
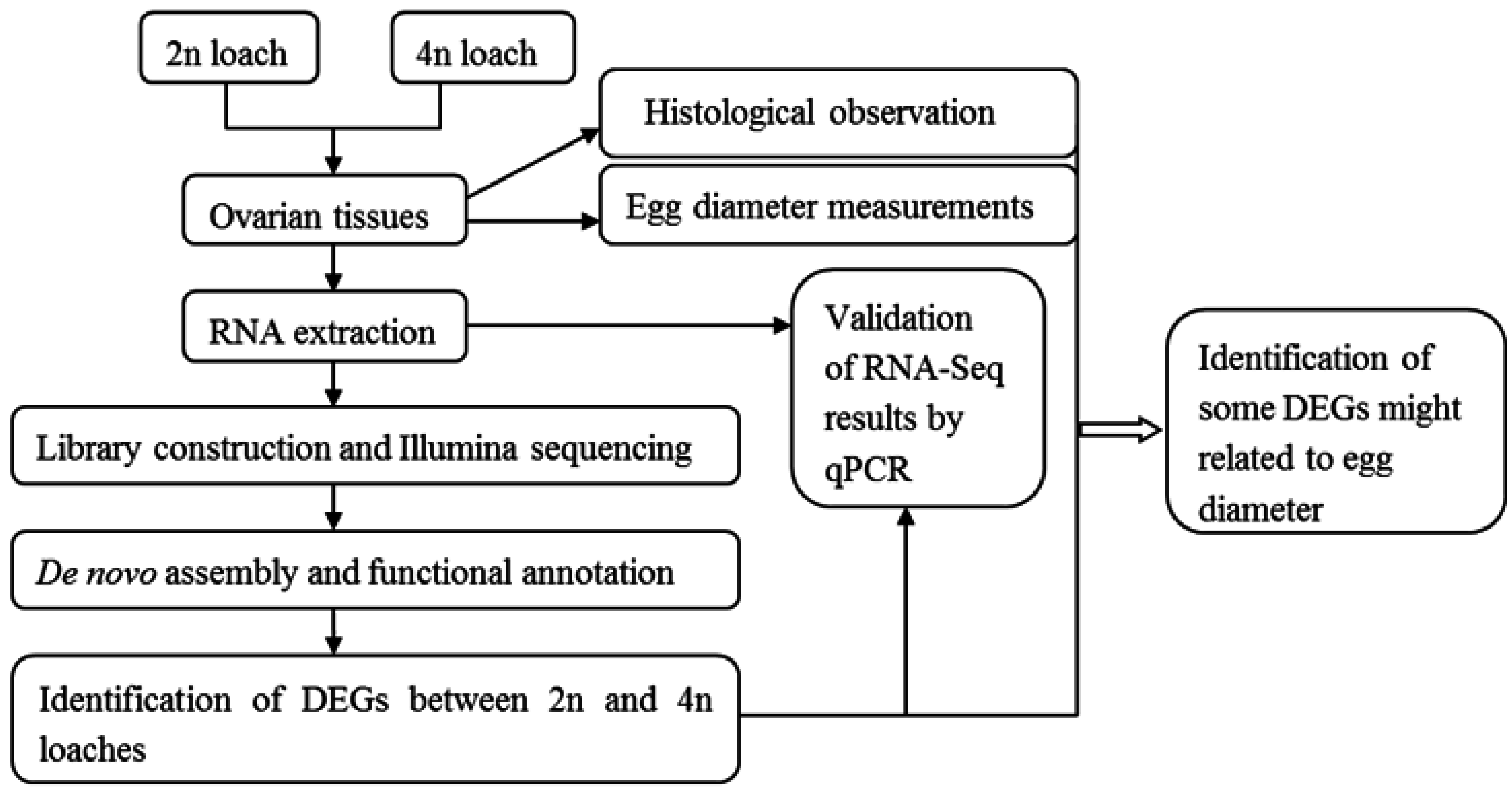
Transcriptome Profile Analysis of Ovarian Tissues from Diploid and Tetraploid Loaches Misgurnus anguillicaudatus

Abstract
:1. Introduction
2. Results
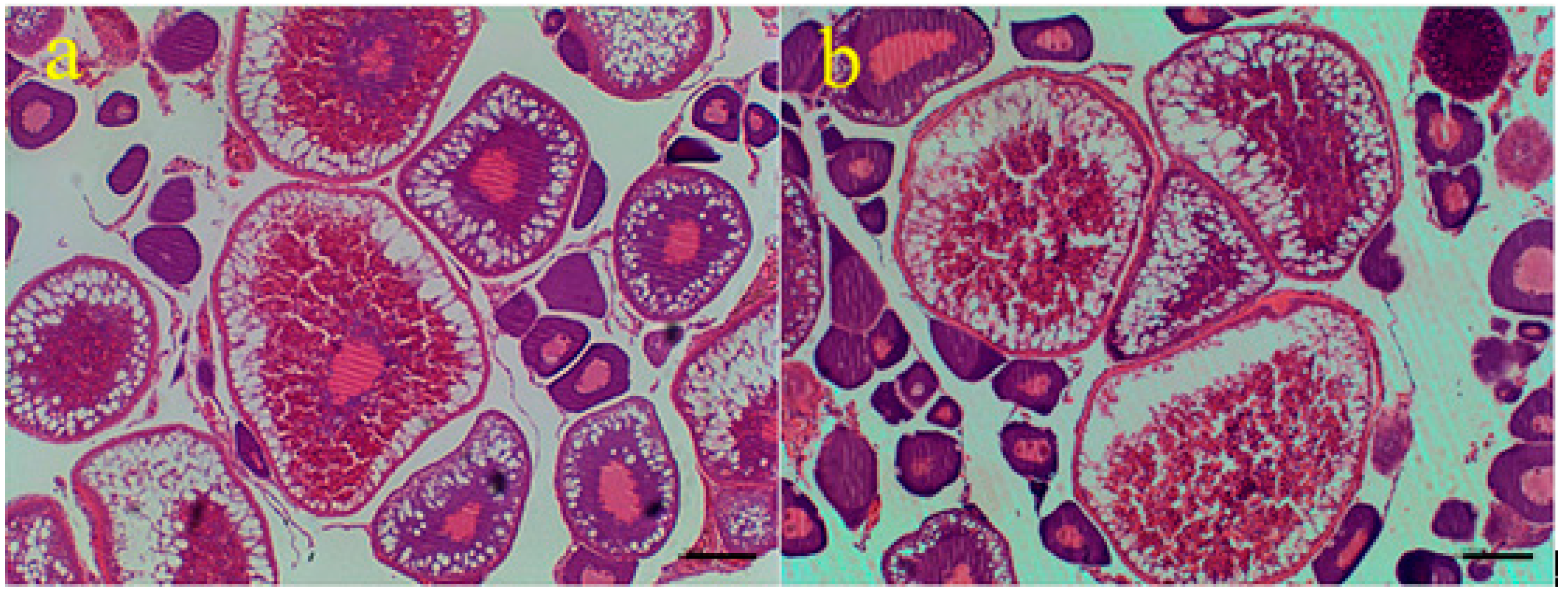
2.1. Egg Diameters of the Three-Year-Old Mature Loaches of Different Ploidy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ploidy | Egg Numbers | Egg Diameters (mm) |
|---|---|---|
| 2n | 340 | 1.17 ± 0.04 a |
| 4n | 340 | 1.37 ± 0.07 b |
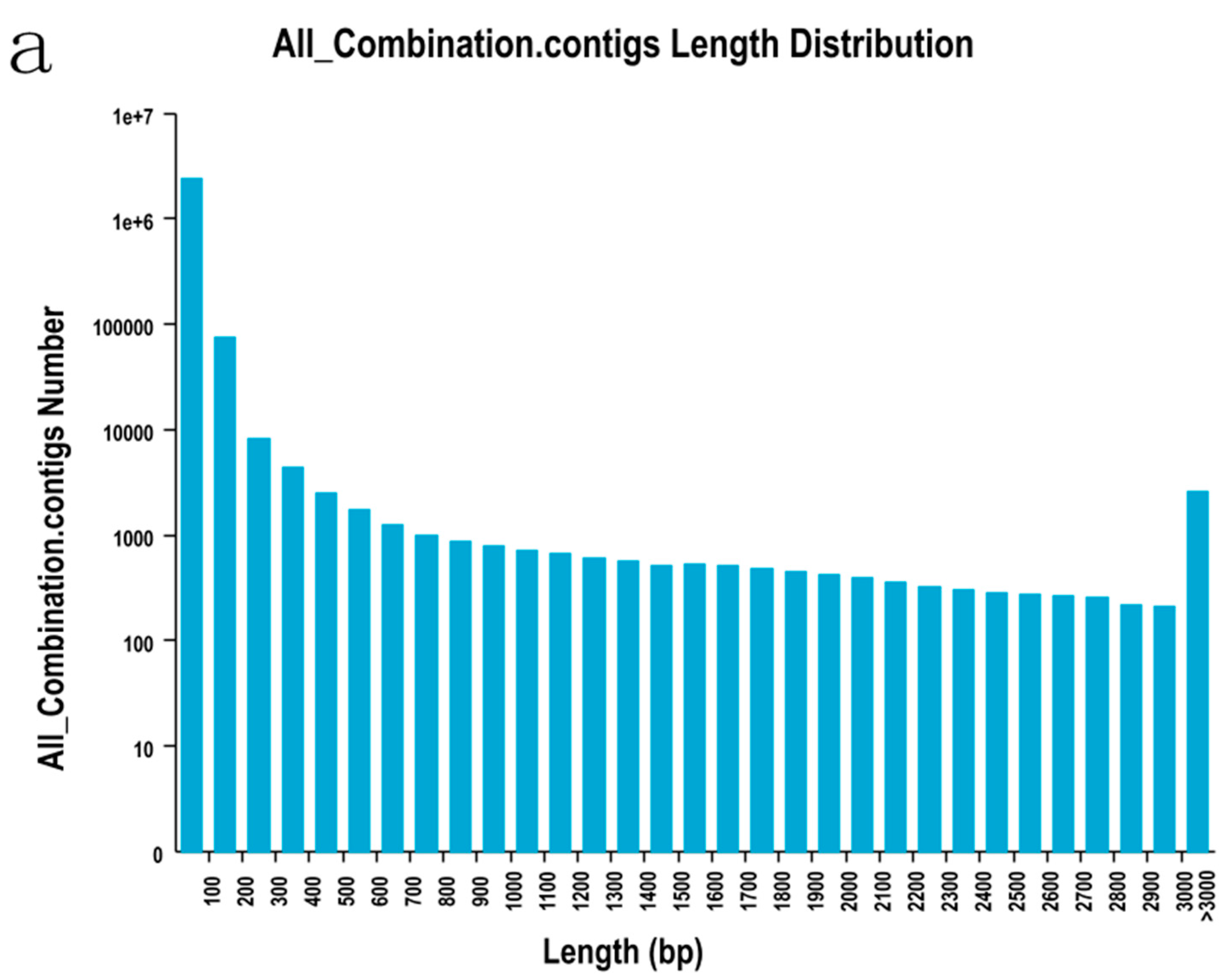
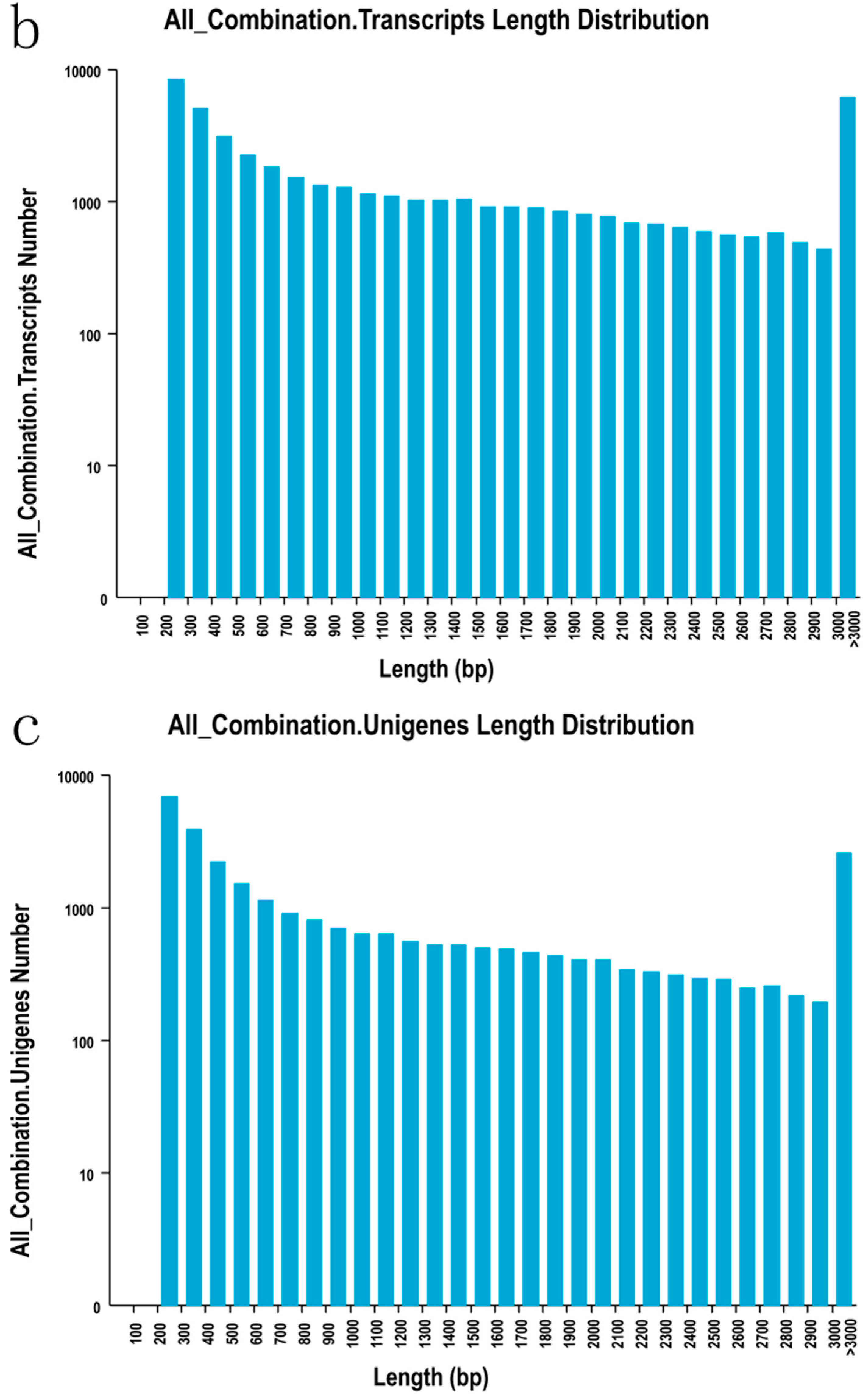
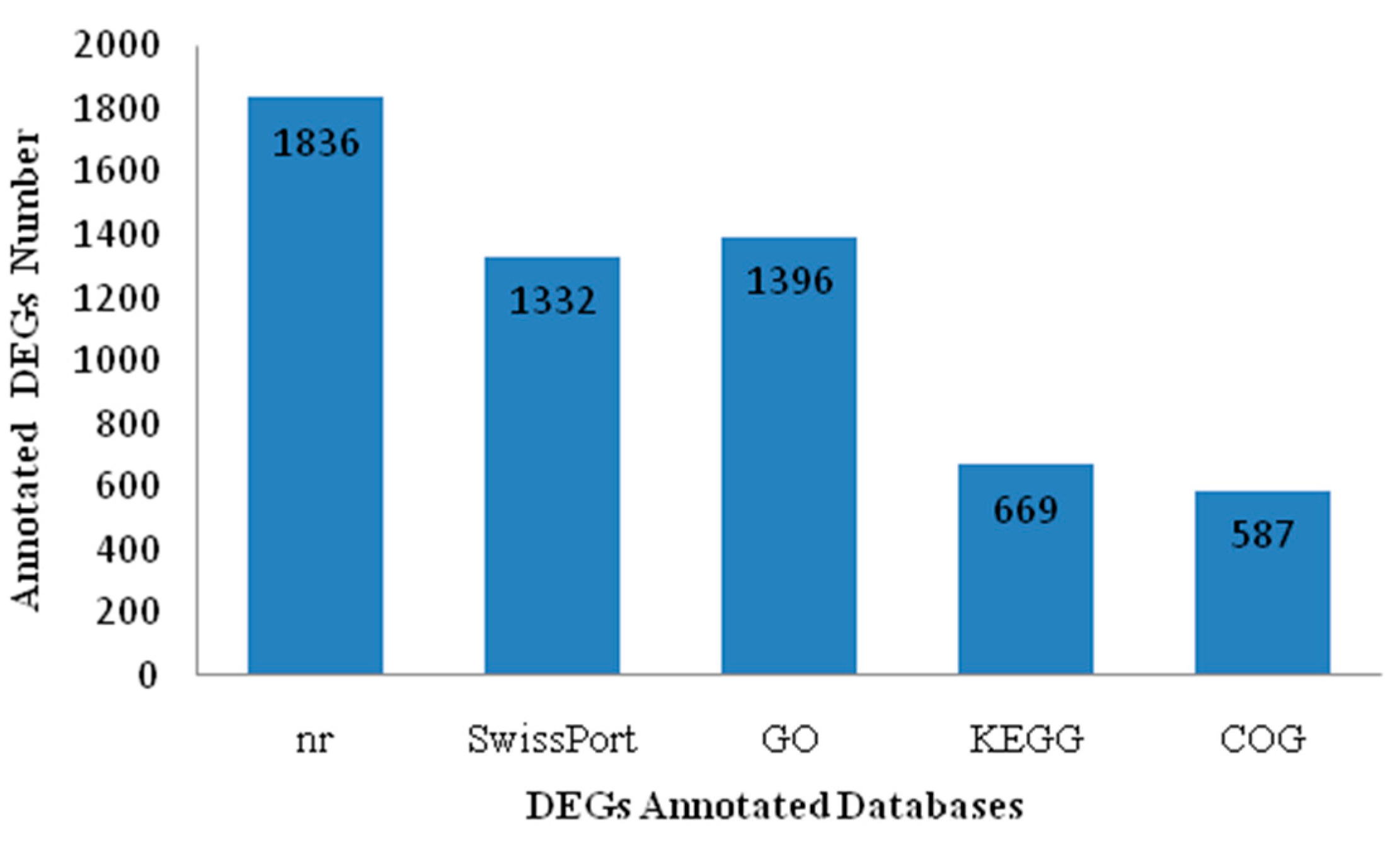
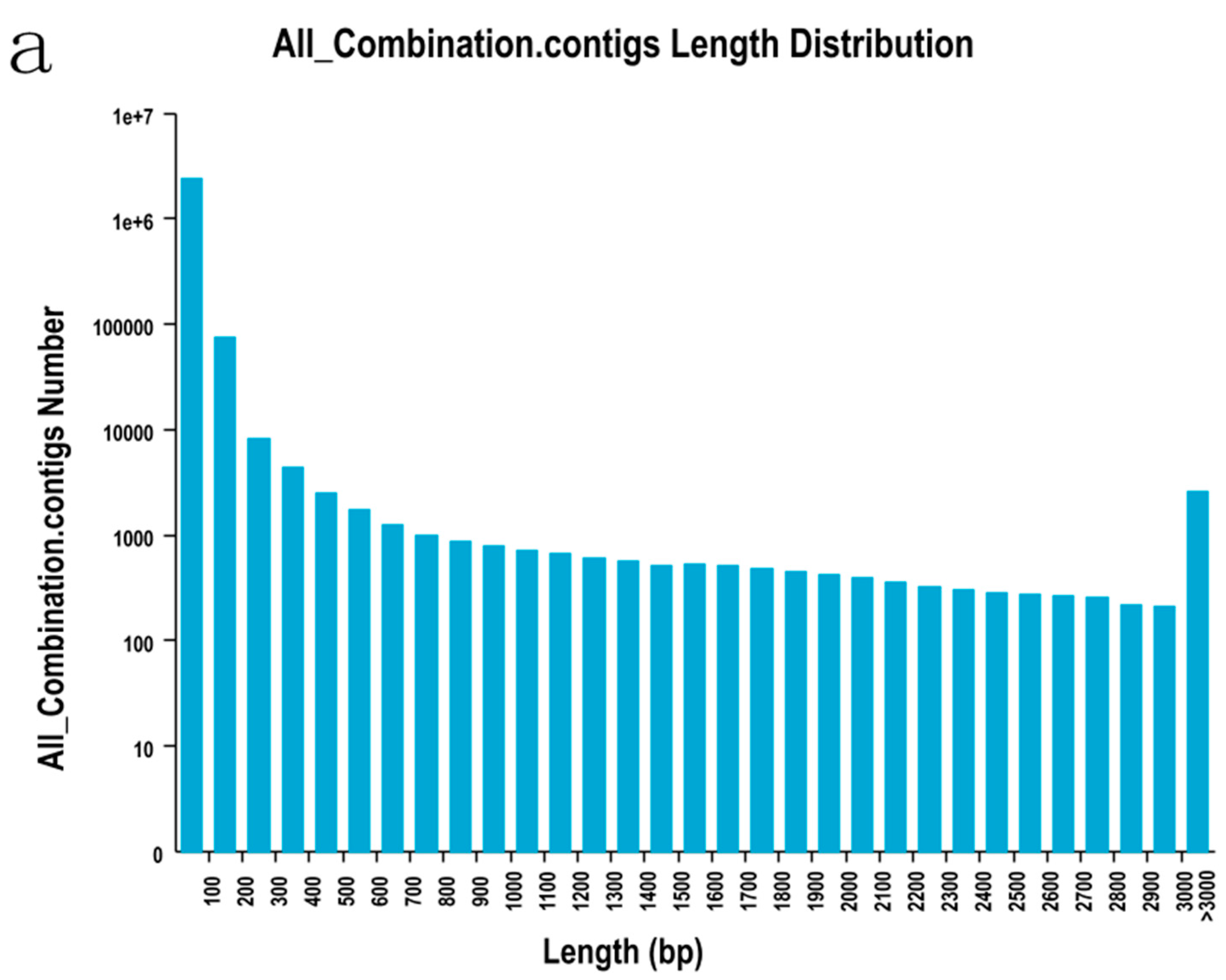
2.2. Sequencing, De Novo Assembly and Functional Annotation
| Parameters | Diploid Loach | Tetraploid Loach | In Total |
|---|---|---|---|
| Number of raw reads | 12,161,766 | 13,262,549 | 25,424,315 |
| Average raw read length (bp) | 101 | 101 | 101 |
| Number of clean reads | 11,416,495 | 12,411,016 | 23,827,511 |
| Percentage retained | 93.87% | 93.58% | 93.72% |
| Average clean read length (bp) | 100.99 | 100.99 | 100.99 |
| Length Range | Contigs | Transcripts | Unigenes |
|---|---|---|---|
| 200–300 | 2,473,738 (99.07%) | 8326 (18.04%) | 6751 (23.80%) |
| 300–500 | 6838 (0.27%) | 8114 (17.58%) | 6034 (21.27%) |
| 500–1000 | 5600 (0.22%) | 8132 (17.62%) | 5038 (17.76%) |
| 1000–2000 | 5433 (0.22%) | 9603 (20.81%) | 5134 (18.10%) |
| >2000 | 5422 (0.22%) | 11,977 (25.95%) | 5412 (19.08%) |
| Total number | 2,497,031 | 46,152 | 28,369 |
| Total length | 134,911,717 | 66,470,417 | 33,401,504 |
| N50 length | 49 | 2549 | 2220 |
| Mean length | 5403 | 1440.25 | 1177.39 |


2.3. The Differentially Expressed Genes between the Two Loaches
| #ID | Gene Name | log2 Ratio (4n/2n) | FDR * |
|---|---|---|---|
| comp118882_c0 | Vitellogenin | 4.441525 | 0.001012 |
| comp20138_c0 | Gonadotropin releasing hormone receptor type A | 4.782572 | 7.43 × 10−9 |
| comp28776_c0 | Steroidogenic acute regulatory protein | 4.097802 | 0.00883243 |
| comp111884_c0 | Mitogen-activated protein kinase 14a | 5.055045 | 6.71 × 10−6 |
| comp28024_c0 | ATP synthase subunit alpha | 6.593683 | 7.22 × 10−15 |
| comp126960_c0 | S-phase kinase-associated protein 1 | 4.516029 | 0.000597 |
| comp33522_c0 | G2/M phase-specific E3 ubiquitin-protein ligase | 7.034851 | 0 |
| comp16585_c0 | Dynein regulatory complex protein 1 | 8.372893 | 0 |
| comp32946_c1 | Cyclin-dependent kinase-like 1 | 4.097802 | 0.00883243 |
| comp38857_c0 | Synaptonemal complex protein 1 | 11.67117 | 0 |
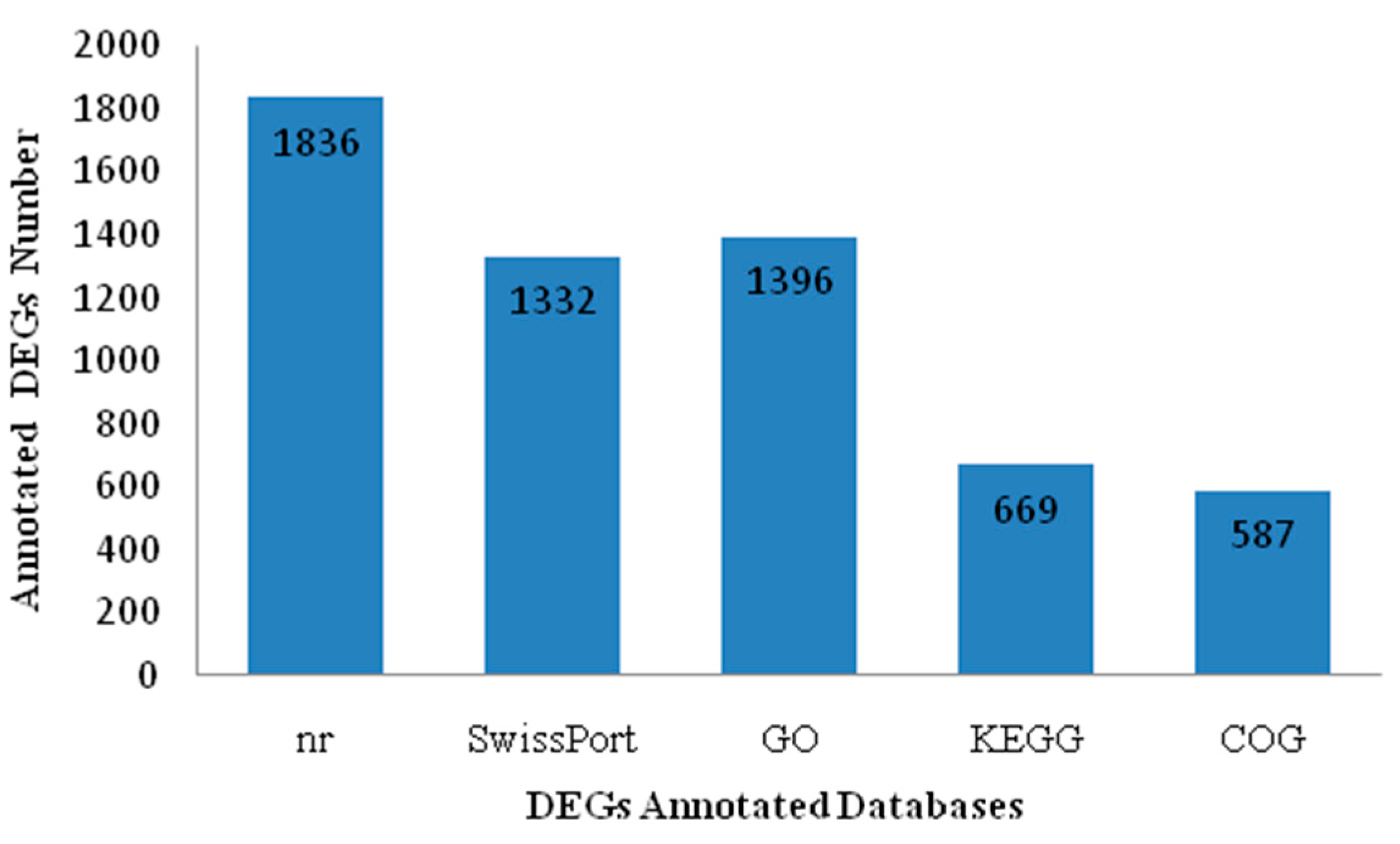
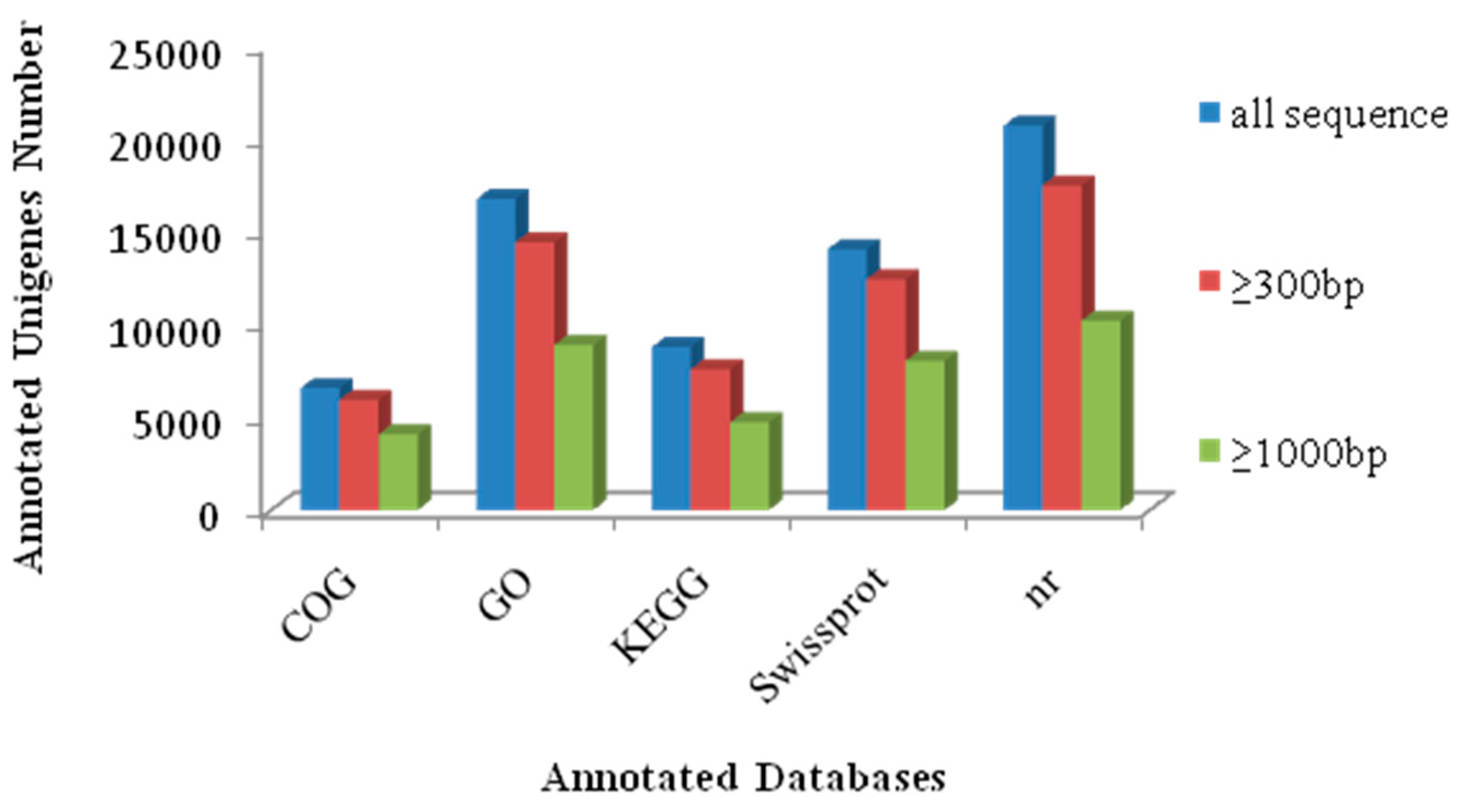
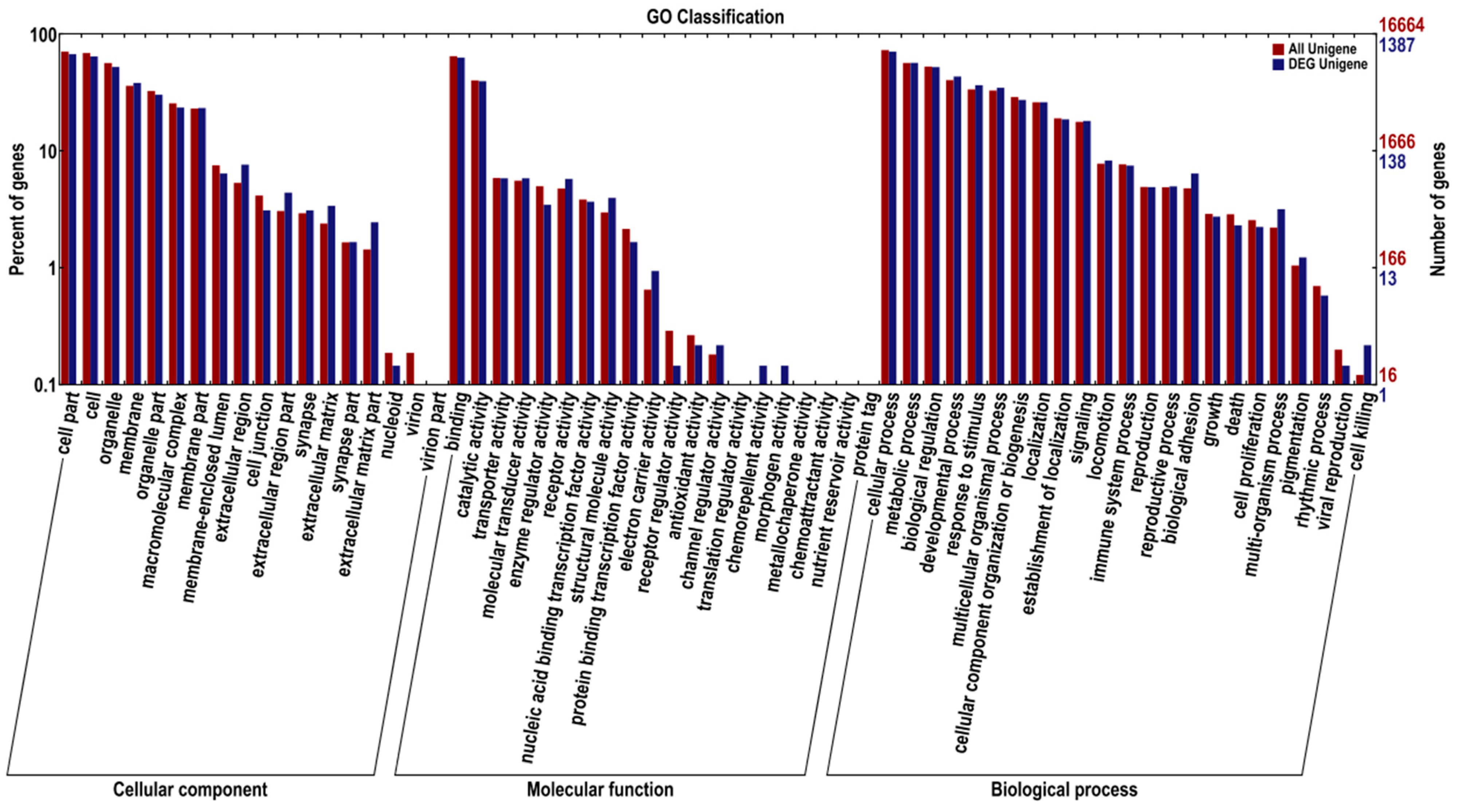
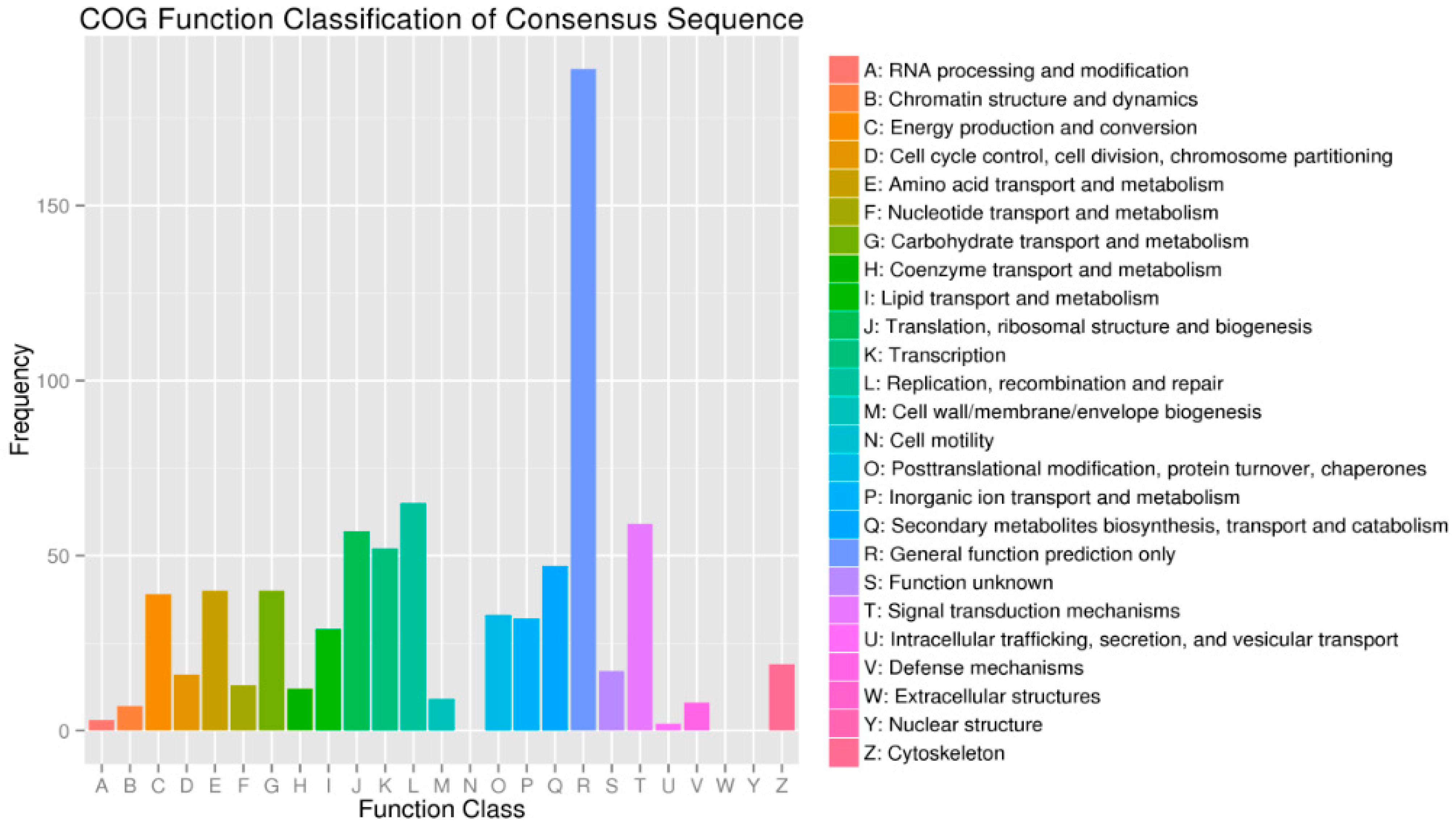
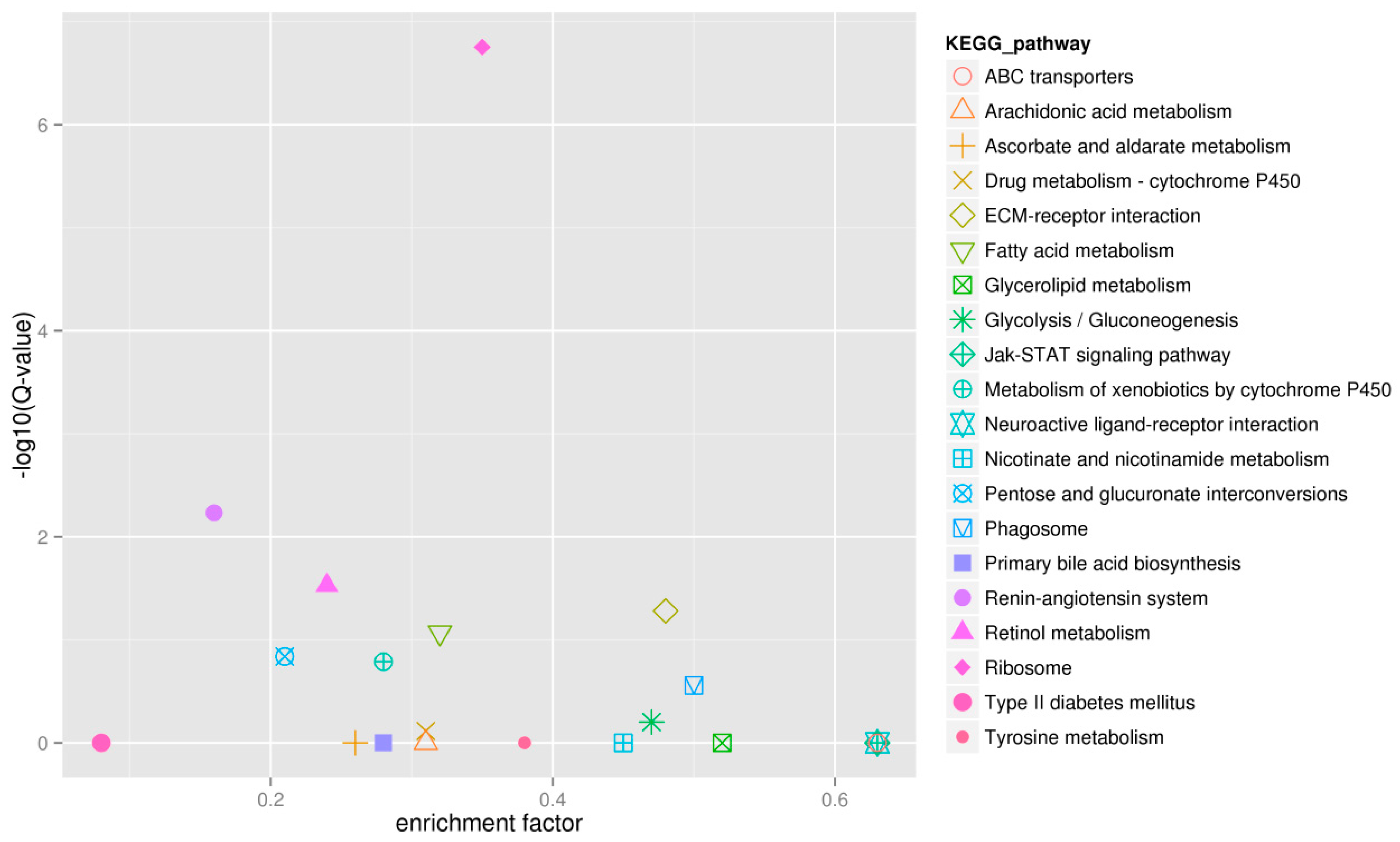
2.4. Functional Enrichment Analysis Results for DEGs



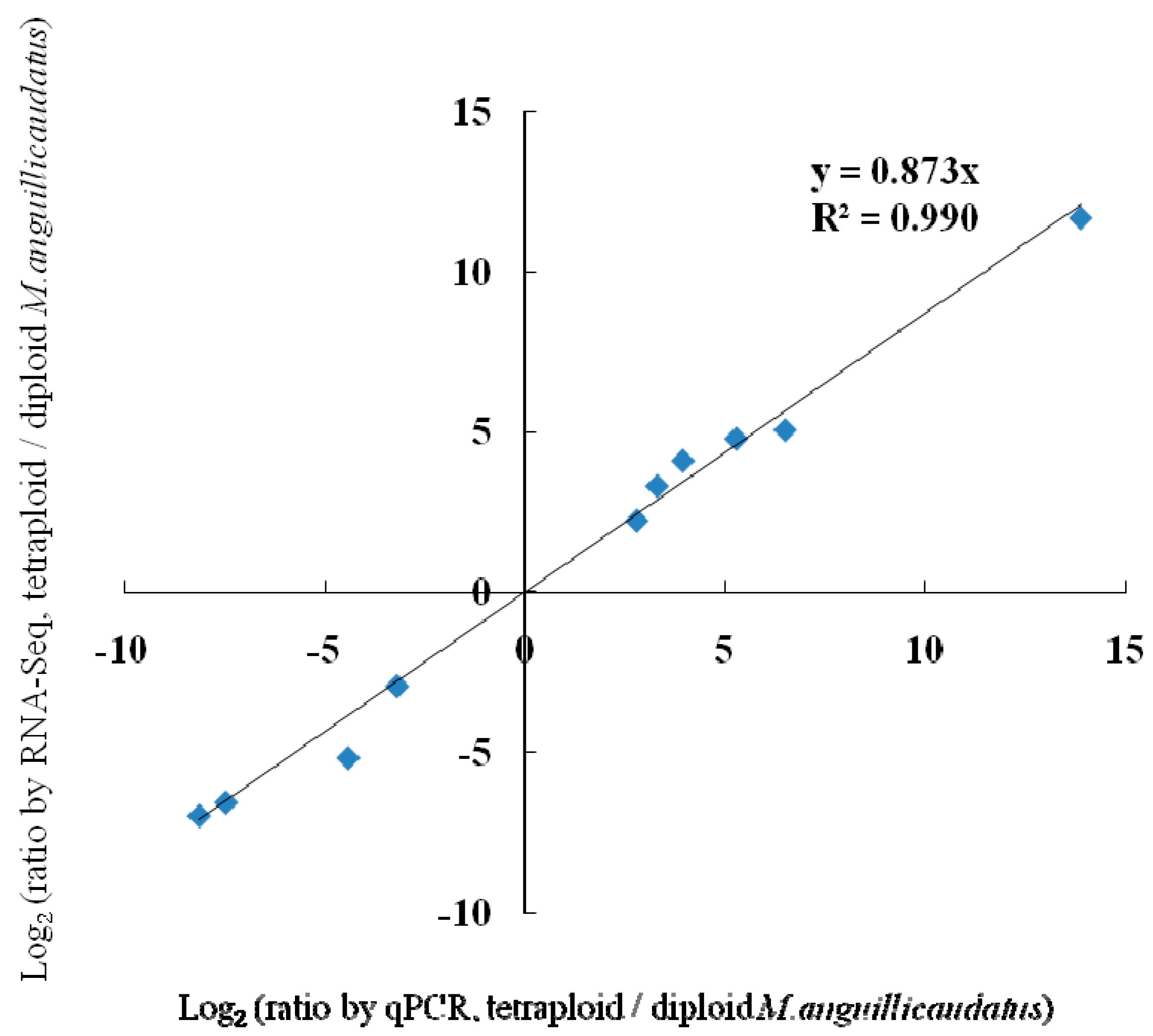
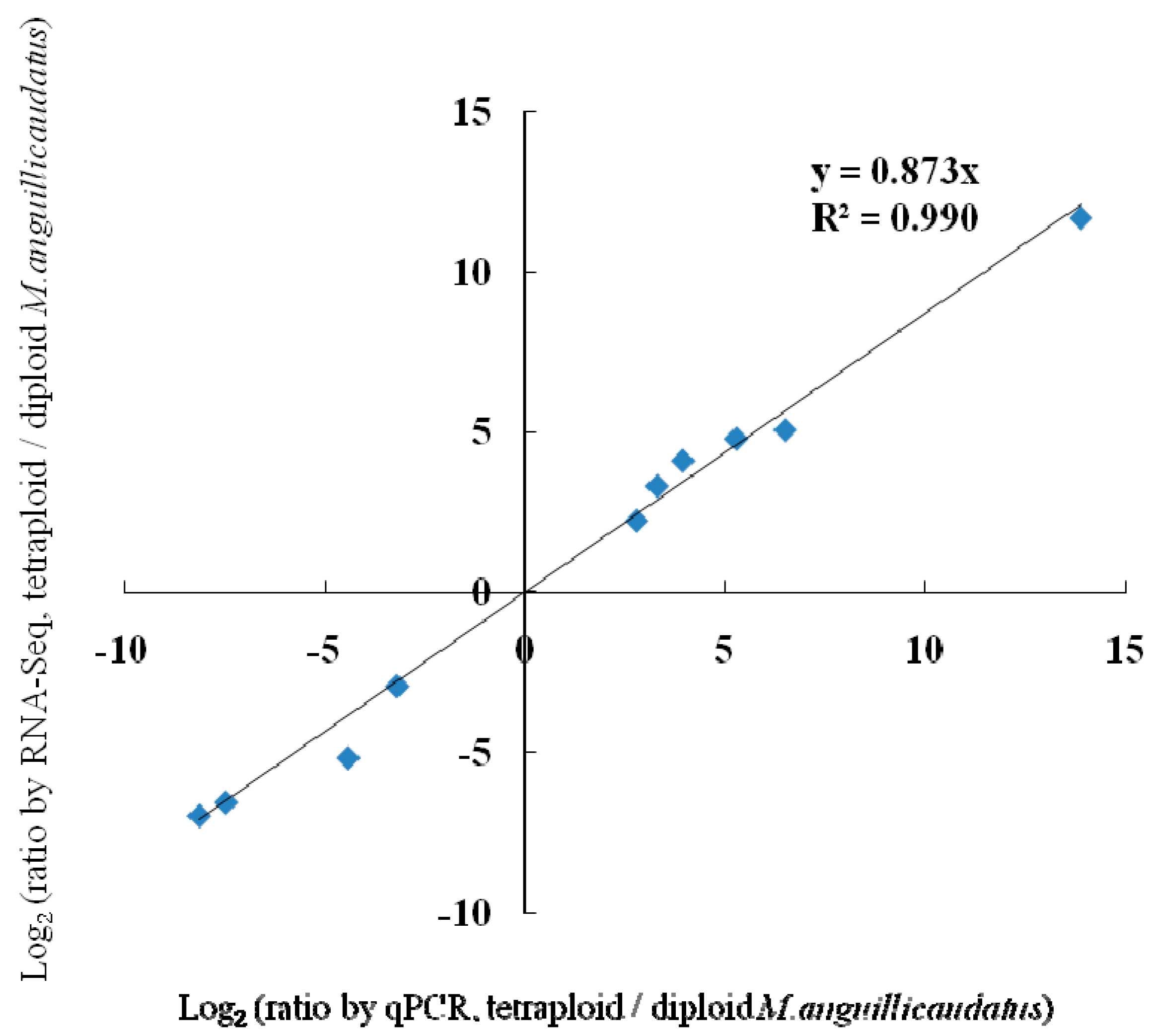
2.5. Validation of Differentially Expressed Genes by Quantitative Real-Time PCR (qPCR)
| Item | Up-Regulated Genes | Down-Regulated Genes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | GnRHRA | CDKL1 | CKb | AHCY | TGFβ2 | Scp1 | ARHGEF3 | GDF7 | Wnt11 | Cyp27A |
| RNA-Seq a | 4.7826 | 4.0978 | 2.2225 | 5.0550 | 3.3047 | 11.6712 | −6.5640 | −6.9797 | −2.9406 | −5.1742 |
| qPCR b | 5.2700 | 3.9425 | 2.7950 | 6.4900 | 3.2950 | 13.8625 | −7.4600 | −8.1300 | −3.1900 | −4.4075 |
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Sample Collection and Preparation
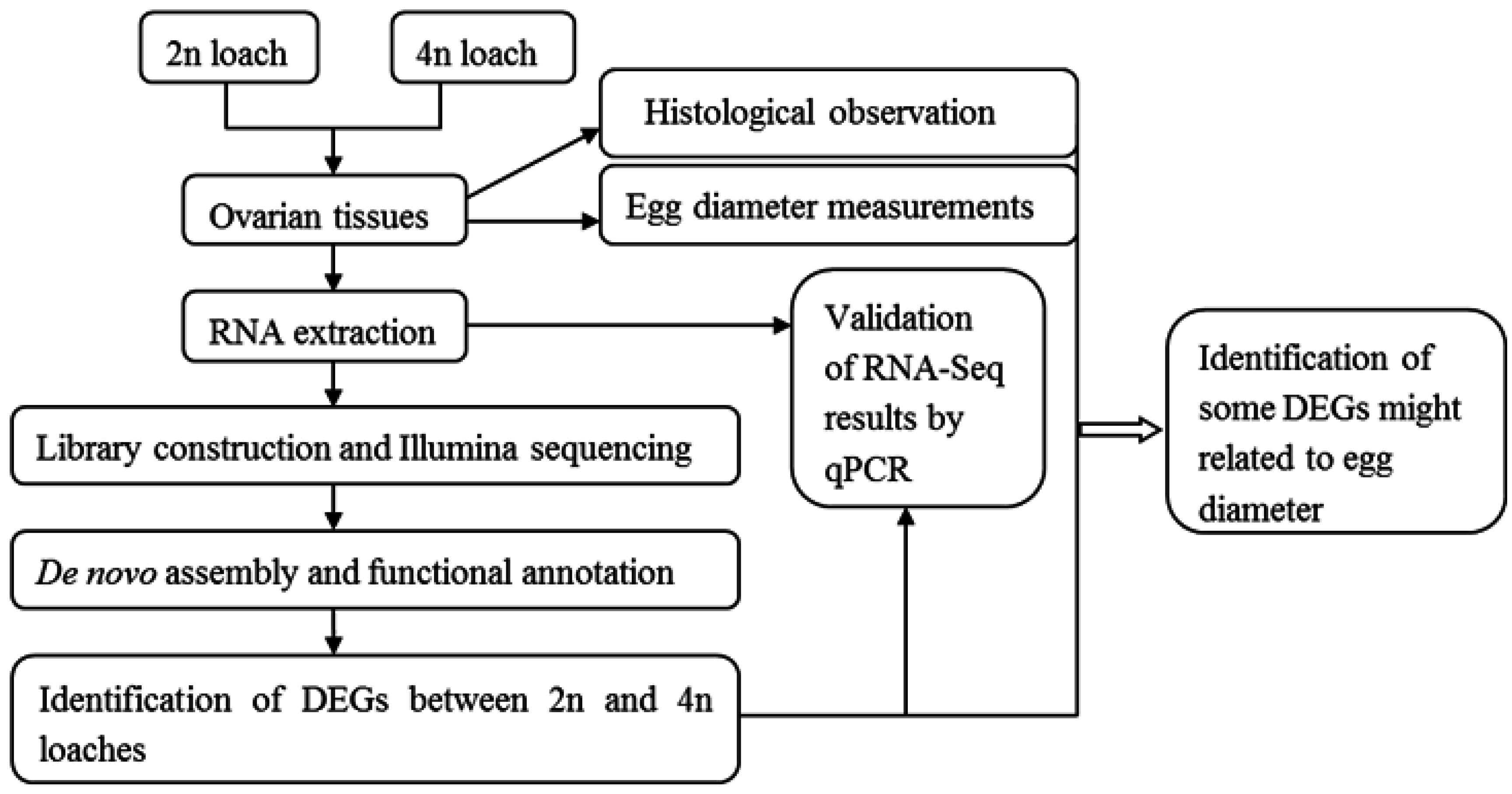
4.3. Library Construction and Illumina Sequencing
4.4. De Novo Assembly and Functional Annotation
4.5. Identification of Differentially Expressed Genes (DEGs)
4.6. Validation of RNA-Seq Results by qPCR
| Gene-ID | Nr_annotation | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Product Size (bp) |
|---|---|---|---|---|
| comp20138_c0 | GnRHRA | CCAGCCAGAGATGTTGAAGGT | GCGGAAGGAAGGAGTGTAAAGA | 118 |
| comp32946_c0 | CDKL1 | AGTCTGCGAGAGGAAAGCGA | GCAAATACCCAGGTGGAAGG | 94 |
| comp42021_c0 | CKb | AGCCCTGTATGAGAAGTTGCG | TATCCTCCATGCCTGTCCCTA | 192 |
| comp34136_c1 | AHCY | GATACGGAGAGGTGGGAAAAGG | TGTAGAGCACAAATGGGGTCAA | 94 |
| comp22497_c0 | ARHGEF3 | CTCAAGACTGGAGAGGCGGA | GGTCGGGTCAAAACCAACACTA | 182 |
| comp34529_c0 | TGFβ2 | GGAGAAGAACGCCTCAAACCT | CGCACCACCTTACTGTCAATG | 162 |
| comp35042_c1 | GDF7 | TTCAAAGAACTGGGTTGGGATG | AGCGTCTGGATGATGGCGT | 131 |
| comp38857_c0 | Scp1 | TCTCCGCCAGTTCAGGGTTA | CTCTGGACTGCCACATTTTGATT | 217 |
| comp38576_c0 | Wnt11 | CCAGAGCACCTTCAGCGACAT | CAGCCACCCCATCTAAAATCC | 231 |
| comp29375_c1 | Cyp27A | GAGACCGTCCTGTTTTTCCCA | GATACATCCCCTCCACCTGCT | 169 |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Y.J.; Hu, M.H.; Wang, W.M.; Cheung, S.G.; Shin, P.K.S.; Li, H.; Cao, L. Effects of the timing of initial feeding on growth and survival of loach (Misgurnus anguillicaudatus) larvae. Aquac. Int. 2010, 18, 135–148. [Google Scholar] [CrossRef]
- Qin, C.G.; Huang, K.X.; Xu, H.B. Protective effect of polysaccharides fromthe loach on the in vitro and in vivo peroxidative damage of hepatocyte. J. Nutr. Biochem. 2002, 13, 592–597. [Google Scholar] [CrossRef]
- Cho, Y.S.; Kim, B.S.; Kim, D.S.; Nam, Y.K. Modulation of warm-temperature-acclimation-associated 65-kDa protein genes (Wap65-1 and Wap65-2) in mud loach (Misgurnusmizolepis Cypriniformes) liver in response to different stimulatory treatments. Fish. Shellfish Immun. 2012, 32, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Zhang, M.Z.; Qian, C.; Gao, M.; Arai, K. Fertility and ploidy of gametes of diploid, triploid and tetraploid loaches, Misgurnus anguillicaudatus, in China. J. Appl. Ichthyol. 2012, 28, 900–905. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Yu, Y.Y.; Li, Y.H.; Wu, J.J.; Zhang, X.J.; Guo, X.W.; Wang, W.M. Comparative analysis of mitochondrial genomes in distinct nuclear ploidy loach Misgurnus anguillicaudatus and its implications for polyploidy evolution. PLoS ONE 2014, 9, e92033. [Google Scholar] [CrossRef] [PubMed]
- Strickler, S.R.; Bombarely, A.; Mueller, L.A. Designing a transcriptome next generation sequencing project for a nonmodel plant species. Am. J. Bot. 2012, 99, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.J.; Yuan, J.; Zhou, L.Y.; Sun, L.N.; Sun, Y.L.; Yang, S.J.; Li, M.H.; Zeng, S.; Huang, B.F.; Wang, D.S. Characterization of gonadal transcriptomes from Nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS ONE 2013, 8, e63604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.P.; Wang, Y.L.; Wang, S.H.; Liu, J.T.; Warren, W.; Mitreva, M.; Walter, R.B. Transcriptome analysis of female and male Xiphophorus Maculatus Jp 163 A. PLoS ONE 2011, 6, e18379. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.F.; You, F.; Wang, L.J.; Weng, S.D.; Wu, Z.H.; Hu, J.W.; Zou, Y.X.; Tan, X.G.; Zhang, P.J. Gonadal transcriptome analysis of male and female olive flounder (Paralichthy solivaceus). Biomed. Res. Int. 2014, 2014, 291067. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, C.; Huang, L.; Wu, M.; Zuo, Z. Transcriptome analysis of male and female Sebastiscus marmoratus. PLoS ONE 2012, 7, e50676. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Rexroad, C.E.; Wang, J.N.; Thorgaard, G.H.; Yao, J.B. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.Y.; Liu, S.K.; Gao, X.Y.; Jiang, Y.L.; Perera, D.; Wang, X.L.; Li, C.; Sun, L.Y.; Zhang, J.R.; Kaltenboeck, L.; et al. Male-Biased genes in catfish as revealed by RNA-seq analysis of the testis transcriptome. PLoS ONE 2013, 8, e68452. [Google Scholar] [CrossRef] [PubMed]
- Ribas, L.; Pardo, B.G.; Fernández, C.; Álvarez-Diós, J.A.; Gómez-Tato, A.; Quiroga, M.I.; Planas, J.V.; Sitjà-Bobadilla, A.; Martínez, P.; Piferrer, F. A combined strategy involving Sanger and 454 pyrosequencing increases genomic resources to aid in the management of reproduction, disease control and genetic selection in the turbot (Scophthalmus maximus). BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reading, B.J.; Chapman, R.W.; Schaff, J.E.; Scholl, E.H.; Opperman, C.H.; Sullivan, C.V. An ovary transcriptome for all maturational stages of the striped bass (Morone saxatilis), a highly advanced perciform fish. BMC Res. Notes 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Bujo, H.; Lindstedt, K.A.; Hermann, M.; Dalmau, L.M.; Nimpf, J.; Schneider, W.J. Chicken oocytes and somatic cells express different splice variants of a multifunctional receptor. J. Biol. Chem. 1995, 270, 23546–23551. [Google Scholar] [CrossRef] [PubMed]
- Davail, B.; Pakdel, F.; Bujo, H.; Perazzolo, L.M.; Waclawek, M.; Schneider, W.J.; Menn, F.L. Evolution of oogenesis: The receptor for vitellogenin from the rainbow trout. J. Lipid Res. 1998, 39, 1929–1937. [Google Scholar] [PubMed]
- Prat, F.; Coward, K.; Sumpter, J.P.; Tyler, C.R. Molecular characterization and expression of two ovarian lipoprotein receptors in the rainbow trout, Oncorhynchus mykiss. Biol. Reprod. 1998, 58, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.B.; Hu, J.Y. Development of a molecular biomarker for detecting intersex after exposure of male medaka fish to synthetic estrogen. Environ. Toxicol. Chem. 2012, 31, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.W.; Reading, B.J.; Sullivan, C.V. Ovary transcriptome profiling via artificial intelligence reveals a transcriptomic fingerprint predicting egg quality in striped bass, Moronesaxatilis. PLoS ONE 2014, 9, e96818. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.M.; Workman, V.L.; Paull, G.C.; Filby, A.L.; Van Look, K.J.W.; Kille, P.; Tyler, C.R. Molecular basis of sex and reproductive status in breeding zebrafish. Physiol. Genomics 2007, 30, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Patino, R.; Suillivan, C.V. Ovarian follicle growth, maturation, and ovulation in teleost fish. Fish. Physiol. Biochem. 2002, 26, 57–70. [Google Scholar] [CrossRef]
- Swanson, P.; Dickey, J.T.; Campbell, B. Biochemistry and physiology of fish gonadotropins. Fish. Physiol. Biochem. 2003, 28, 53–59. [Google Scholar] [CrossRef]
- Tinikul, Y.; Poljaroen, J.; Tinikul, R.; Anuracpreeda, P.; Chotwiwatthanakun, C.; Senin, N.; Poomtong, T.; Hanna, P.J.; Sobhon, P. Effects of gonadotropin-releasing hormones and dopamine on ovarian maturation in the Pacific white shrimp, Litopenaeus vannamei, and their presence in the ovary during ovarian development. Aquaculture 2014, 420, 79–88. [Google Scholar] [CrossRef]
- Nunez, B.S.; Evans, A.N. Hormonal regulation of the steroidogenic acute regulatory protein (StAR) in gonadal tissues of the Atlantic croaker (Micropogonias undulatus). Gen. Comp. Endocr. 2007, 150, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Krens, S.F.; He, S.; Spaink, H.P.; Snaar-Jagalska, B.E. Characterization and expression patterns of the MAPK family in zebrafish. Gene Expr. Patterns 2006, 6, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Knoll-Gellida, A.; Andre, M.; Gattegno, T.; Forgue, J.; Admon, A.; Babin, P.J. Molecular phenotype of zebrafish ovarian follicle by serial analysis of gene expression and proteomic profiling, and comparison with the transcriptomes of other animals. BMC Genomics 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Song, Y.L.; Zhang, S.X.; Zhao, X.L.; Wang, J.M.; Niu, N.; Zhang, G.S. The analysis of Skp1 gene expression in physiological male sterility induced by chemical hybridizing agent SQ-1in Wheat (Triticum aestivum L.). Cereal Res. Commun. 2015, 43, 204–212. [Google Scholar] [CrossRef]
- Brooks, W.S.; Helton, E.S.; Banerjee, S.; Venable, M.; Johnson, L.; Schoeb, T.R.; Kesterson, R.A.; Crawford, D.F. G2E3 is a dual function ubiquitin ligase required for early embryonic development. J. Biol. Chem. 2008, 283, 22304–22315. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.A.; Medema, R.H. Function and regulation of dynein in mitotic chromosome segregation. Chromosoma 2014, 123, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.S.; Liang, C.J.; Tseng, C.Y.; Yeh, C.W.; Tsai, J.N. Zebrafish cyclin-dependent protein kinase-Like 1 (zcdkl1): Identification and functional characterization. Int. J. Mol. Sci. 2011, 12, 3606–3617. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Yoshii, A.; Yokota, T.; Sakai, C.; Hori, H.; Kanamori, A.; Yamashita, M. Structural components of the synaptonemal complex, SYCP1and SYCP3, in the medaka fish Oryzias latipes. Exp. Cell Res. 2006, 312, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.M.; Song, W.; Yang, K.; Cao, X.J.; Gul, Y.; Wang, W.M. Flow cytometric determination of genome size for eight commercially important fish species in China. Vitro Cell. Dev. Anim. 2012, 48, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Q.; Wang, Y.K.; Zheng, X.H.; Wang, W.M.; Cao, X.J. Comparative analysis of three methods of making scale specimens for small fish. Environ. Biol. Fish. 2015, 98, 697–703. [Google Scholar] [CrossRef]
- Zhao, J.; Bian, X.D.; Sakurat, Y.; Zhang, X.M. Observation on morphological structure of the early development of Theragra chalcogramma. J. Fisher. China 2012, 36, 247–261. [Google Scholar] [CrossRef]
- Cao, X.J.; Wang, W.M. Histology and mucin histochemistry of the digestive tract of yellow catfish, Pelteobagrus fulvidraco. Anat. Histol. Embryol. 2009, 38, 254–261. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, W.; Liu, C.; Cao, X.; Huang, S.; Wang, W.; Wang, Y. Transcriptome Profile Analysis of Ovarian Tissues from Diploid and Tetraploid Loaches Misgurnus anguillicaudatus. Int. J. Mol. Sci. 2015, 16, 16017-16033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160716017
Luo W, Liu C, Cao X, Huang S, Wang W, Wang Y. Transcriptome Profile Analysis of Ovarian Tissues from Diploid and Tetraploid Loaches Misgurnus anguillicaudatus. International Journal of Molecular Sciences. 2015; 16(7):16017-16033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160716017
Chicago/Turabian StyleLuo, Weiwei, Chuanshu Liu, Xiaojuan Cao, Songqian Huang, Weimin Wang, and Yeke Wang. 2015. "Transcriptome Profile Analysis of Ovarian Tissues from Diploid and Tetraploid Loaches Misgurnus anguillicaudatus" International Journal of Molecular Sciences 16, no. 7: 16017-16033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160716017