
Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals?


{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Mitochondrial Retrograde Responses
1.1.1. Retrograde Signaling Involving Increased Cytosolic Calcium Concentration
1.1.2. Retrograde Response Associated with ROS Production and Signaling
1.1.3. Energy Deprivation and Retrograde Response
1.1.4. Beyond Regulation by Transcription Factors
1.2. The Mitochondrial Unfolded Protein Response (UPRmt)
1.2.1. The UPRmt in C. elegans
Stress Models for the Study of UPRmt
Signaling Pathway of the UPRmt
Crosstalk between the UPRmt and Other Stress Pathways in C. elegans
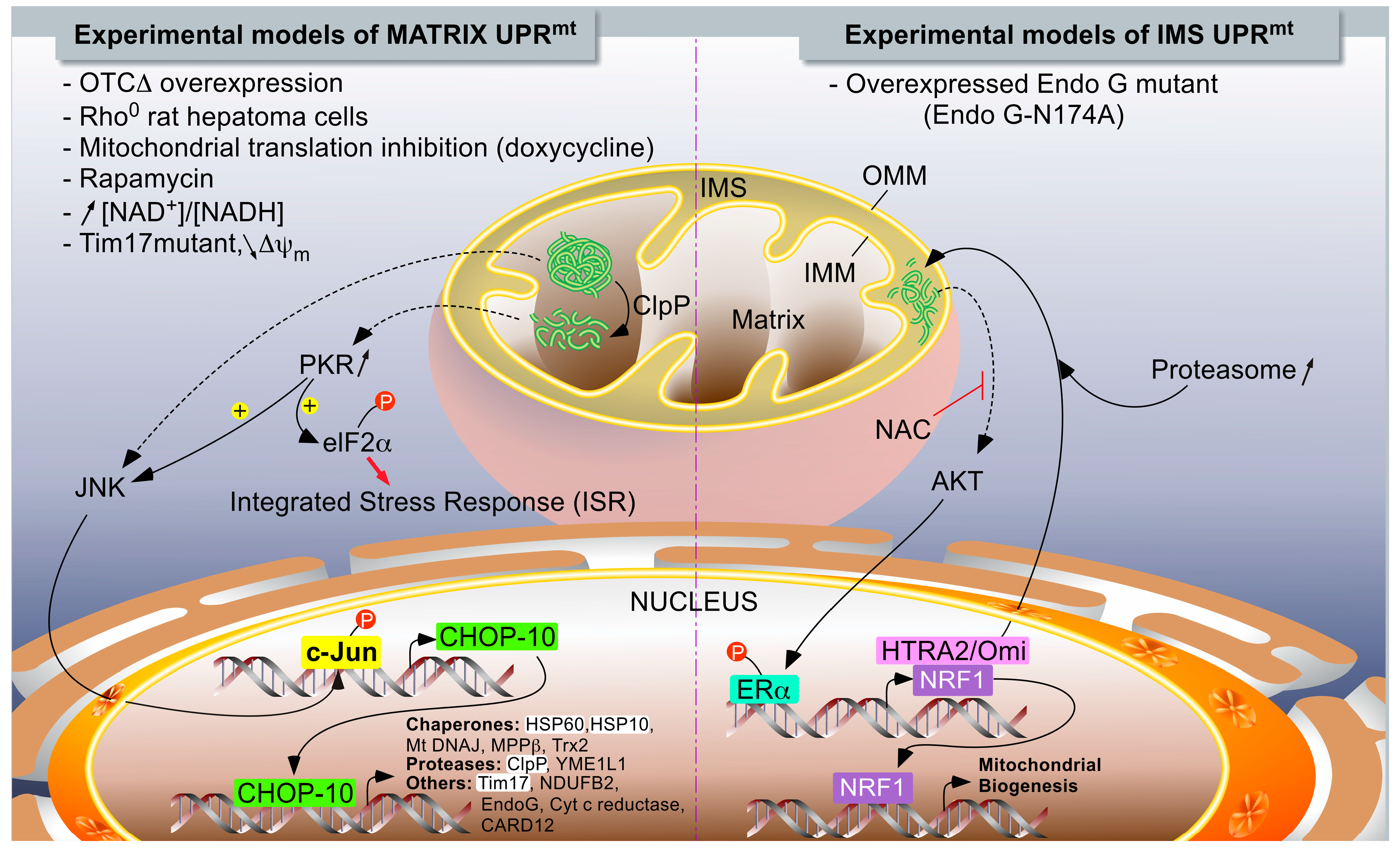
1.2.2. The UPRmt Models in Mammals

Accumulation of Unfolded Proteins within the Mitochondrial Matrix
IMS-Associated mtUPR
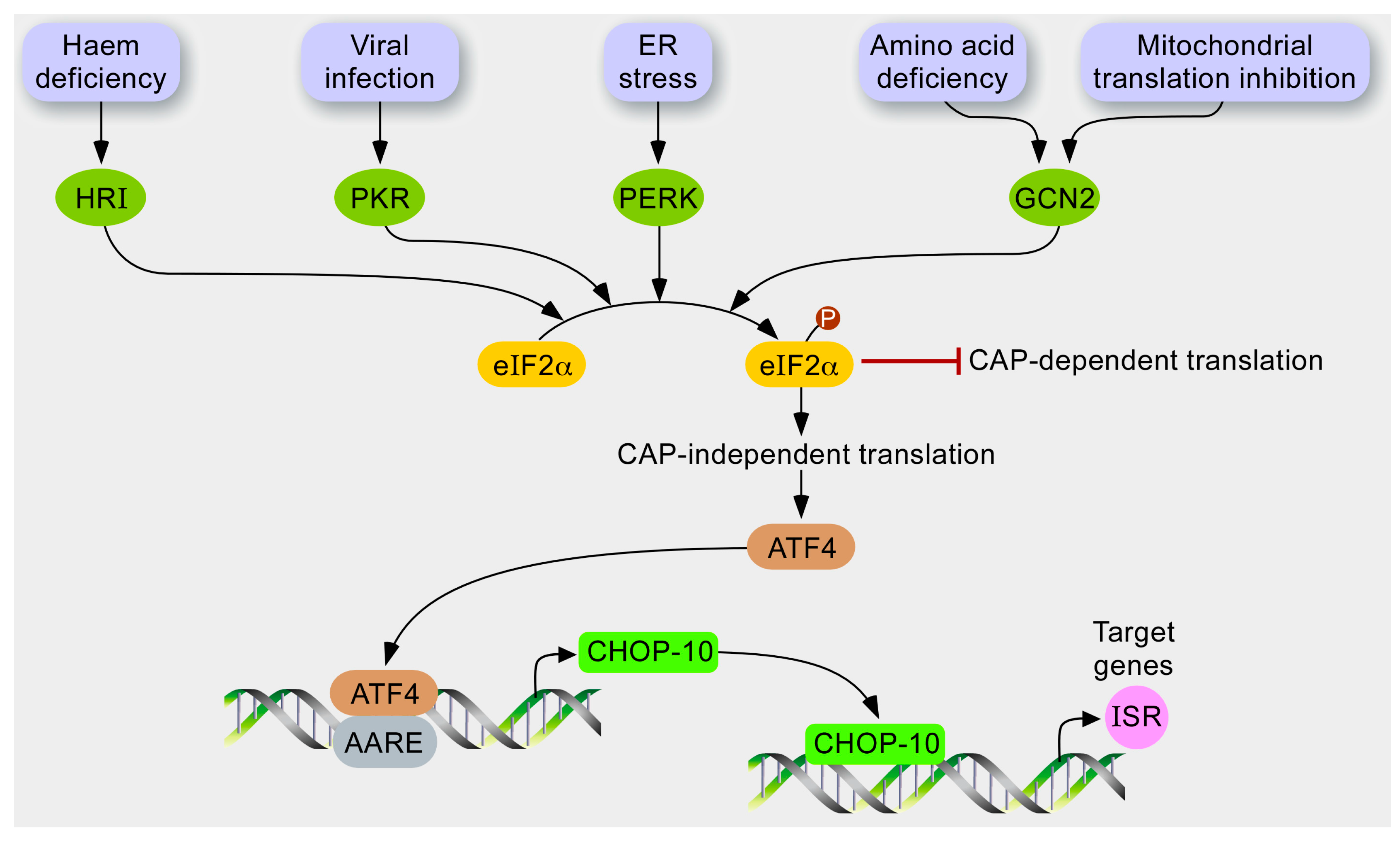
1.3. The Integrated Stress Response (ISR)

1.4. What Is the (Patho)Physiological Relevance of a UPRmt in Mammals?
1.4.1. The UPRmt and Neurodegenerative Diseases
1.4.2. The UPRmt in Aging
1.4.3. The UPRmt and Stem Cells
1.4.4. The UPRmt in Innate Immunity
2. Conclusions
2.1. The UPRmt versus ISR or Both in Response to a Mitochondrial Stress?
2.2. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goffart, S.; Wiesner, R.J. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp. Physiol. 2003, 88, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L. Shaping the dynamic mitochondrial network. BMC Biol. 2014, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 2008, 7, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Wanet, A.; de Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell. Physiol. 2012, 227, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Amuthan, G.; Biswas, G.; Ananadatheerthavarada, H.K.; Vijayasarathy, C.; Shephard, H.M.; Avadhani, N.G. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 2002, 21, 7839–7849. [Google Scholar] [CrossRef] [PubMed]
- Arnould, T.; Vankoningsloo, S.; Renard, P.; Houbion, A.; Ninane, N.; Demazy, C.; Remacle, J.; Raes, M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002, 21, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.P.; Zuckerbraun, B.S. Mitochondrial signaling: Forwards, backwards, and in between. Oxid. Med. Cell. Longev. 2013, 2013, 351613. [Google Scholar] [CrossRef] [PubMed]
- Cagin, U.; Enriquez, J.A. The complex crosstalk between mitochondria and the nucleus: What goes in between? Int. J. Biochem. Cell Biol. 2015, 63, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Nakashima-Kamimura, N.; Asoh, S.; Ishibashi, Y.; Mukai, Y.; Shidara, Y.; Oda, H.; Munakata, K.; Goto, Y.; Ohta, S. MIDAS/GPP34, a nuclear gene product, regulates total mitochondrial mass in response to mitochondrial dysfunction. J. Cell Sci. 2005, 118, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.S.; Morgan, M.M.; Scott, R.; Clements, L.S.; Butow, R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science 1987, 235, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.S.; Conrad-Webb, H.; Docherty, R.; Butow, R.A. Interaction between the yeast mitochondrial and nuclear genomes influences the abundance of novel transcripts derived from the spacer region of the nuclear ribosomal DNA repeat. Mol. Cell. Biol. 1989, 9, 1897–1907. [Google Scholar] [PubMed]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Butow, R.A. RTG1 and RTG2: Two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 1993, 72, 61–71. [Google Scholar] [CrossRef]
- Biswas, G.; Anandatheerthavarada, H.K.; Zaidi, M.; Avadhani, N.G. Mitochondria to nucleus stress signaling: A distinctive mechanism of NF-κB/Rel activation through calcineurin-mediated inactivation of IκBβ. J. Cell Biol. 2003, 161, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Vankoningsloo, S.; de Pauw, A.; Houbion, A.; Tejerina, S.; Demazy, C.; de Longueville, F.; Bertholet, V.; Renard, P.; Remacle, J.; Holvoet, P.; et al. CREB activation induced by mitochondrial dysfunction triggers triglyceride accumulation in 3T3-L1 preadipocytes. J. Cell Sci. 2006, 119, 1266–1282. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Pan, H.; Fang, J.K.; Avadhani, N.G. Heterogeneous nuclear ribonucleoprotein A2 is a common transcriptional coactivator in the nuclear transcription response to mitochondrial respiratory stress. Mol. Biol. Cell 2009, 20, 4107–4119. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Engel, J.C.; Correia, M.A. Hepatic CYP3A suppression by high concentrations of proteasomal inhibitors: A consequence of endoplasmic reticulum (ER) stress induction, activation of RNA-dependent protein kinase-like ER-bound eukaryotic initiation factor 2α (eIF2α)-kinase (PERK) and general control nonderepressible-2 eIF2α kinase (GCN2), and global translational shutoff. Mol. Pharmacol. 2009, 76, 503–515. [Google Scholar] [PubMed]
- Arnould, T.; Mercy, L.; Houbion, A.; Vankoningsloo, S.; Renard, P.; Pascal, T.; Ninane, N.; Demazy, C.; Raes, M. mtCLIC is up-regulated and maintains a mitochondrial membrane potential in mtDNA-depleted L929 cells. FASEB J. 2003, 17, 2145–2147. [Google Scholar] [CrossRef] [PubMed]
- Rohas, L.M.; St-Pierre, J.; Uldry, M.; Jager, S.; Handschin, C.; Spiegelman, B.M. A fundamental system of cellular energy homeostasis regulated by PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 7933–7938. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Tang, W.; Sondheimer, N.; Avadhani, N.G. Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim. Biophys. Acta 2010, 1797, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial transcription factor a induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 2006, 281, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Favre, C.; Zhdanov, A.; Leahy, M.; Papkovsky, D.; O’Connor, R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 2010, 29, 3964–3976. [Google Scholar] [CrossRef] [PubMed]
- Perez-de-Arce, K.; Foncea, R.; Leighton, F. Reactive oxygen species mediates homocysteine-induced mitochondrial biogenesis in human endothelial cells: Modulation by antioxidants. Biochem. Biophys. Res. Commun. 2005, 338, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell. Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1340–E1346. [Google Scholar] [PubMed]
- Holmuhamedov, E.; Jahangir, A.; Bienengraeber, M.; Lewis, L.D.; Terzic, A. Deletion of mtDNA disrupts mitochondrial function and structure, but not biogenesis. Mitochondrion 2003, 3, 13–19. [Google Scholar] [CrossRef]
- Mercy, L.; Pauw, A.; Payen, L.; Tejerina, S.; Houbion, A.; Demazy, C.; Raes, M.; Renard, P.; Arnould, T. Mitochondrial biogenesis in mtDNA-depleted cells involves a Ca2+-dependent pathway and a reduced mitochondrial protein import. FEBS J. 2005, 272, 5031–5055. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.H.; Lee, B.; Hwang, D.; Park, K.S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signal. 2013, 6, rs4. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, U.; Bertrand, L.; Hue, L. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur. J. Biochem. 2002, 269, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, P.; Pande, S.V.; Prentki, M.; Madiraju, S.R. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 2009, 4, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Ribas, F.; Villarroya, J.; Hondares, E.; Giralt, M.; Villarroya, F. FGF21 expression and release in muscle cells: Involvement of MyoD and regulation by mitochondria-driven signalling. Biochem. J. 2014, 463, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zheng, J.; Lv, J.; Xu, J.; Ji, X.; Luo, Y.B.; Li, W.; Zhao, Y.; Yan, C. Skeletal muscle increase FGF21 expression in mitochondrial disorder to compensate for the energy metabolic insufficiency by activating mTOR-YY1-PGC1α pathway. Free Radic. Biol. Med. 2015, 84, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.M.; Haynes, C.M. UPRmt-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Kaeberlein, M. The mitochondrial unfolded protein response and increased longevity: Cause, consequence, or correlation? Exp. Gerontol. 2014, 56, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Jasper, H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FoxO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. Elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria—A regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Hartl, F.U.; Horwich, A.L. The mitochondrial chaperonin HSP60 is required for its own assembly. Nature 1990, 348, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef] [PubMed]
- Cairns, B.R. The logic of chromatin architecture and remodelling at promoters. Nature 2009, 461, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Canonne, M.; Arnould, T.; Renard, P. Inhibition of mitochondrial genome expression triggers the activation of chop-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 2015, 21, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, W.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ito, M.; Nozawa, Y.; Yoneda, M.; Oshida, Y.; Tanaka, M. CHOP (C/EBP homologous protein) and ASNS (asparagine synthetase) induction in cybrid cells harboring MELAS and NARP mitochondrial DNA mutations. Mitochondrion 2007, 7, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Akimoto, T.; Yamamoto, H.; Araki, Y.; Yoshie, T.; Mori, K.; Hayashi, H.; Nose, K.; Shibanuma, M. Gene expression profiling identifies a role for CHOP during inhibition of the mitochondrial respiratory chain. J. Biochem. 2009, 146, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Fu, W.; Zhang, P.; Cheng, A.; Lee, J.; Kokame, K.; Mattson, M.P. Herp stabilizes neuronal Ca2+ homeostasis and mitochondrial function during endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 28733–28743. [Google Scholar] [CrossRef] [PubMed]
- Prudente, S.; Sesti, G.; Pandolfi, A.; Andreozzi, F.; Consoli, A.; Trischitta, V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr. Rev. 2012, 33, 526–546. [Google Scholar] [CrossRef] [PubMed]
- Padyana, A.K.; Qiu, H.; Roll-Mecak, A.; Hinnebusch, A.G.; Burley, S.K. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2α protein kinase GCN2. J. Biol. Chem. 2005, 280, 29289–29299. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, R.C. GCN2 phosphorylation of eIF2α activates NF-κB in response to UV irradiation. Biochem. J. 2005, 385, 371–380. [Google Scholar] [PubMed]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Habener, J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992, 6, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Kilberg, M.S. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J. Biol. Chem. 2008, 283, 35106–35117. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014, 26, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [PubMed]
- Park, H.J.; Kim, S.S.; Seong, Y.M.; Kim, K.H.; Goo, H.G.; Yoon, E.J.; Min, D.S.; Kang, S.; Rhim, H. β-Amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J. Biol. Chem. 2006, 281, 34277–34287. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, J.; Milojevic, J.; Melacini, G.; Ortega, J. A new function of human HtrA2 as an amyloid-β oligomerization inhibitor. J. Alzheimer’s Dis. 2009, 17, 281–294. [Google Scholar]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Shavali, S.; Brown-Borg, H.M.; Ebadi, M.; Porter, J. Mitochondrial localization of α-synuclein protein in α-synuclein overexpressing cells. Neurosci. Lett. 2008, 439, 125–128. [Google Scholar] [CrossRef] [PubMed]
- De Castro, I.P.; Costa, A.C.; Lam, D.; Tufi, R.; Fedele, V.; Moisoi, N.; Dinsdale, D.; Deas, E.; Loh, S.H.; Martins, L.M. Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ. 2012, 19, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; van Remmen, H. Mitochondrial stress signaling in longevity: A new role for mitochondrial function in aging. Redox Biol. 2014, 2, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Lobet, E.; Letesson, J.J.; Arnould, T. Mitochondria: A target for bacteria. Biochem. Pharmacol. 2015, 94, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Haller, D. Unfolded protein responses in the intestinal epithelium: Sensors for the microbial and metabolic environment. J. Clin. Gastroenterol. 2012, 46, S3–S5. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Song, B.; Kaufman, R.J. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm. Bowel Dis. 2012, 18, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogan, S.A.; Pujol, C.; Maiti, P.; Kukat, A.; Wang, S.; Hermans, S.; Senft, K.; Wibom, R.; Rugarli, E.I.; Trifunovic, A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014, 19, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Blackinton, J.G.; Keene, J.D. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin. Cell Dev. Biol. 2014, 34, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houten, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Waldschmitt, N.; Berger, E.; Rath, E.; Sartor, R.B.; Weigmann, B.; Heikenwalder, M.; Gerhard, M.; Janssen, K.P.; Haller, D. C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol. 2014, 7, 1452–1466. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Deval, C.; Maurin, A.C.; Parry, L.; Cherasse, Y.; Chaveroux, C.; Lefloch, R.; Lenormand, P.; Bruhat, A.; Fafournoux, P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J. Biol. Chem. 2007, 282, 15851–15861. [Google Scholar] [CrossRef] [PubMed]
- Frechin, M.; Enkler, L.; Tetaud, E.; Laporte, D.; Senger, B.; Blancard, C.; Hammann, P.; Bader, G.; Clauder-Munster, S.; Steinmetz, L.M.; et al. Expression of nuclear and mitochondrial genes encoding ATP synthase is synchronized by disassembly of a multisynthetase complex. Mol. Cell 2014, 56, 763–776. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224-18251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818224
Arnould T, Michel S, Renard P. Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals? International Journal of Molecular Sciences. 2015; 16(8):18224-18251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818224
Chicago/Turabian StyleArnould, Thierry, Sébastien Michel, and Patricia Renard. 2015. "Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals?" International Journal of Molecular Sciences 16, no. 8: 18224-18251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160818224