
De Novo Transcriptome Sequencing of Low Temperature-Treated Phlox subulata and Analysis of the Genes Involved in Cold Stress

Abstract
:1. Introduction
2. Results and Discussion
2.1. Phlox subulata (P. subulata) Transcriptome Sequencing and de Novo Assembly


{kind=link}
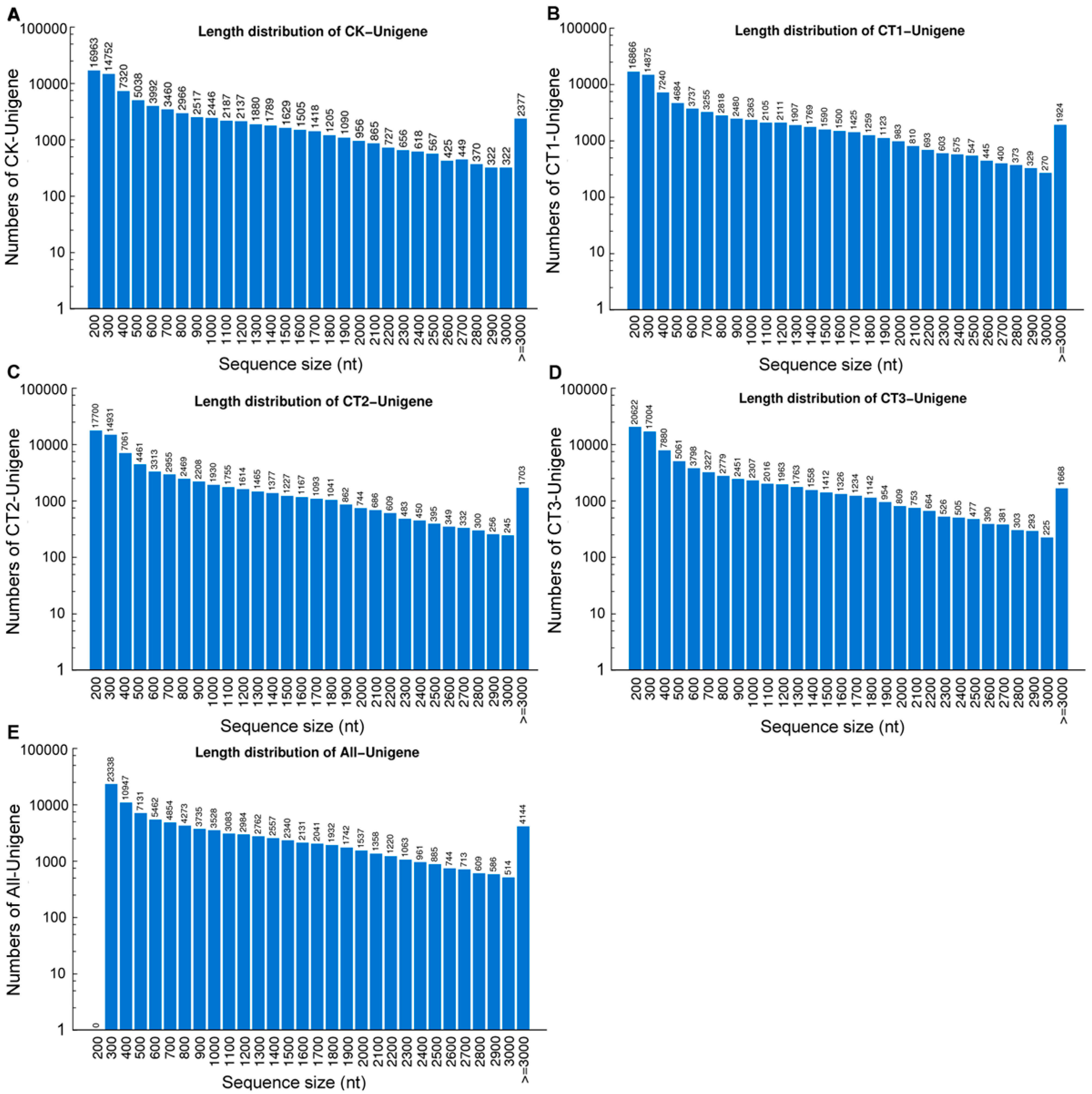{kind=link}
{kind=link}
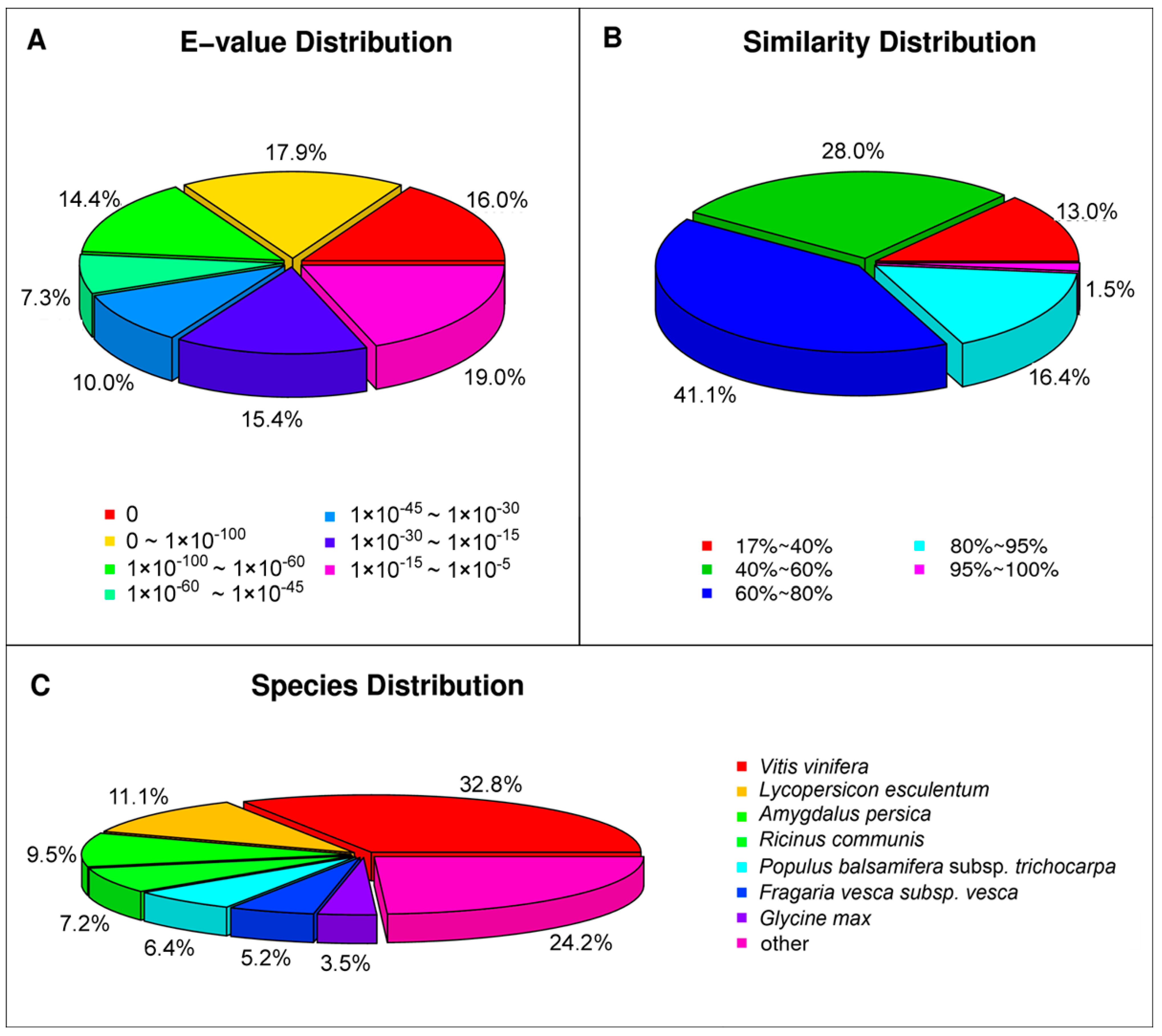{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | CK (20 °C) | CT1 (4 °C) | CT2 (0 °C) | CT3 (−10 °C) | Total |
|---|---|---|---|---|---|
| Total Raw Reads | 59,580,728 | 59,263,032 | 55,328,170 | 55,583,970 | – |
| Total Clean Reads | 55,528,592 | 55,319,076 | 51,409,114 | 51,736,252 | – |
| Total Clean Nucleotides (nt) | 4,997,573,280 | 4,978,716,840 | 4,626,820,260 | 4,656,262,680 | – |
| Q20 percentage | 97.50% | 97.53% | 97.41% | 97.51% | – |
| N percentage | 0.01% | 0.01% | 0.01% | 0.01% | – |
| GC percentage | 46.40% | 46.32% | 46.85% | 46.95% | – |
| Contig | |||||
| Total Number | 126,166 | 125,583 | 120,123 | 140,329 | – |
| Total Length (nt) | 48,095,639 | 47,695,976 | 42,622,577 | 48,874,606 | – |
| Mean Length (nt) | 381 | 380 | 355 | 348 | – |
| N50 | 820 | 857 | 714 | 705 | – |
| Unigene | |||||
| Total Number | 82,948 | 81,059 | 75,181 | 85,491 | 99,174 |
| Total Length (nt) | 67,146,823 | 63,741,994 | 54,402,959 | 60,415,665 | 98,892,318 |
| Mean Length (nt) | 810 | 786 | 724 | 707 | 997 |
| N50 | 1470 | 1434 | 1369 | 1322 | 1622 |
| Total Consensus Sequences | 82,948 | 81,059 | 75,181 | 85,491 | 99,174 |
| Distinct Clusters | 30,546 | 29,615 | 23,876 | 27,448 | 42,007 |
| Distinct Singletons | 52,402 | 51,444 | 51,305 | 58,043 | 57,167 |
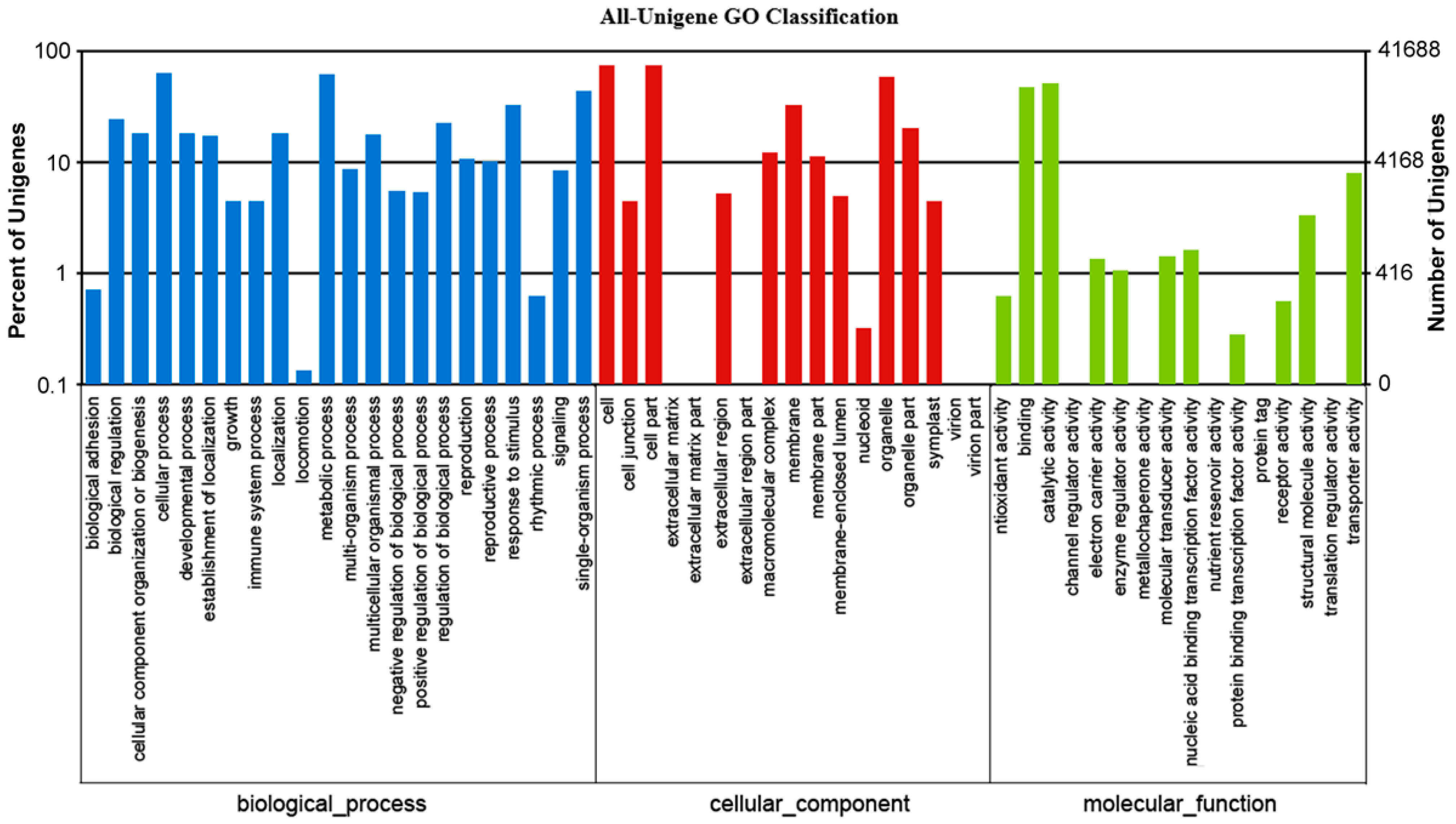
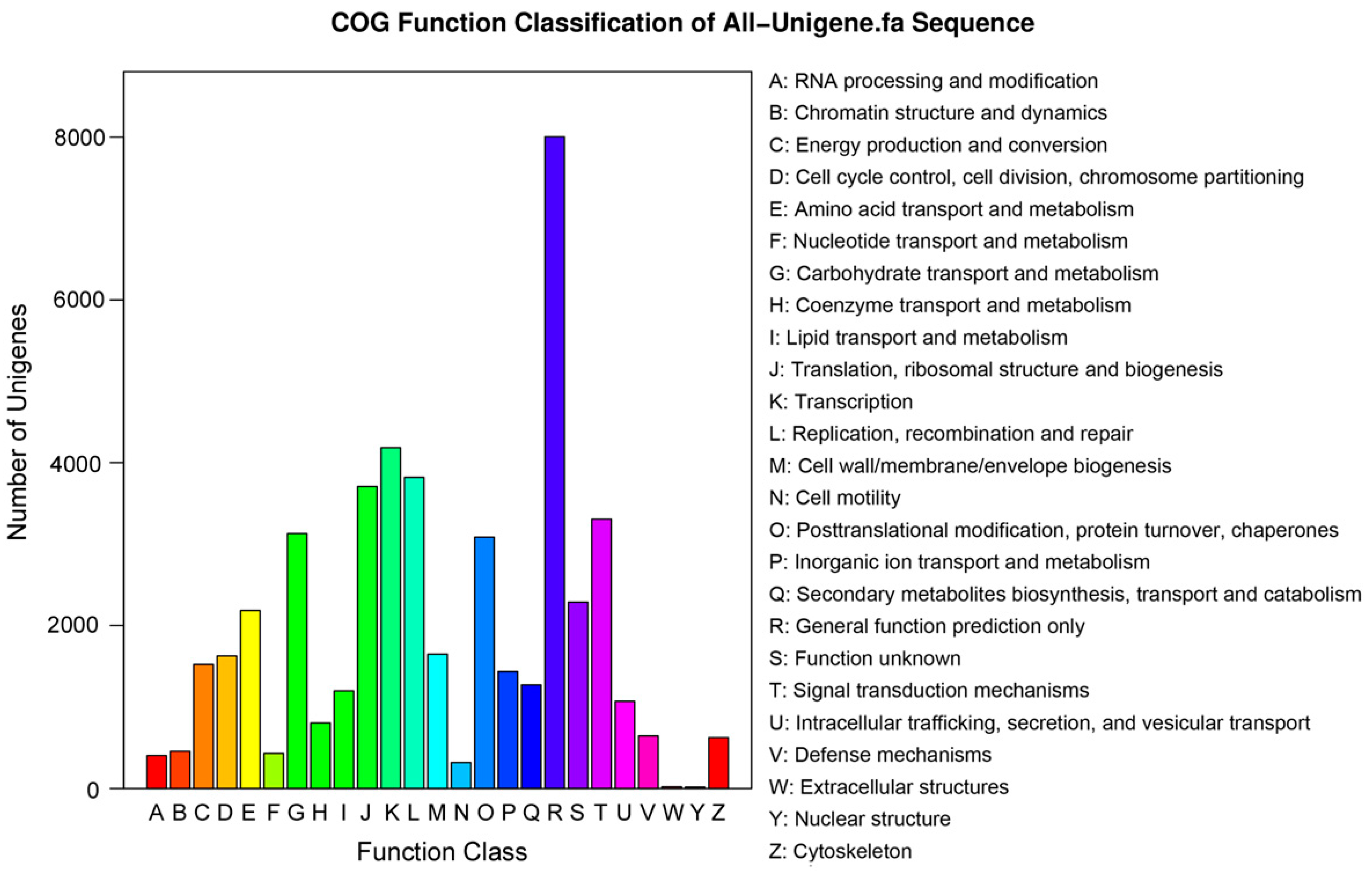
2.2. Functional Annotation and Classification of the Assembled Unigenes

| Public Protein Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| NR | 55,996 | 56.5 |
| Swiss-Prot | 39,519 | 39.8 |
| KEGG | 36,150 | 36.5 |
| COG | 24,872 | 25.1 |
| GO | 41,668 | 42.0 |
| All | 59,994 | 60.5 |
| Unmapped | 39,180 | 39.5 |




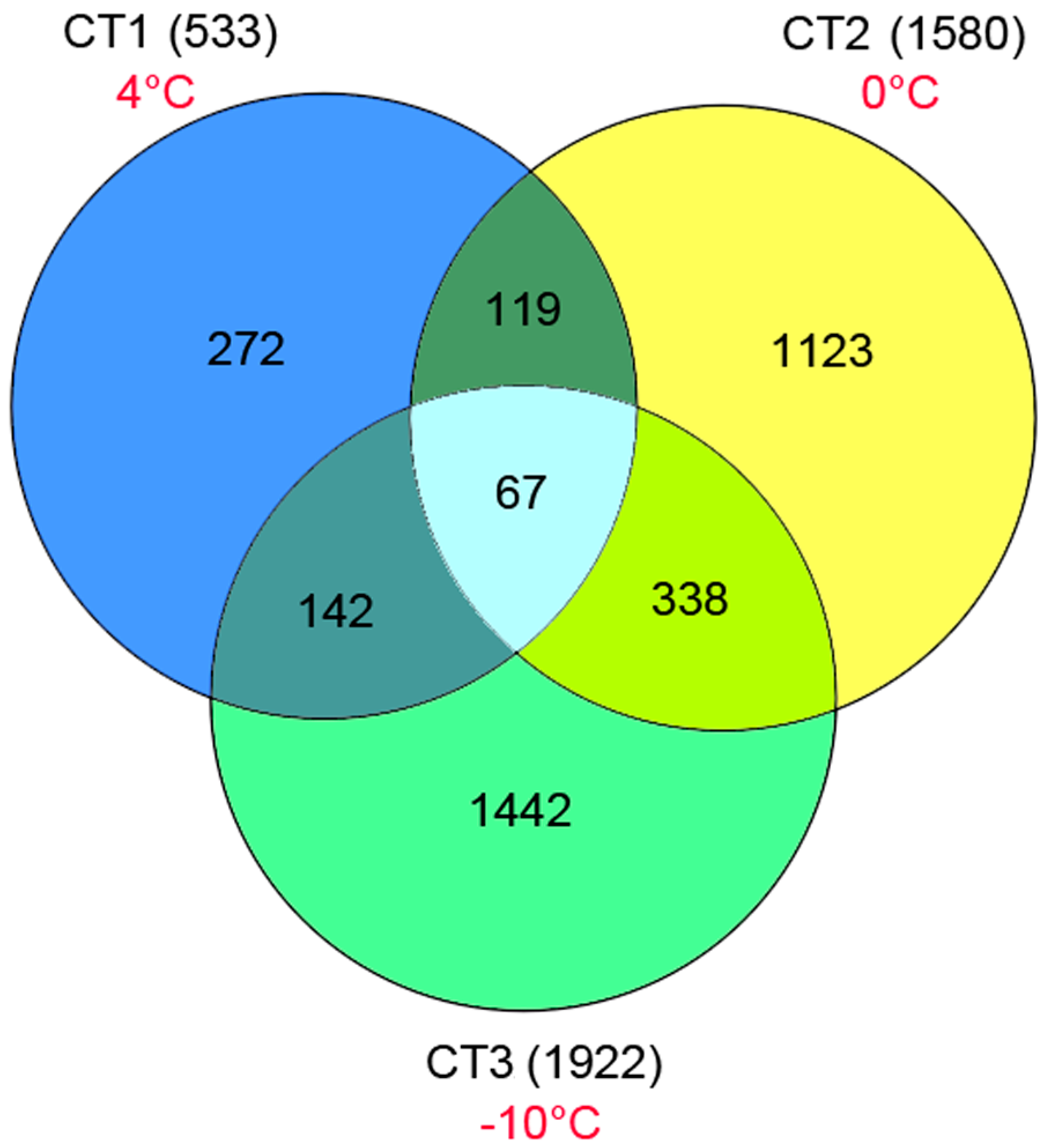
2.3. Analysis of Potential Differentially Expressed Genes (DEGs)


2.4. Analysis of DEGs Related to Metabolism
| Sample | Pathway ID | Pathway | All Genes with Pathway Annotation | DEGs Genes with Pathway Annotation | p Value |
|---|---|---|---|---|---|
| CT1/CK | ko04626 | Plant-pathogen interaction | 2388 (6.61%) | 691 (9.14%) | 4.63 × 10−22 |
| ko00565 | Ether lipid metabolism | 791 (2.19%) | 278 (3.68%) | 6.63 × 10−21 | |
| ko01110 | Biosynthesis of secondary metabolites | 4516 (12.49%) | 1171 (15.49%) | 2.76 × 10−18 | |
| ko00564 | Glycerophospholipid metabolism | 1034 (2.86%) | 332 (4.39%) | 9.88 × 10−18 | |
| ko00945 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 319 (0.88%) | 134 (1.77%) | 1.06 × 10−17 | |
| ko04144 | Endocytosis | 1097 (3.03%) | 346 (4.58%) | 3.95 × 10−17 | |
| ko00941 | Flavonoid biosynthesis | 391 (1.08%) | 153 (2.02%) | 1.23 × 10−16 | |
| ko01100 | Metabolic pathways | 8740 (24.18%) | 2088 (27.62%) | 4.49 × 10−15 | |
| ko04075 | Plant hormone signal transduction | 1752 (4.85%) | 499 (6.6%) | 8.37 × 10−15 | |
| ko03050 | Proteasome | 166 (0.46%) | 79 (1.04%) | 1.60 × 10−14 | |
| CT2/CK | ko03010 | Ribosome | 1440 (3.98%) | 593 (5.23%) | 6.57 × 10−16 |
| ko04626 | Plant-pathogen interaction | 2388 (6.61%) | 881 (7.77%) | 1.47 × 10−9 | |
| ko00908 | Zeatin biosynthesis | 384 (1.06%) | 176 (1.55%) | 1.71 × 10−9 | |
| ko00460 | Cyanoamino acid metabolism | 355 (0.98%) | 163 (1.44%) | 5.71 × 10−9 | |
| ko00945 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 319 (0.88%) | 145 (1.28%) | 8.29 × 10−8 | |
| ko00940 | Phenylpropanoid biosynthesis | 812 (2.25%) | 320 (2.82%) | 5.48 × 10−7 | |
| ko00904 | Diterpenoid biosynthesis | 188 (0.52%) | 91 (0.8%) | 7.60 × 10−7 | |
| ko00900 | Terpenoid backbone biosynthesis | 284 (0.79%) | 128 (1.13%) | 8.04 × 10−7 | |
| ko00944 | Flavone and flavonol biosynthesis | 179 (0.5%) | 87 (0.77%) | 1.06 × 10−6 | |
| ko01110 | Biosynthesis of secondary metabolites | 4516 (12.49%) | 1552 (13.69%) | 1.97 × 10−6 | |
| CT3/CK | ko01110 | Biosynthesis of secondary metabolites | 4516 (12.49%) | 1521 (16.37%) | 9.61 × 10−38 |
| ko01100 | Metabolic pathways | 8740 (24.18%) | 2704 (29.11%) | 3.08 × 10−37 | |
| ko00940 | Phenylpropanoid biosynthesis | 812 (2.25%) | 351 (3.78%) | 4.43 × 10−28 | |
| ko00565 | Ether lipid metabolism | 791 (2.19%) | 325 (3.5%) | 9.99 × 10−22 | |
| ko00564 | Glycerophospholipid metabolism | 1034 (2.86%) | 403 (4.34%) | 1.47 × 10−21 | |
| ko00500 | Starch and sucrose metabolism | 986 (2.73%) | 376 (4.05%) | 2.23 × 10−18 | |
| ko00941 | Flavonoid biosynthesis | 391 (1.08%) | 179 (1.93%) | 5.66 × 10−18 | |
| ko04144 | Endocytosis | 1097 (3.03%) | 386 (4.16%) | 9.57 × 10−13 | |
| ko00908 | Zeatin biosynthesis | 384 (1.06%) | 161 (1.73%) | 2.54 × 10−12 | |
| ko00945 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 319 (0.88%) | 137 (1.47%) | 1.38 × 10−11 |
2.5. Identification of Major Genes Involved in Cold Stress
| Gene ID | Annotation | Expression Difference Analysis (Log2(CT_RPKM/CK_RPKM)) | ||
|---|---|---|---|---|
| CT1/CK | CT2/CK | CT3/CK | ||
| Unigene933_All | Dehydration-responsive element-binding protein 1B | 0.49 | 4.23 | 5.12 |
| Unigene30882_All | Dehydration-responsive element-binding protein 1D | 1.49 | 4.22 | 5.23 |
| Unigene13555_All | AP2/ERF domain-containing transcription factor | 1.56 | 3.50 | 4.03 |
| CL11425.Contig1_All | Transcription factor MYB1R1 | 1.91 | 0.47 | 1.49 |
| CL10927.Contig2_All | Transcription factor ICE1 | 0.56 | 1.88 | 2.18 |
| CL12801.Contig1_All | Cold-inducible protein | 3.46 | 7.43 | 4.37 |
| CL3749.Contig3_All | Cold-inducible protein | 5.33 | 10.29 | 9.68 |
| CL10279.Contig1_All | Cold acclimation protein COR413PM2 | 2.55 | 1.36 | 1.03 |
| Unigene18542_All | Cold acclimation protein COR413IM2 | 1.80 | 1.56 | 1.41 |
| CL6095.Contig8_All | Cold regulated COR18 | 2.41 | 4.13 | 2.24 |
| Unigene52767_All | Cold shock protein CS66 | 6.88 | 9.67 | 12.23 |
| CL10842.Contig2_All | Cold-inducible RNA-binding protein | 3.56 | 5.61 | 3.58 |
| CL12984.Contig6_All | Low-temperature-induced 65 kDa protein | 1.56 | 6.35 | 5.07 |
| CL10225.Contig1_All | Heat shock protein | 0.60 | 1.26 | 2.75 |
| Unigene52380_All | Superoxide dismutase | 4.03 | 3.65 | 11.15 |
| Unigene9176_All | Peroxidase 52 | 1.02 | 2.48 | 1.26 |
| Unigene701_All | Peroxidase 57 | 1.53 | 2.51 | 2.75 |
| CL12499.Contig1_All | Catalase isozyme B | 1.47 | 3.21 | 5.69 |
| CL6576.Contig2_All | Glutathione peroxidase GPX6 | 1.52 | 2.68 | 1.68 |
| CL11774.Contig1_All | Water stress-induced protein ER5 | 3.46 | 6.19 | 4.78 |
| Unigene8303_All | Bidirectional sugar transporter N3 | 7.75 | 6.29 | 9.03 |
| CL7748.Contig2_All | Bidirectional sugar transporter SWEET11 | 0.66 | 2.49 | 2.04 |
| CL1443.Contig6_All | Abscisic acid-hydroxylase | 0.80 | 3.33 | 2.93 |
| Unigene58_All | Calcium-dependent protein kinase | 0.46 | 2.05 | 1.26 |
3. Experimental Section
3.1. Plant Materials and Cold Treatments
3.2. RNA Extraction, cDNA Library Construction, and Transcriptome Sequencing
3.3. De Novo Assembly and Functional Annotation
3.4. Differential Expression Analysis of Unigenes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, W.; Yu, K.; He, T.; Li, F.; Zhang, D.; Liu, J. The low temperature induced physiological responses of Avena nuda L., a cold-tolerant plant species. Sci. World J. 2013, 2013, 658793. [Google Scholar]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Takeda, M.; Kidokoro, S.; Yamada, K.; Sakuma, Y.; Urano, K.; Fujita, M.; Yoshiwara, K.; Matsukura, S.; Morishita, Y.; et al. Metabolic pathways involved in cold acclimation identified by integrated analysis of metabolites and transcripts regulated by DREB1A and DREB2A. Plant Physiol. 2009, 150, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Jin, J.B.; Lee, J.; Yoo, C.Y.; Stirm, V.; Miura, T.; Ashworth, E.N.; Bressan, R.A.; Yun, D.J.; Hasegawa, P.M. SIZ1-mediated sumoylation of ICE1 controls CBF3/DREB1A expression and freezing tolerance in Arabidopsis. Plant Cell 2007, 19, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, N.A. CAMTA proteins: A direct link between calcium signals and cold acclimation? Plant Cell 2009, 21, 697. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.J.; van Buskirk, H.A.; Myers, S.J.; Thomashow, M.F. Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell 2009, 21, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.H.; Agarwal, M.; Zhang, Y.; Xie, Q.; Zhu, J.K. The negative regulator of plant cold responses, HOS1, is a RING E3 ligase that mediates the ubiquitination and degradation of ICE1. Proc. Natl. Acad. Sci. USA 2006, 103, 8281–8286. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Xu, L.L.; Tong, Z.K.; Lin, E.P.; Liu, Q.P.; Cheng, L.J.; Zhu, M.Y. De novo characterization of the Chinese fir (Cunninghamia lanceolata) transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis. BMC Genomics 2012, 13, 648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Y.; Liu, Z.; Zhu, X.; Zhai, L.; Xu, L.; Yu, R.; Gong, Y.; Liu, L. De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 2013, 14, 836. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Ye, C.Y.; Xia, X.; Yin, W. De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genomics 2013, 14, 488. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.Q.; Pan, X.Y.; Yu, Y.M.; Wang, W.Y.; Zhang, F.; Ge, Y.Y.; Shen, X.L.; Shen, F.Q.; Liu, X.J. De novo characterization of the Anthurium. transcriptome and analysis of its digital gene expression under cold stress. BMC Genomics 2013, 14, 827. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Chen, B.; Zhou, M.; Zhao, X.; Xia, H.; Wang, M. De novo sequencing and transcriptome analysis of Wolfiporia cocos to reveal genes related to biosynthesis of triterpenoids. PLoS ONE 2013, 8, e71350. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Winfield, M.O.; Lu, C.; Wilson, I.D.; Coghill, J.A.; Edwards, K.J. Plant responses to cold: Transcriptome analysis of wheat. Plant Biotechnol. J. 2010, 8, 749–771. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Yang, J.; Zhang, P. Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genomics 2012, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Fowler, S.; Fiehn, O.; Thomashow, M.F. A prominent role for the CBF cold response pathway in configuring the low-temperature metabolome of Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 15243–15248. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. So what’s new in the field of plant cold acclimation? Lots! Plant Physiol. 2001, 125, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Danyluk, J.; Charron, J.B.; Sarhan, F. Expression profiling and bioinformatic analyses of a novel stress-regulated multispanning transmembrane protein family from cereals and Arabidopsis. Plant Physiol. 2003, 132, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.P. The DNA-binding activity of an AP2 transcriptional activator HvCBF2 involved in regulation of low-temperature responsive genes in barley is modulated by temperature. Plant J. 2003, 33, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Airaki, M.; Leterrier, M.; Mateos, R.M.; Valderrama, R.; Chaki, M.; Barroso, J.B.; del Rio, L.A.; Palma, J.M.; Corpas, F.J. Metabolism of reactive oxygen species and reactive nitrogen species in pepper (Capsicum annuum L.) plants under low temperature stress. Plant Cell Environ. 2012, 35, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Khaper, N.; Kaur, K.; Li, T.; Farahmand, F.; Singal, P.K. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem. 2003, 251, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 1, 138–148. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Zhang, H.; Huang, H.; Folta, K.M.; Lu, J. Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using Solexa sequencing technology. BMC Plant Biol. 2010, 10, 234. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, Y.; Zhou, A.; Zhang, X.; Tang, H.; Liang, M.; Han, H.; Zuo, Y. De Novo Transcriptome Sequencing of Low Temperature-Treated Phlox subulata and Analysis of the Genes Involved in Cold Stress. Int. J. Mol. Sci. 2015, 16, 9732-9748. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16059732
Qu Y, Zhou A, Zhang X, Tang H, Liang M, Han H, Zuo Y. De Novo Transcriptome Sequencing of Low Temperature-Treated Phlox subulata and Analysis of the Genes Involved in Cold Stress. International Journal of Molecular Sciences. 2015; 16(5):9732-9748. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16059732
Chicago/Turabian StyleQu, Yanting, Aimin Zhou, Xing Zhang, Huanwei Tang, Ming Liang, Hui Han, and Yuhu Zuo. 2015. "De Novo Transcriptome Sequencing of Low Temperature-Treated Phlox subulata and Analysis of the Genes Involved in Cold Stress" International Journal of Molecular Sciences 16, no. 5: 9732-9748. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16059732