
Metastatic Melanoma Cells Evade Immune Detection by Silencing STAT1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results
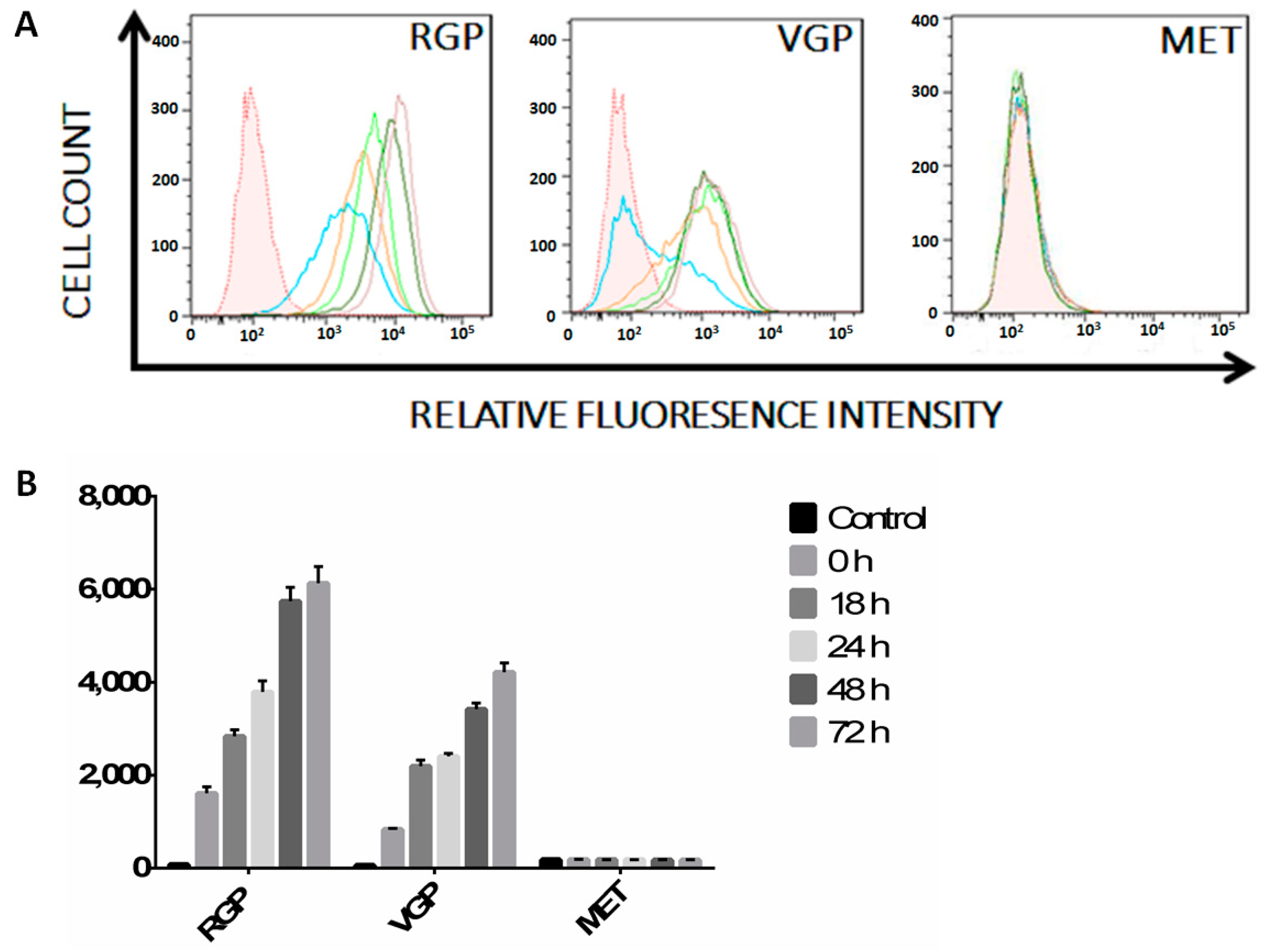
2.1. MHC II Is Increasingly Suppressed in RGP, VGP and MET Cells
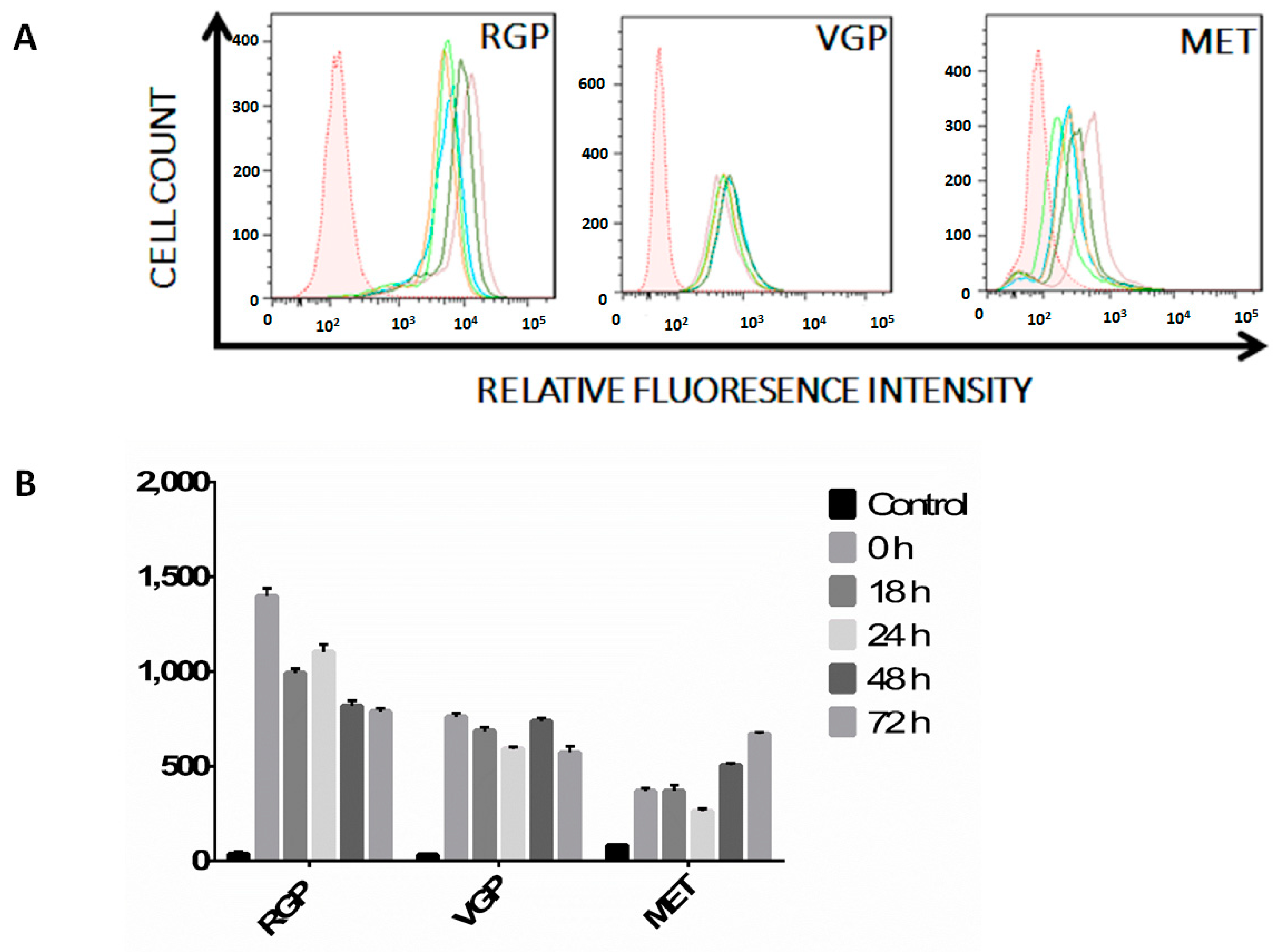
2.2. Melanoma Cells Remain IFN-γ Responsive throughout Disease Progression

2.3. MET Cells Express Both Janus Kinase 1 (JAK1) and JAK2


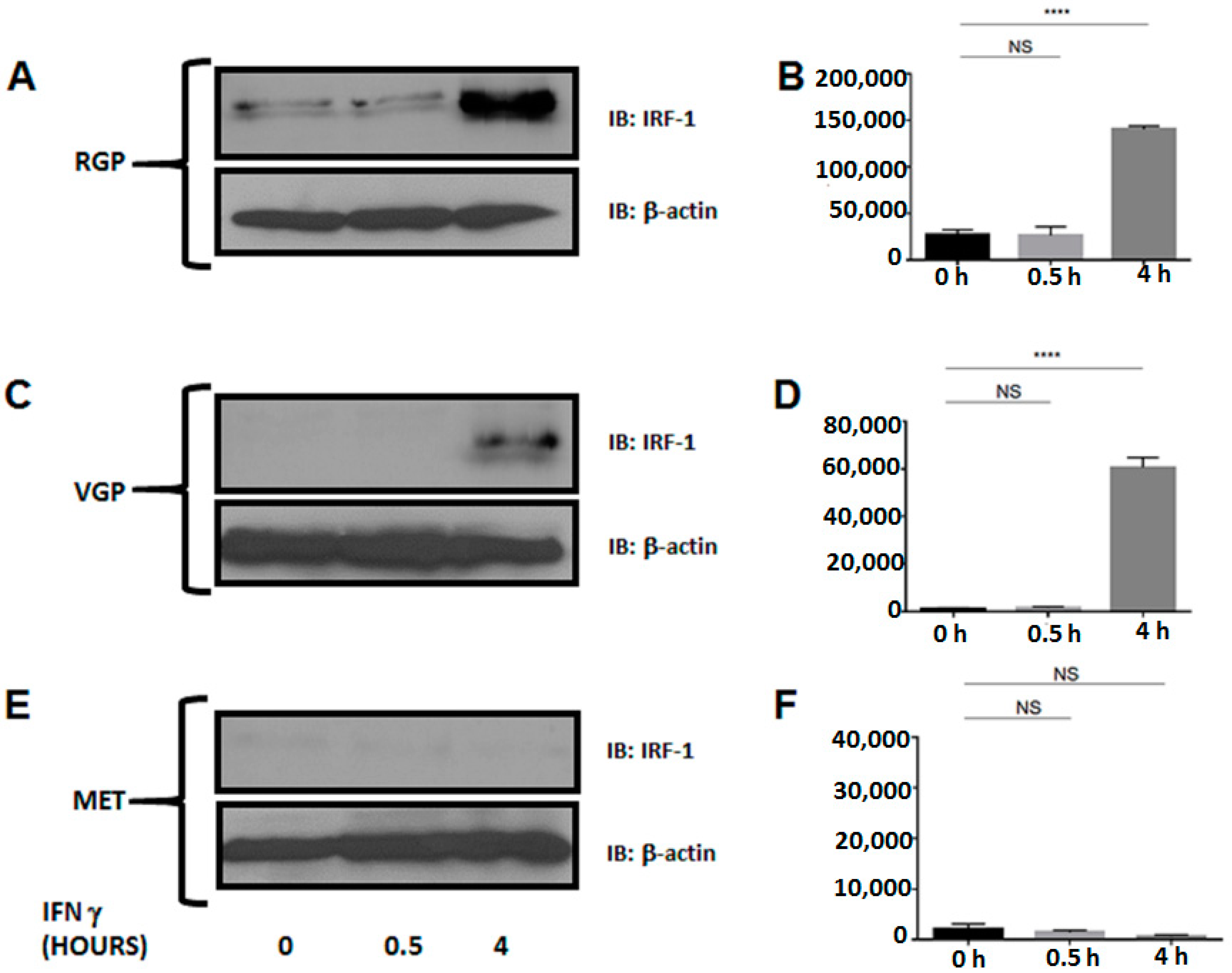
2.4. Metastatic Melanoma Cells Lack the Interferon Response Factor, IRF-1



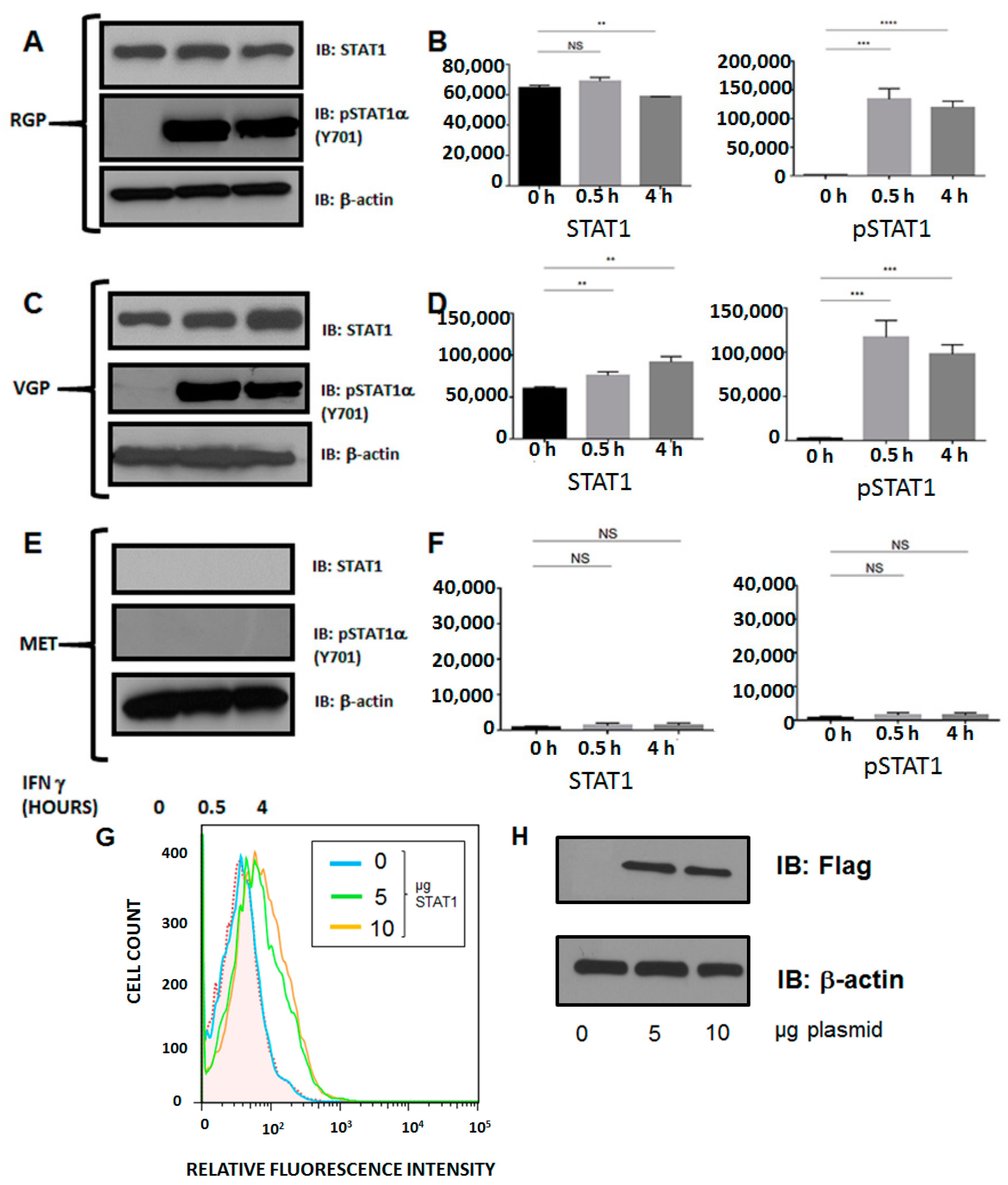
2.5. Silencing of MHC II in Metastatic Melanoma Is the Result of Dysregulation of Basal STAT1α Expression
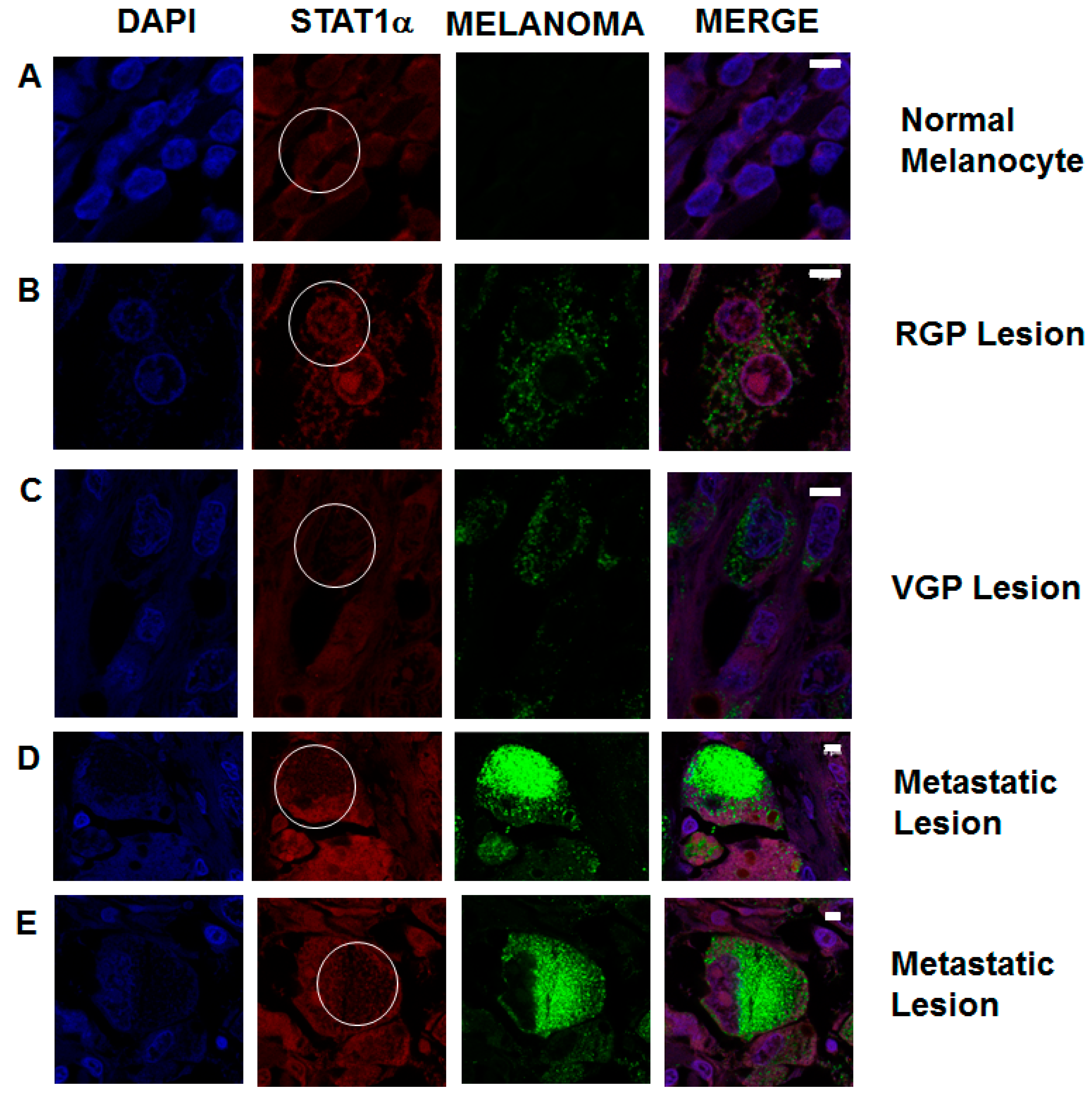
2.6. STAT1 Expression Is Decreased in Patient Metastatic Melanoma Samples
3. Discussion
4. Experimental Section
4.1. Cell Lines
4.2. Western Blots
4.3. Flow Cytometry
4.4. Immunofluorescence
4.5. Overexpression Assays
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clark, W.H., Jr.; Ainsworth, A.M.; Bernardino, E.A.; Yang, C.H.; Mihm, C.M., Jr.; Reed, R.J. The developmental biology of primary human malignant melanomas. Semin. Oncol. 1975, 2, 83–103. [Google Scholar] [PubMed]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tyminska, A. Skin melanocytes: Biology and development. Postepy Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Crutcher, W.A. Dysplastic Nevi—Markers and precursors of malignant melanoma. West. J. Med. 1987, 147, 459. [Google Scholar] [PubMed]
- Ciarletta, P.; Foret, L.; Ben Amar, M. The radial growth phase of malignant melanoma: Multi-phase modelling, numerical simulations and linear stability analysis. J. R. Soc. Interface 2011, 8, 345–368. [Google Scholar] [CrossRef] [PubMed]
- Herlyn, M.; Thurin, J.; Balaban, G.; Bennicelli, J.L.; Herlyn, D.; Elder, D.E.; Bondi, E.; Guerry, D.; Nowell, P.; Clark, W.H.; et al. Characteristics of cultured human melanocytes isolated from different stages of tumor progression. Cancer Res. 1985, 45, 5670–5676. [Google Scholar] [PubMed]
- Laga, A.C.; Murphy, G.F. Cellular heterogeneity in vertical growth phase melanoma. Arch. Pathol. Lab. Med. 2010, 134, 1750–1757. [Google Scholar] [PubMed]
- Rak, J.W.; Hegmann, E.J.; Lu, C.; Kerbel, R.S. Progressive loss of sensitivity to endothelium-derived growth inhibitors expressed by human melanoma cells during disease progression. J. Cell. Physiol. 1994, 159, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Guirguis, R.; Stracke, M. Biology of melanoma invasion and metastasis. Pigment Cell Res. 1987, 1, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Pages, F.; Galon, J. Tumor immunosurveillance in human cancers. Cancer Metastasis Rev. 2011, 30, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; McDermott, D. Update on benefit of immunotherapy and targeted therapy in melanoma: The changing landscape. Cancer Manag. Res. 2014, 6, 279–289. [Google Scholar] [PubMed]
- Gajewski, T.F. Identifying and overcoming immune resistance mechanisms in the melanoma tumor microenvironment. Clin. Cancer Res. 2006, 12, 2326s–2330s. [Google Scholar] [CrossRef] [PubMed]
- Braciale, T.J.; Morrison, L.A.; Sweetser, M.T.; Sambrook, J.; Gething, M.J.; Braciale, V.L. Antigen presentation pathways to class I and class II MHC-restricted T lymphocytes. Immunol. Rev. 1987, 98, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Gerloni, M.; Zanetti, M. CD4 T cells in tumor immunity. Springer Semin. Immunopathol. 2005, 27, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.G.; Khammanivong, V.; Liu, W.J.; Leggatt, G.R.; Frazer, I.H.; Fernando, G.J. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res. 2002, 62, 6438–6441. [Google Scholar] [PubMed]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Thakur, A.; Clements, V. Rejection of mouse sarcoma cells after transfection of MHC class II genes. J. Immunol. 1990, 144, 4068–4071. [Google Scholar] [PubMed]
- Ostrand-Rosenberg, S.; Roby, C.A.; Clements, V.K. Abrogation of tumorigenicity by MHC class II antigen expression requires the cytoplasmic domain of the class II molecule. J. Immunol. 1991, 147, 2419–2422. [Google Scholar] [PubMed]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Lundin, K.U.; Lorvik, K.B.; Hofgaard, P.O.; Bogen, B. Secretion of tumor-specific antigen by myeloma cells is required for cancer immunosurveillance by CD4+ T cells. Cancer Res. 2009, 69, 5901–5907. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.K.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.; Lorvik, K.B.; Hammarstrom, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Rosjo, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary antitumor immune response mediated by CD4+ T cells. Immunity 2005, 22, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Fukuzawa, M.; Yoshioka, T.; Nakajima, H.; Hamaoka, T. The role of tumor-specific Lyt-1+2- T cells in eradicating tumor cells in vivo. I. Lyt-1+2- T cells do not necessarily require recruitment of host’s cytotoxic T cell precursors for implementation of in vivo immunity. J. Immunol. 1984, 133, 1671–1676. [Google Scholar] [PubMed]
- Greenberg, P.D.; Kern, D.E.; Cheever, M.A. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+2-T cells. Tumor eradication does not require participation of cytotoxic T cells. J. Exp. Med. 1985, 161, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Lauritzsen, G.F.; Weiss, S.; Dembic, Z.; Bogen, B. Naive idiotype-specific CD4+ T cells and immunosurveillance of B-cell tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 5700–5704. [Google Scholar] [CrossRef] [PubMed]
- Rudensky, A.; Preston-Hurlburt, P.; Hong, S.C.; Barlow, A.; Janeway, C.A., Jr. Sequence analysis of peptides bound to MHC class II molecules. Nature 1991, 353, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Sartoris, S.; Scupoli, M.T.; Scarpellino, L.; Paiola, F.; Jotterand-Bellomo, M.; Tridente, G.; Accolla, R.S. Inducible and constitutive MHC class II gene expression. Distinct tissue-specific genetic controls. J. Immunol. 1990, 145, 1960–1967. [Google Scholar] [PubMed]
- Billiau, A.; Matthys, P. Interferon-γ: A historical perspective. Cytokine Growth Factor Rev. 2009, 20, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukocyte Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Garotta, G.; Ozmen, L.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Larner, A.C.; Finbloom, D.S. Interferon-γ induces tyrosine phosphorylation of interferon-γ receptor and regulated association of protein tyrosine kinases, JAK1 and JAK2, with its receptor. J. Biol. Chem. 1994, 269, 14333–14336. [Google Scholar] [PubMed]
- Sakatsume, M.; Igarashi, K.; Winestock, K.D.; Garotta, G.; Larner, A.C.; Finbloom, D.S. The JAK kinases differentially associate with the α and β (accessory factor) chains of the interferon γ receptor to form a functional receptor unit capable of activating STAT transcription factors. J. Biol. Chem. 1995, 270, 17528–17534. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Horvath, C.M.; Huang, L.H.; Qureshi, S.A.; Cowburn, D.; Darnell, J.E., Jr. Interferon activation of the transcription factor STAT91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 1994, 76, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Masternak, K.; Muhlethaler-Mottet, A.; Villard, J.; Zufferey, M.; Steimle, V.; Reith, W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000, 14, 1156–1166. [Google Scholar] [PubMed]
- Zika, E.; Greer, S.F.; Zhu, X.S.; Ting, J.P. Histone deacetylase 1/mSin3a disrupts γ interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol. Cell. Biol. 2003, 23, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Spilianakis, C.; Kretsovali, A.; Agalioti, T.; Makatounakis, T.; Thanos, D.; Papamatheakis, J. CIITA regulates transcription onset viaSer5-phosphorylation of RNA Pol II. EMBO J. 2003, 22, 5125–5136. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Huang, J.; Greshock, J.; Horiates, G.; Nathanson, K.; Yang, X.; Herlyn, M.; Weber, B. Distinct MHC gene expression patterns during progression of melanoma. Genes Chromosom. Cancer 2010, 49, 144–154. [Google Scholar] [PubMed]
- Groth, A.; Kloss, S.; von Strandmann, E.P.; Koehl, U.; Koch, J. Mechanisms of tumor and viral immune escape from natural killer cell-mediated surveillance. J. Innate Immun. 2011, 3, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Enderling, H.; Hlatky, L.; Hahnfeldt, P. Immunoediting: Evidence of the multifaceted role of the immune system in self-metastatic tumor growth. Theor. Biol. Med. Model. 2012, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Boss, J.M. Regulation of transcription of MHC class II genes. Curr. Opin. Immunol. 1997, 9, 107–113. [Google Scholar] [CrossRef] [PubMed]
- De Paus, R.A.; Kilic, S.S.; van Dissel, J.T.; van de Vosse, E. Effect of amino acid substitutions in the human IFN-γ2 on IFN-γ responsiveness. Genes Immun. 2011, 12, 136–144. [Google Scholar]
- Gros, E.; Petzold, S.; Maintz, L.; Bieber, T.; Novak, N. Reduced IFN-γ receptor expression and attenuated IFN-γ response by dendritic cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Srisatjaluk, R.; Kotwal, G.J.; Hunt, L.A.; Justus, D.E. Modulation of γ interferon-induced major histocompatibility complex class II gene expression by porphyromonas gingivalis membrane vesicles. Infect. Immun. 2002, 70, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.C.; Beresford, G.W.; Mooney, M.R.; Boss, J.M. Kinetics of a γ interferon response: Expression and assembly of CIITA promoter IV and inhibition by methylation. Mol. Cell. Biol. 2002, 22, 4781–4791. [Google Scholar] [CrossRef] [PubMed]
- Truax, A.D.; Thakkar, M.; Greer, S.F. Dysregulated recruitment of the histone methyltransferase EZH2 to the class II transactivator (CIITA) promoter IV in breast cancer cells. PLoS One 2012, 7, e36013. [Google Scholar] [CrossRef] [PubMed]
- Reinsbach, S.; Nazarov, P.V.; Philippidou, D.; Schmitt, M.; Wienecke-Baldacchino, A.; Muller, A.; Vallar, L.; Behrmann, I.; Kreis, S. Dynamic regulation of microRNA expression following interferon-γ-induced gene transcription. RNA Biol. 2012, 9, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, T.; Mendez, R.; Del Campo, A.; Jimenez, P.; Aptsiauri, N.; Garrido, F.; Ruiz-Cabello, F. Distinct mechanisms of loss of IFN-γ mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer 2007, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Tanzarella, S.; Lionello, I.; Mendez, R.; Traversari, C.; Ruiz-Cabello, F.; Garrido, F. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2'-deoxycytidine treatment. Int. J. Cancer 2001, 94, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Respa, A.; Bukur, J.; Ferrone, S.; Pawelec, G.; Zhao, Y.; Wang, E.; Marincola, F.M.; Seliger, B. Association of IFN-γ signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clin. Cancer Res. 2011, 17, 2668–2678. [Google Scholar] [CrossRef] [PubMed]
- Radosevich, M.; Song, Z.; Gorga, J.C.; Ksander, B.; Ono, S.J. Epigenetic silencing of the CIITA gene and posttranscriptional regulation of class II MHC genes in ocular melanoma cells. Investig. Ophthalmol. Visual Sci. 2004, 45, 3185–3195. [Google Scholar] [CrossRef]
- Amalraj, J.; Cutler, S.J.; Ghazawi, I.; Boyle, G.M.; Ralph, S.J. Rest negatively and ISGF3 positively regulate the human STAT1 gene in melanoma. Mol. Cancer Ther. 2013, 12, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.J.; Paquette, A.J.; Anderson, D.J. Identification of potential target genes for the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 1996, 93, 9881–9886. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Golzio, M.; Sersa, G.; Hojman, P.; Kranjc, S.; Mesojednik, S.; Rols, M.P.; Teissie, J. Control by pulse parameters of DNA electrotransfer into solid tumors in mice. Gene Ther. 2009, 16, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Shen, X.; Kichaev, G.; Mendoza, J.M.; Yang, M.; Armendi, P.; Yan, J.; Kobinger, G.P.; Bello, A.; Khan, A.S.; et al. Optimization of electroporation-enhanced intradermal delivery of DNA vaccine using a minimally invasive surface device. Hum. Gene Ther. Methods 2012, 23, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Tavernier, G.; Bavarsad, N.; Demeester, J.; de Smedt, S.C. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control. Release 2010, 147, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Tuxen, I.V.; Jonson, L.; Santoni-Rugiu, E.; Hasselby, J.P.; Nielsen, F.C.; Lassen, U. Personalized oncology: Genomic screening in phase 1. APMIS 2014, 122, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.; Savage, K.; Reis-Filho, J.S.; Isacke, C.M. Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biol. 2008, 9, 13. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osborn, J.L.; Greer, S.F. Metastatic Melanoma Cells Evade Immune Detection by Silencing STAT1. Int. J. Mol. Sci. 2015, 16, 4343-4361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024343
Osborn JL, Greer SF. Metastatic Melanoma Cells Evade Immune Detection by Silencing STAT1. International Journal of Molecular Sciences. 2015; 16(2):4343-4361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024343
Chicago/Turabian StyleOsborn, JoDi Lynn, and Susanna F. Greer. 2015. "Metastatic Melanoma Cells Evade Immune Detection by Silencing STAT1" International Journal of Molecular Sciences 16, no. 2: 4343-4361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16024343