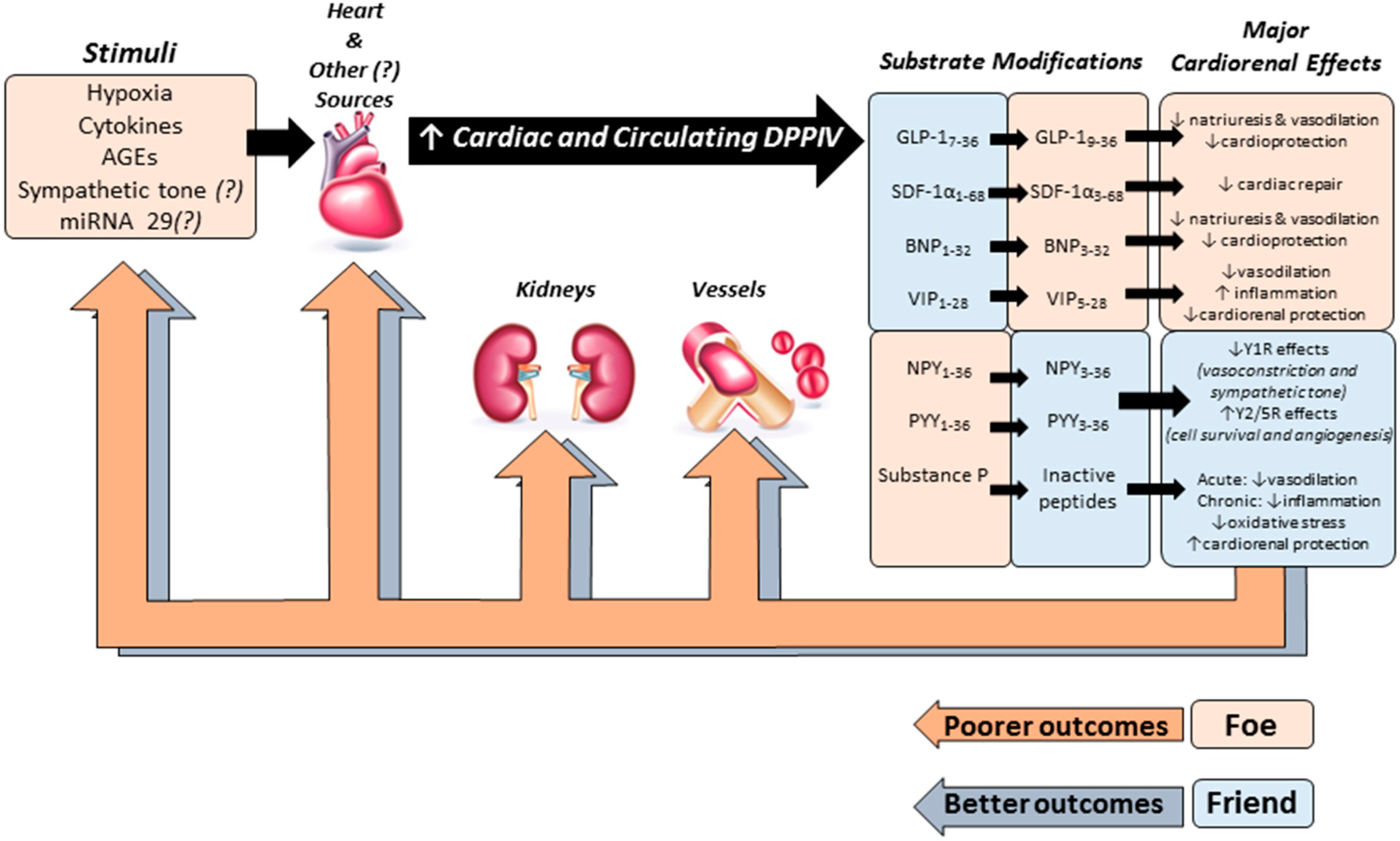
3. Cardiorenal Effects of DPPIV Substrates
3.1. Glucagon-Like Peptide-1 (GLP-1)
3.2. Brain Natriuretic Peptide
3.3. Stromal Cell-Derived Factor 1-α (SDF-1α)
3.4. Other DPPIV Substrates and Potential Effects on the Cardiovascular System
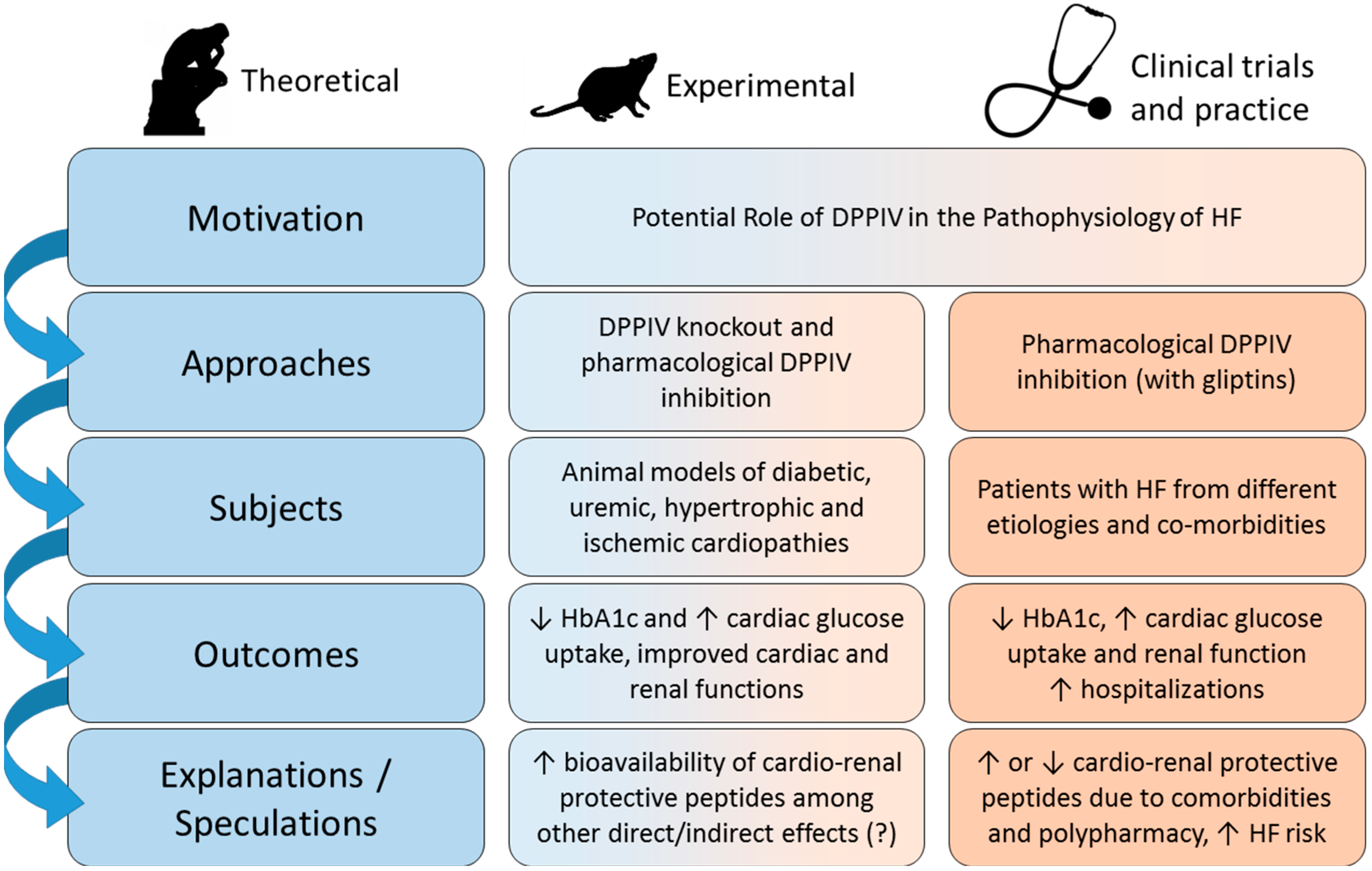
4. DPPIV Inhibitors and HF: Preclinical Studies
5. DPPIV Inhibitors and HF: Clinical Studies
6. Summary and Perspectives

Acknowledgments
Conflicts of Interest
References
- Mill, J.G.; Stefanon, I.; dos Santos, L.; Baldo, M.P. Remodeling in the ischemic heart: The stepwise progression for heart failure. Braz. J. Med. Biol. Res. 2011, 44, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S. Pathophysiology of chronic heart failure. Am. J. Med. 2001, 110, 37S–46S. [Google Scholar] [CrossRef] [PubMed]
- MacIver, D.H.; Dayer, M.J.; Harrison, A.J. A general theory of acute and chronic heart failure. Int. J. Cardiol. 2013, 165, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–239. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.J.; Petell, J.K.; Swank, D.; Sanford, J.; Hixson, D.C.; Doyle, D. Expression of dipeptidyl peptidase IV in rat tissues is mainly regulated at the mRNA levels. Exp. Cell Res. 1989, 182, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Kenny, A.J.; Booth, A.G.; George, S.G.; Ingram, J.; Kershaw, D.; Wood, E.J.; Young, A.R. Dipeptidyl peptidase IV, a kidney brush-border serine peptidase. Biochem. J. 1976, 157, 169–182. [Google Scholar] [PubMed]
- Lambeir, A.M.; Durinx, C.; Scharpe, S.; de Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef] [PubMed]
- Matheeussen, V.; Baerts, L.; de Meyer, G.; de Keulenaer, G.; van der Veken, P.; Augustyns, K.; Dubois, V.; Scharpe, S.; de Meester, I. Expression and spatial heterogeneity of dipeptidyl peptidases in endothelial cells of conduct vessels and capillaries. Biol. Chem. 2011, 392, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Steeg, C.; Hartwig, U.; Fleischer, B. Unchanged signaling capacity of mutant CD26/dipeptidylpeptidase IV molecules devoid of enzymatic activity. Cell. Immunol. 1995, 164, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Duke-Cohan, J.S.; Kameoka, J.; Yaron, A.; Lee, I.; Schlossman, S.F.; Morimoto, C. Enhancement of antigen-induced T-cell proliferation by soluble cd26/dipeptidyl peptidase IV. Proc. Natl. Acad. Sci. USA 1994, 91, 3082–3086. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Takahashi, N.; Yamochi, T.; Hosono, O.; Dang, N.H.; Morimoto, C. Role of CD26/dipeptidyl peptidase IV in human T cell activation and function. Front. Biosci. 2008, 13, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Marguet, D.; Dobers, J.; Reutter, W.; Fan, H. Deficiency of CD26 results in a change of cytokine and immunoglobulin secretion after stimulation by pokeweed mitogen. Eur. J. Immunol. 2003, 33, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Durinx, C.; Lambeir, A.M.; Bosmans, E.; Falmagne, J.B.; Berghmans, R.; Haemers, A.; Scharpe, S.; de Meester, I. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-pro dipeptides. Eur. J. Biochem. 2000, 267, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.J.; Salgado, F.J.; Nogueira, M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol. Immunother. 2009, 58, 1723–1747. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Somogyi, A.; Barna, G.; Wichmann, B.; Nagy, G.; Racz, K.; Selmeci, L.; Firneisz, G. Higher serum DPP-4 enzyme activity and decreased lymphocyte CD26 expression in type 1 diabetes. Pathol. Oncol. Res. 2011, 17, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Eric-Nikolic, A.; Matic, I.Z.; Dordevic, M.; Milovanovic, Z.; Markovic, I.; Dzodic, R.; Inic, M.; Srdic-Rajic, T.; Jevric, M.; Gavrilovic, D.; et al. Serum DPPIV activity and CD26 expression on lymphocytes in patients with benign or malignant breast tumors. Immunobiology 2011, 216, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Lamers, D.; Famulla, S.; Wronkowitz, N.; Hartwig, S.; Lehr, S.; Ouwens, D.M.; Eckardt, K.; Kaufman, J.M.; Ryden, M.; Muller, S.; et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011, 60, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Bluher, M.; Kloting, N.; Schlich, R.; Willems, M.; Ruppe, F.; Knoefel, W.T.; Dietrich, A.; Fielding, B.A.; Arner, P.; et al. Adipose dipeptidyl peptidase-4 and obesity: Correlation with insulin resistance and depot-specific release from adipose tissue in vivo and in vitro. Diabetes Care 2013, 36, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Rohrborn, D.; Eckel, J.; Sell, H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett. 2014, 588, 3870–3877. [Google Scholar] [CrossRef] [PubMed]
- Hopsu-Havu, V.K.; Sarimo, S.R. Purification and characterization of an aminopeptidase hydrolyzing glycyl-proline-naphthylamide. Hoppe Seylers Z. Physiol. Chem. 1967, 348, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Marguet, D.; Baggio, L.; Kobayashi, T.; Bernard, A.M.; Pierres, M.; Nielsen, P.F.; Ribel, U.; Watanabe, T.; Drucker, D.J.; Wagtmann, N. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc. Natl. Acad. Sci. USA 2000, 97, 6874–6879. [Google Scholar] [CrossRef] [PubMed]
- Gutniak, M.; Orskov, C.; Holst, J.J.; Ahren, B.; Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Degray, B.C.; Nagy, T.; Biemesderfer, D.; Aronson, P.S. Association of Na+-H+ exchanger isoform NHE3 and dipeptidyl peptidase IV in the renal proximal tubule. J. Biol. Chem. 2001, 276, 46671–46677. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, P.J.; Clarke, L.L.; Meneton, P.; Miller, M.L.; Soleimani, M.; Gawenis, L.R.; Riddle, T.M.; Duffy, J.J.; Doetschman, T.; Wang, T.; et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998, 19, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Knauf, F.; Demuth, H.U.; Aronson, P.S. Role of dipeptidyl peptidase IV in regulating activity of Na+/H+ exchanger isoform nhe3 in proximal tubule cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1238–C1245. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Fukuda, L.E.; Rossoni, L.V.; Malnic, G.; Reboucas, N.A. Dipeptidyl peptidase IV inhibition downregulates Na+-H+ exchanger NHE3 in rat renal proximal tubule. Am. J. Physiol. Ren. Physiol. 2008, 294, F414–F422. [Google Scholar] [CrossRef]
- Bauvois, B. A collagen-binding glycoprotein on the surface of mouse fibroblasts is identified as dipeptidyl peptidase IV. Biochem. J. 1988, 252, 723–731. [Google Scholar] [PubMed]
- Loster, K.; Zeilinger, K.; Schuppan, D.; Reutter, W. The cysteine-rich region of dipeptidyl peptidase IV (CD 26) is the collagen-binding site. Biochem. Biophys. Res. Commun. 1995, 217, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Callanan, H.M.; Mowery, J.; Hixson, D.C. Evidence for a role of dipeptidyl peptidase IV in fibronectin-mediated interactions of hepatocytes with extracellular matrix. Biochem. J. 1989, 262, 327–334. [Google Scholar] [PubMed]
- Cheng, H.C.; Abdel-Ghany, M.; Elble, R.C.; Pauli, B.U. Lung endothelial dipeptidyl peptidase IV promotes adhesion and metastasis of rat breast cancer cells via tumor cell surface-associated fibronectin. J. Biol. Chem. 1998, 273, 24207–24215. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, G.; Dong, H.; Goldstein, L.A.; Yeh, Y.; Hakkinen, L.; Larjava, H.S.; Chen, W.T. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J. Biol. Chem. 2002, 277, 29231–29241. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Asakura, M.; Ito, S.; Min, K.D.; Shindo, K.; Yan, Y.; Liao, Y.; Yamazaki, S.; Sanada, S.; Asano, Y.; et al. Dipeptidyl-peptidase IV inhibition improves pathophysiology of heart failure and increases survival rate in pressure-overloaded mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1361–H1369. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, L.; Salles, T.A.; Arruda-Junior, D.F.; Campos, L.C.; Pereira, A.C.; Barreto, A.L.; Antonio, E.L.; Mansur, A.J.; Tucci, P.J.; Krieger, J.E.; et al. Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ. Heart Fail. 2013, 6, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, T.; Aoyama, M.; Bando, Y.K.; Monji, A.; Mitsui, T.; Takatsu, M.; Cheng, X.W.; Okumura, T.; Hirashiki, A.; Nagata, K.; et al. Dipeptidyl peptidase-4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis-dependent and -independent actions. Circulation 2012, 126, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Chaykovska, L.; von Websky, K.; Rahnenfuhrer, J.; Alter, M.; Heiden, S.; Fuchs, H.; Runge, F.; Klein, T.; Hocher, B. Effects of DPP-4 inhibitors on the heart in a rat model of uremic cardiomyopathy. PLoS One 2011, 6, e27861. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Habibi, J.; Ma, L.; Aroor, A.; Rehmer, N.; Hayden, M.R.; Sowers, J.R. Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of western diet induced obesity. Metabolism 2014, 63, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Bowskill, B.B.; Advani, S.L.; Thai, K.; Chen, L.H.; Kabir, M.G.; Gilbert, R.E.; Advani, A. Dipeptidyl peptidase-4 inhibition improves left ventricular function in chronic kidney disease. Clin. Investig Med. 2014, 37, 172–185. [Google Scholar]
- Miyoshi, T.; Nakamura, K.; Yoshida, M.; Miura, D.; Oe, H.; Akagi, S.; Sugiyama, H.; Akazawa, K.; Yonezawa, T.; Wada, J.; et al. Effect of vildagliptin, a dipeptidyl peptidase 4 inhibitor, on cardiac hypertrophy induced by chronic β-adrenergic stimulation in rats. Cardiovasc. Diabetol. 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, B.P.; Crajoinas, R.O.; Couto, G.K.; Davel, A.P.; Lessa, L.M.; Rossoni, L.V.; Girardi, A.C. Dipeptidyl peptidase IV inhibition attenuates blood pressure rising in young spontaneously hypertensive rats. J. Hypertens. 2011, 29, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; di Sole, F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am. J. Physiol. Cell Physiol. 2012, 302, C1569–C1587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Baskota, A.; Gao, Y.; Tian, H.; Yang, F. Increased plasma dipeptidyl peptidase 4 activities predict new-onset microalbuminuria in association with its proinflammatory effects in chinese without diabetes: A four-year prospective study. Nephrol. Dial. Transplant. 2014. [Google Scholar] [CrossRef]
- Agrawal, N.K.; Kant, S. Targeting inflammation in diabetes: Newer therapeutic options. World J. Diabetes 2014, 5, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.J.; Lamarche, B.; Deacon, C.F.; Weisnagel, S.J.; Couture, P. Effects of sitagliptin therapy on markers of low-grade inflammation and cell adhesion molecules in patients with type 2 diabetes. Metabolism 2014, 63, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, P.; Frioes, F.; Silva, N.; Guimaraes, J.T.; Bettencourt, P. Dipeptidyl peptidase IV and mortality after an acute heart failure episode. J. Cardiovasc. Pharmacol. 2013, 62, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.; Matheeussen, V.; Damoiseaux, C.; Tamborini, A.; Merveille, A.C.; Jespers, P.; Michaux, C.; Clercx, C.; de Meester, I.; Mc Entee, K. Effect of heart failure on dipeptidyl peptidase IV activity in plasma of dogs. J. Vet. Intern. Med. 2012, 26, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.F.; Fernandes, T.; Barauna, V.G.; Matos, K.C.; Santos, A.A.; Tucci, P.J.; Oliveira, E.M. Expression of microRNA-29 and collagen in cardiac muscle after swimming training in myocardial-infarcted rats. Cell. Physiol. Biochem. 2014, 33, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of mir-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Czuriga, D.; Paulus, W.J.; Czuriga, I.; Edes, I.; Papp, Z.; Borbely, A. Cellular mechanisms for diastolic dysfunction in the human heart. Curr. Pharm. Biotechnol. 2012, 13, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Schmidt, A.M. Receptor for advanced glycation end products (rage) and implications for the pathophysiology of heart failure. Curr. Heart Fail. Rep. 2012, 9, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.J.; Salgado, F.J.; Vinuela, J.E.; Nogueira, M. Interleukin-12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology 1997, 197, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, V.; Ardaillou, N.; Vlahovic, P.; Placier, S.; Ronco, P.; Ardaillou, R. Interferon-γ induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology 1993, 80, 465–470. [Google Scholar] [PubMed]
- Riemann, D.; Kehlen, A.; Langner, J. Stimulation of the expression and the enzyme activity of aminopeptidase N/CD13 and dipeptidylpeptidase IV/CD26 on human renal cell carcinoma cells and renal tubular epithelial cells by T cell-derived cytokines, such as IL-4 and IL-13. Clin. Exp. Immunol. 1995, 100, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Yamabe, T.; Takakura, K.; Sugie, K.; Kitaoka, Y.; Takeda, S.; Okubo, Y.; Teshigawara, K.; Yodoi, J.; Hori, T. Induction of the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural killer cells by IL-2, IL-12 or IL-15. Immunology 1997, 91, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12. [Google Scholar] [CrossRef]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Tahara, A.; Kaifu, K.; Ueda, S.; Okuda, S.; Imaizumi, T. Serum levels of advanced glycation end products (ages) are independently correlated with circulating levels of dipeptidyl peptidase-4 (DPP-4) in humans. Clin. Biochem. 2013, 46, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Faigle, M.; Knapp, S.; Karhausen, J.; Ibla, J.; Rosenberger, P.; Odegard, K.C.; Laussen, P.C.; Thompson, L.F.; Colgan, S.P. Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26. Blood 2006, 108, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Matheeussen, V.; Jungraithmayr, W.; de Meester, I. Dipeptidyl peptidase 4 as a therapeutic target in ischemia/reperfusion injury. Pharmacol. Ther. 2012, 136, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Biologic actions and therapeutic potential of the proglucagon-derived peptides. Nat. Clin. Pract. Endocrinol. Metab. 2005, 1, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Knop, F.K.; Vilsboll, T. Glucagon-like peptide-1 receptor agonists for the treatment of type 2 diabetes: Differences and similarities. Eur. J. Int. Med. 2014, 25, 407–414. [Google Scholar] [CrossRef]
- Scheen, A.J. A review of gliptins for 2014. Expert Opin. Pharmacother. 2015, 16, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Anderson, C.; Broyde, A.; Polizzi, C.; Fernandez, R.; Baron, A.; Parkes, D.G. Glucagon-like peptide-1 and the exenatide analogue AC 3174 improve cardiac function, cardiac remodeling, and survival in rats with chronic heart failure. Cardiovasc. Diabetol. 2010, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Poornima, I.; Brown, S.B.; Bhashyam, S.; Parikh, P.; Bolukoglu, H.; Shannon, R.P. Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circulation 2008, 1, 153–160. [Google Scholar] [PubMed]
- Ravassa, S.; Zudaire, A.; Carr, R.D.; Diez, J. Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am. J. Physiol. 2011, 300, H1361–H1372. [Google Scholar]
- Timmers, L.; Henriques, J.P.; de Kleijn, D.P.; Devries, J.H.; Kemperman, H.; Steendijk, P.; Verlaan, C.W.; Kerver, M.; Piek, J.J.; Doevendans, P.A.; et al. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J. Am. Coll. Cardiol. 2009, 53, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Mistry, M.; Roman, R.J. Renal effects of glucagon-like peptide in rats. Eur. J. Pharmacol. 2002, 434, 163–167. [Google Scholar] [CrossRef]
- Crajoinas, R.O.; Oricchio, F.T.; Pessoa, T.D.; Pacheco, B.P.; Lessa, L.M.; Malnic, G.; Girardi, A.C. Mechanisms mediating the diuretic and natriuretic actions of the incretin hormone glucagon-like peptide-1. Am. J. Physiol. Ren. Physiol. 2011, 301, F355–F363. [Google Scholar] [CrossRef]
- Rieg, T.; Gerasimova, M.; Murray, F.; Masuda, T.; Tang, T.; Rose, M.; Drucker, D.J.; Vallon, V. Natriuretic effect by exendin-4, but not the DPP-4 inhibitor alogliptin, is mediated via the GLP-1 receptor and preserved in obese type 2 diabetic mice. Am. J. Physiol. Ren. Physiol. 2012, 303, F963–F971. [Google Scholar] [CrossRef]
- Thomson, S.C.; Kashkouli, A.; Singh, P. Glucagon-like peptide-1 receptor stimulation increases GFR and suppresses proximal reabsorption in the rat. Am. J. Physiol. Ren. Physiol. 2013, 304, F137–F144. [Google Scholar] [CrossRef]
- Skov, J.; Holst, J.J.; Gotze, J.P.; Frokiaer, J.; Christiansen, J.S. Glucagon-like peptide-1: Effect on pro-atrial natriuretic peptide in healthy males. Endocr. Connect. 2014, 3, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Gutzwiller, J.P.; Hruz, P.; Huber, A.R.; Hamel, C.; Zehnder, C.; Drewe, J.; Gutmann, H.; Stanga, Z.; Vogel, D.; Beglinger, C. Glucagon-like peptide-1 is involved in sodium and water homeostasis in humans. Digestion 2006, 73, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Gutzwiller, J.P.; Tschopp, S.; Bock, A.; Zehnder, C.E.; Huber, A.R.; Kreyenbuehl, M.; Gutmann, H.; Drewe, J.; Henzen, C.; Goeke, B.; et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J. Clin. Endocrinol. Metab. 2004, 89, 3055–3061. [Google Scholar] [CrossRef] [PubMed]
- Skov, J.; Dejgaard, A.; Frokiaer, J.; Holst, J.J.; Jonassen, T.; Rittig, S.; Christiansen, J.S. Glucagon-like peptide-1 (GLP-1): Effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J. Clin. Endocrinol. Metab. 2013, 98, E664–E671. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Platt, M.J.; Shibasaki, T.; Quaggin, S.E.; Backx, P.H.; Seino, S.; Simpson, J.A.; Drucker, D.J. GLP-1 receptor activation and epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 2013, 19, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Okamoto, K.; Negishi, K.; Nangaku, M.; Noiri, E. Protection of glucagon-like peptide-1 in cisplatin-induced renal injury elucidates gut-kidney connection. J. Am. Soc. Nephrol. 2013, 24, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Mather, A.; Pollock, C. Role of GLP-1 and DPP-4 in diabetic nephropathy and cardiovascular disease. Clin. Sci. 2013, 124, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Higashijima, Y.; Wada, T.; Nangaku, M. The potential for renoprotection with incretin-based drugs. Kidney Int. 2014, 86, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Cavusoglu, T.; Erbas, O.; Karadeniz, T.; Akdemir, O.; Acikgoz, E.; Karadeniz, M.; Tuglu, M.I.; Ates, U. Comparison of nephron-protective effects of enalapril and GLP analogues (exenatide) in diabetic nephropathy. Exp. Clin. Endocrinol. Diabetes 2014, 122, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Tsai, T.H.; Yang, C.C.; Sun, C.K.; Chang, L.T.; Chen, H.H.; Chang, C.L.; Sung, P.H.; Zhen, Y.Y.; Leu, S.; et al. Exendin-4 and sitagliptin protect kidney from ischemia-reperfusion injury through suppressing oxidative stress and inflammatory reaction. J. Transl. Med. 2013, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.K.; Mocanu, M.M.; Carr, R.D.; Brand, C.L.; Yellon, D.M. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2005, 54, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Noyan-Ashraf, M.H.; Momen, M.A.; Ban, K.; Sadi, A.M.; Zhou, Y.Q.; Riazi, A.M.; Baggio, L.L.; Henkelman, R.M.; Husain, M.; Drucker, D.J. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009, 58, 975–983. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, M.; Du, J.; Wang, Z.; Yano, N.; Zhang, L.; Wang, Y.; Qin, G.; Zhuang, S.; Zhao, T.C. Stimulation of glucagon-like peptide-1 receptor through exendin-4 preserves myocardial performance and prevents cardiac remodeling in infarcted myocardium. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E630–E643. [Google Scholar] [CrossRef] [PubMed]
- Lonborg, J.; Vejlstrup, N.; Kelbaek, H.; Nepper-Christensen, L.; Jorgensen, E.; Helqvist, S.; Holmvang, L.; Saunamaki, K.; Botker, H.E.; Kim, W.Y.; et al. Impact of acute hyperglycemia on myocardial infarct size, area at risk, and salvage in patients with stemi and the association with exenatide treatment: Results from a randomized study. Diabetes 2014, 63, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Vigili de Kreutzenberg, S.; Fadini, G.P. Cardiovascular actions of GLP-1 and incretin-based pharmacotherapy. Curr. Diabetes Rep. 2014, 14, 483. [Google Scholar] [CrossRef]
- Picatoste, B.; Ramirez, E.; Caro-Vadillo, A.; Iborra, C.; Ares-Carrasco, S.; Egido, J.; Tunon, J.; Lorenzo, O. Sitagliptin reduces cardiac apoptosis, hypertrophy and fibrosis primarily by insulin-dependent mechanisms in experimental type-II diabetes. Potential roles of GLP-1 isoforms. PLoS One 2013, 8, e78330. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T.; Gonon, A.T.; Sjoholm, A.; Pernow, J. Glucagon-like peptide-1 relaxes rat conduit arteries via an endothelium-independent mechanism. Regul. Pept. 2005, 125, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T.; Gutniak, M.K.; Zhang, Q.; Zhang, F.; Holst, J.J.; Ahren, B.; Sjoholm, A. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E1209–E1215. [Google Scholar] [CrossRef] [PubMed]
- Crajoinas, R.O.; Savignano, F.A.; Nakamuta, J.; Girardi, A.C. Renal vascular relaxation response to glucagon-like peptide-1 (GLP-1) is impaired in spontaneously hypertensive rats (SHR). FASEB J. 2012, 26. (1_MeetingAbstracts): 688.2. [Google Scholar] [CrossRef]
- Limei, L.; Jian, L.; Yu, H. Protective effects of glucagon-like peptide-1 on endothelial function in hypertension. J. Cardiovasc. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Shimizu, H.; Masuta, K.; Aono, K.; Asada, H.; Sasakura, K.; Tamaki, M.; Sugita, K.; Yamada, K. Molecular forms of human brain natriuretic peptide in plasma. Clin. Chim. Acta 2002, 316, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Rubattu, S.; Burnett, J. Natriuretic peptides in cardiovascular diseases: Current use and perspectives. Eur. Heart J. 2013, 35, 419–425. [Google Scholar] [CrossRef] [PubMed]
- McGregor, A.; Richards, M.; Espiner, E.; Yandle, T.; Ikram, H. Brain natriuretic peptide administered to man: Actions and metabolism. J. Clin. Endocrinol. Metab. 1990, 70, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Gunning, M.; Ballermann, B.J.; Silva, P.; Brenner, B.M.; Zeidel, M.L. Brain natriuretic peptide: Interaction with renal anp system. Am. J. Physiol. 1990, 258, F467–F472. [Google Scholar] [PubMed]
- Morita, H.; Nishida, Y.; Motochigawa, H.; Kangawa, K.; Minamino, N.; Matsuo, H.; Hosomi, H. Effects of brain natriuretic peptide on renal nerve activity in conscious rabbits. Am. J. Physiol. 1989, 256, R792–R796. [Google Scholar] [PubMed]
- Remes, J. Neuroendocrine activation after myocardial infarction. Br. Heart J. 1994, 72, S65–S69. [Google Scholar] [CrossRef] [PubMed]
- Riegger, G.A.; Kromer, E.P.; Kochsiek, K. Human atrial natriuretic peptide: Plasma levels, hemodynamic, hormonal, and renal effects in patients with severe congestive heart failure. J. Cardiovasc. Pharmacol. 1986, 8, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Cody, R.J.; Atlas, S.A.; Laragh, J.H.; Kubo, S.H.; Covit, A.B.; Ryman, K.S.; Shaknovich, A.; Pondolfino, K.; Clark, M.; Camargo, M.J.; et al. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J. Clin. Investig. 1986, 78, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Yasue, H.; Yoshimura, M.; Ogawa, H.; Jougasaki, M.; Matsumura, T.; Mukoyama, M.; Nakao, K. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 1993, 88, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ledwidge, M.; Gallagher, J.; Conlon, C.; Tallon, E.; O’Connell, E.; Dawkins, I.; Watson, C.; O’Hanlon, R.; Bermingham, M.; Patle, A.; et al. Natriuretic peptide-based screening and collaborative care for heart failure: The stop-HF randomized trial. JAMA 2013, 310, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Eiskjaer, H.; Carstens, J.; Pedersen, E.B. Renal effects of brain natriuretic peptide in patients with congestive heart failure. Clin. Sci. 1999, 96, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Charloux, A.; Piquard, F.; Doutreleau, S.; Brandenberger, G.; Geny, B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur. J. Clin. Investig. 2003, 33, 769–778. [Google Scholar] [CrossRef]
- Cadnapaphornchai, M.A.; Gurevich, A.K.; Weinberger, H.D.; Schrier, R.W. Pathophysiology of sodium and water retention in heart failure. Cardiology 2001, 96, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Inoue, B.H.; dos Santos, L.; Pessoa, T.D.; Antonio, E.L.; Pacheco, B.P.; Savignano, F.A.; Carraro-Lacroix, L.R.; Tucci, P.J.; Malnic, G.; Girardi, A.C. Increased NHE3 abundance and transport activity in renal proximal tubule of rats with heart failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R166–R174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkridge, A.M.; Heublein, D.M.; Bergen, H.R., 3rd; Cataliotti, A.; Burnett, J.C.; Muddiman, D.C. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 17442–17447. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Phelps, M.A.; Wood, C.M.; Schellenberger, U.; van Le, A.; Perichon, R.; Jaffe, A.S. Comparison of mass spectrometry and clinical assay measurements of circulating fragments of b-type natriuretic peptide in patients with chronic heart failure. Circ. Heart Fail. 2011, 4, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Brandt, I.; Lambeir, A.M.; Ketelslegers, J.M.; Vanderheyden, M.; Scharpe, S.; de Meester, I. Dipeptidyl-peptidase IV converts intact b-type natriuretic peptide into its des-serpro form. Clin. Chem. 2006, 52, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Costello-Boerrigter, L.C.; Harty, G.J.; Lapp, H.; Burnett, J.C., Jr. Des-serine-proline brain natriuretic peptide 3–32 in cardiorenal regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R897–R901. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.; Touihri, K.; Matheeussen, V.; Mendes Da Costa, A.; Mahmoudabady, M.; Mathieu, M.; Baerts, L.; Peace, A.; Lybaert, P.; Scharpe, S.; et al. Dipeptidyl peptidase IV inhibition improves cardiorenal function in overpacing-induced heart failure. Eur. J. Heart Fail. 2012, 14, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Pawig, L.; Weber, C.; Noels, H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front. Physiol. 2014, 5, 212. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. Role of the SDF-1/CXCR4 system in myocardial infarction. Circ. J. 2010, 74, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Kanki, S.; Segers, V.F.; Wu, W.; Kakkar, R.; Gannon, J.; Sys, S.U.; Sandrasagra, A.; Lee, R.T. Stromal cell-derived factor-1 retention and cardioprotection for ischemic myocardium. Circ. Heart Fail. 2011, 4, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Salvucci, O.; Yao, L.; Villalba, S.; Sajewicz, A.; Pittaluga, S.; Tosato, G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood 2002, 99, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.; Revin, V.; Wu, W.; Qiu, H.; Yan, Z.; Lee, R.T.; Sandrasagra, A. Protease-resistant stromal cell-derived factor-1 for the treatment of experimental peripheral artery disease. Circulation 2011, 123, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.; Tokunou, T.; Higgins, L.J.; MacGillivray, C.; Gannon, J.; Lee, R.T. Local delivery of protease-resistant stromal cell derived factor-1 for stem cell recruitment after myocardial infarction. Circulation 2007, 116, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Zaruba, M.M.; Theiss, H.D.; Vallaster, M.; Mehl, U.; Brunner, S.; David, R.; Fischer, R.; Krieg, L.; Hirsch, E.; Huber, B.; et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell 2009, 4, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Theiss, H.D.; Vallaster, M.; Rischpler, C.; Krieg, L.; Zaruba, M.M.; Brunner, S.; Vanchev, Y.; Fischer, R.; Grobner, M.; Huber, B.; et al. Dual stem cell therapy after myocardial infarction acts specifically by enhanced homing via the SDF-1/CXCR4 axis. Stem Cell Res. 2011, 7, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, O.; Spinetti, G.; Specchia, C.; Cangiano, E.; Valgimigli, M.; Madeddu, P. Migratory activity of circulating progenitor cells and serum SDF-1α predict adverse events in patients with myocardial infarction. Cardiovasc. Res. 2013, 100, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Liu, C.; Aviv, A.; Ho, J.E.; Courchesne, P.; Muntendam, P.; Larson, M.G.; Cheng, S.; Wang, T.J.; Mehta, N.N.; et al. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Boscaro, E.; Albiero, M.; Menegazzo, L.; Frison, V.; de Kreutzenberg, S.; Agostini, C.; Tiengo, A.; Avogaro, A. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: Possible role of stromal-derived factor-1α. Diabetes Care 2010, 33, 1607–1609. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J.; Sawmiller, D.R. Vasoactive intestinal peptide: Cardiovascular effects. Cardiovasc. Res. 2001, 49, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.C. Cardioprotective role of the VIP signaling system. Drug News Perspect 2005, 18, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Vacas, E.; Bajo, A.M.; Schally, A.V.; Sanchez-Chapado, M.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide induces oxidative stress and suppresses metastatic potential in human clear cell renal cell carcinoma. Mol. Cell. Endocrinol. 2012, 365, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.M.; Silva, P.; Stoff, J.S.; Epstein, F.H. Effect of vasoactive intestinal peptide on isolated perfused rat kidney. Am. J. Physiol. 1985, 249, E494–E497. [Google Scholar] [PubMed]
- Lambeir, A.M.; Durinx, C.; Proost, P.; van Damme, J.; Scharpe, S.; de Meester, I. Kinetic study of the processing by dipeptidyl-peptidase IV/CD26 of neuropeptides involved in pancreatic insulin secretion. FEBS Lett. 2001, 507, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, H.M.; Levick, S.P. Substance P in heart failure: The good and the bad. Int. J. Cardiol. 2014, 170, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Tentolouris, C.; Crake, T.; Katsimaglis, G.; Stefanadis, C.; Toutouzas, P.; Davies, G.J. Effects of l- and d-arginine on the basal tone of human diseased coronary arteries and their responses to substance P. Heart 1999, 81, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.Y.; Song, J.X.; Lu, M.Y.; Chen, H. Cardioprotection by ischemic postconditioning is lost in isolated perfused heart from diabetic rats: Involvement of transient receptor potential vanilloid 1, calcitonin gene-related peptide and substance P. Regul. Pept. 2011, 169, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chiao, H.; Caldwell, R.W. The role of substance P in myocardial dysfunction during ischemia and reperfusion. Naunyn Schmiedebergs Arch. Pharmacol. 1996, 353, 400–407. [Google Scholar] [PubMed]
- Mentlein, R.; Dahms, P.; Grandt, D.; Kruger, R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul. Pept. 1993, 49, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.; Cavadas, C.; Grouzmann, E. Neuropeptide y and its receptors as potential therapeutic drug targets. Clin. Chim. Acta 2002, 326, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Zukowska, Z.; Grant, D.S.; Lee, E.W. Neuropeptide y: A novel mechanism for ischemic angiogenesis. Trends Cardiovasc. Med. 2003, 13, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Hobson, J.P.; Spiegel, S.; Kleinman, H.K.; Zukowska, Z. Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. FASEB J. 2006, 20, 1924–1926. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Scott, N.A.; Motulsky, H.J.; Michel, M.C.; Boublik, J.H.; Rivier, J.E.; Ziegler, M.; Allen, R.S.; Brown, M.R. Elevation of plasma neuropeptide Y levels in congestive heart failure. Am. J. Med. 1989, 86, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Hulting, J.; Sollevi, A.; Ullman, B.; Franco-Cereceda, A.; Lundberg, J.M. Plasma neuropeptide Y on admission to a coronary care unit: Raised levels in patients with left heart failure. Cardiovasc. Res. 1990, 24, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.A.; Gullion, C.M.; Koro, C.E.; Ephross, S.A.; Brown, J.B. The incidence of congestive heart failure in type 2 diabetes: An update. Diabetes Care 2004, 27, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Sauve, M.; Ban, K.; Momen, M.A.; Zhou, Y.Q.; Henkelman, R.M.; Husain, M.; Drucker, D.J. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 2010, 59, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Sillje, H.H.; Meissner, M.; van Gilst, W.H.; de Boer, R.A. Early and late effects of the DPP-4 inhibitor vildagliptin in a rat model of post-myocardial infarction heart failure. Cardiovasc. Diabetol. 2011, 10. [Google Scholar] [CrossRef]
- Arruda-Junior, D.F.; Socas, L.J.; Dariolli, R.; Antonio, E.L.; Tucci, P.J.; dos Santos, L.; Girardi, A.C. Dipeptidyl Peptidase IV Inhibition Ameliorates Cardiorenal Function in Experimental Heart Failure; Experimental Biology: San Diego, CA, USA, 2014. [Google Scholar]
- Read, P.A.; Khan, F.Z.; Heck, P.M.; Hoole, S.P.; Dutka, D.P. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ. Cardiovasc. Imaging 2010, 3, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Witteles, R.M.; Keu, K.V.; Quon, A.; Tavana, H.; Fowler, M.B. Dipeptidyl peptidase 4 inhibition increases myocardial glucose uptake in nonischemic cardiomyopathy. J. Card. Fail. 2012, 18, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Giannocco, G.; Oliveira, K.C.; Crajoinas, R.O.; Venturini, G.; Salles, T.A.; Fonseca-Alaniz, M.H.; Maciel, R.M.; Girardi, A.C. Dipeptidyl peptidase IV inhibition upregulates GLUT4 translocation and expression in heart and skeletal muscle of spontaneously hypertensive rats. Eur. J. Pharmacol. 2013, 698, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Braunwald, E.; Raz, I.; Cavender, M.A.; Morrow, D.A.; Jarolim, P.; Udell, J.A.; Mosenzon, O.; Im, K.; Umez-Eronini, A.A.; et al. Heart failure, saxagliptin, and diabetes mellitus: Observations from the savor-timi 53 randomized trial. Circulation 2014, 130, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Zannad, F. Saxagliptin, alogliptin, and cardiovascular outcomes. N. Engl. J. Med. 2014, 370, 483–484. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V. Vildagliptin Shows no Adverse Effect on Ejection Fraction in Diabetic Patients with HF. In Proceedings of the Heart Failure Congress 2013, Lisbon, Portugal, 25–28 May 2013.
- Krum, H.; Lukashevich, V.; Bolli, G.B.; Kozlovski, P.; Kothny, W.; Ponikowski, P. No Significant Difference in Risk of Heart Failure Hospitalization with Vildagliptin in Diabetic Patients with Systolic Chronic Heart Failure: Vividd Study; American Diabetes Association, 74th Scientific Sessions: San Francisco, CA, USA, 2014. [Google Scholar]
- ClinicalTrials.gov. CAROLINA: Cardiovascular Outcome Study of Linagliptin vs. Glimepiride in Patients With Type 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT01243424 (accessed on 4 February 2015).
- ClinicalTrials.gov. Sitagliptin Cardiovascular Outcome Study (MK-0431-082) (TECOS). Available online: https://clinicaltrials.gov/ct2/show/NCT00790205 (accessed on 28 January 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salles, T.A.; Dos Santos, L.; Barauna, V.G.; Girardi, A.C.C. Potential Role of Dipeptidyl Peptidase IV in the Pathophysiology of Heart Failure. Int. J. Mol. Sci. 2015, 16, 4226-4249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024226
Salles TA, Dos Santos L, Barauna VG, Girardi ACC. Potential Role of Dipeptidyl Peptidase IV in the Pathophysiology of Heart Failure. International Journal of Molecular Sciences. 2015; 16(2):4226-4249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024226
Chicago/Turabian StyleSalles, Thiago A., Leonardo Dos Santos, Valério G. Barauna, and Adriana C. C. Girardi. 2015. "Potential Role of Dipeptidyl Peptidase IV in the Pathophysiology of Heart Failure" International Journal of Molecular Sciences 16, no. 2: 4226-4249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024226