
Cisplatin Targeting of Bacterial Ribosomal RNA Hairpins

Abstract
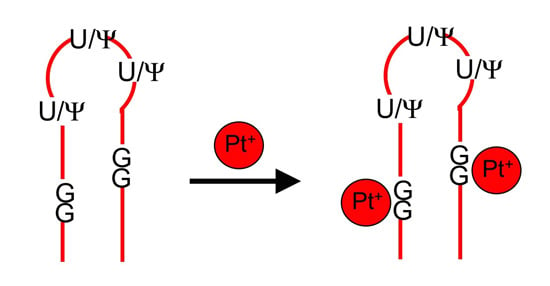
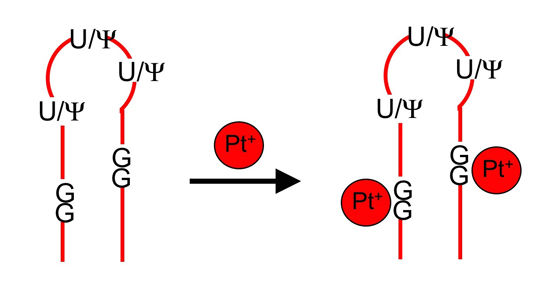
:
1. Introduction

2. Results and Discussion
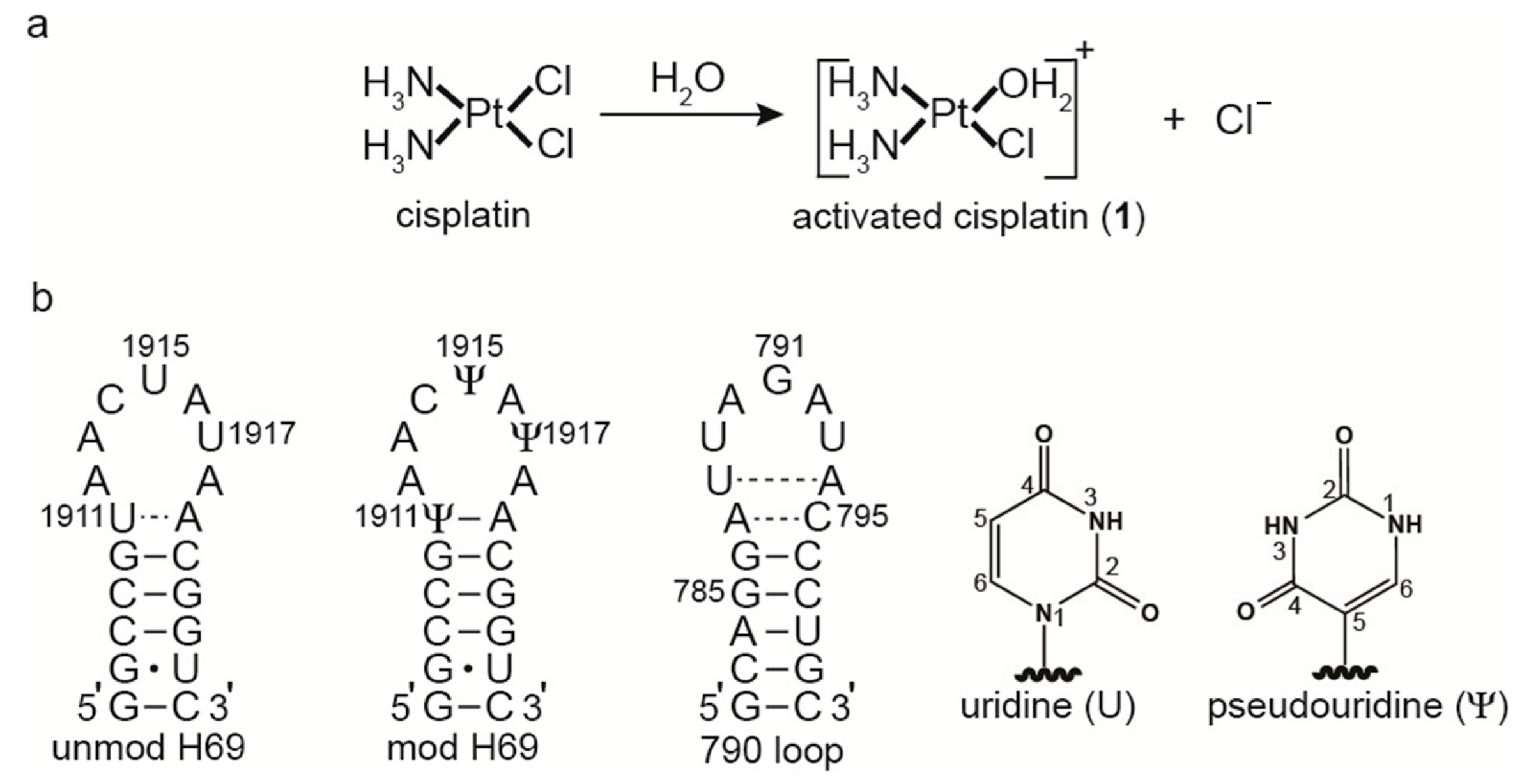
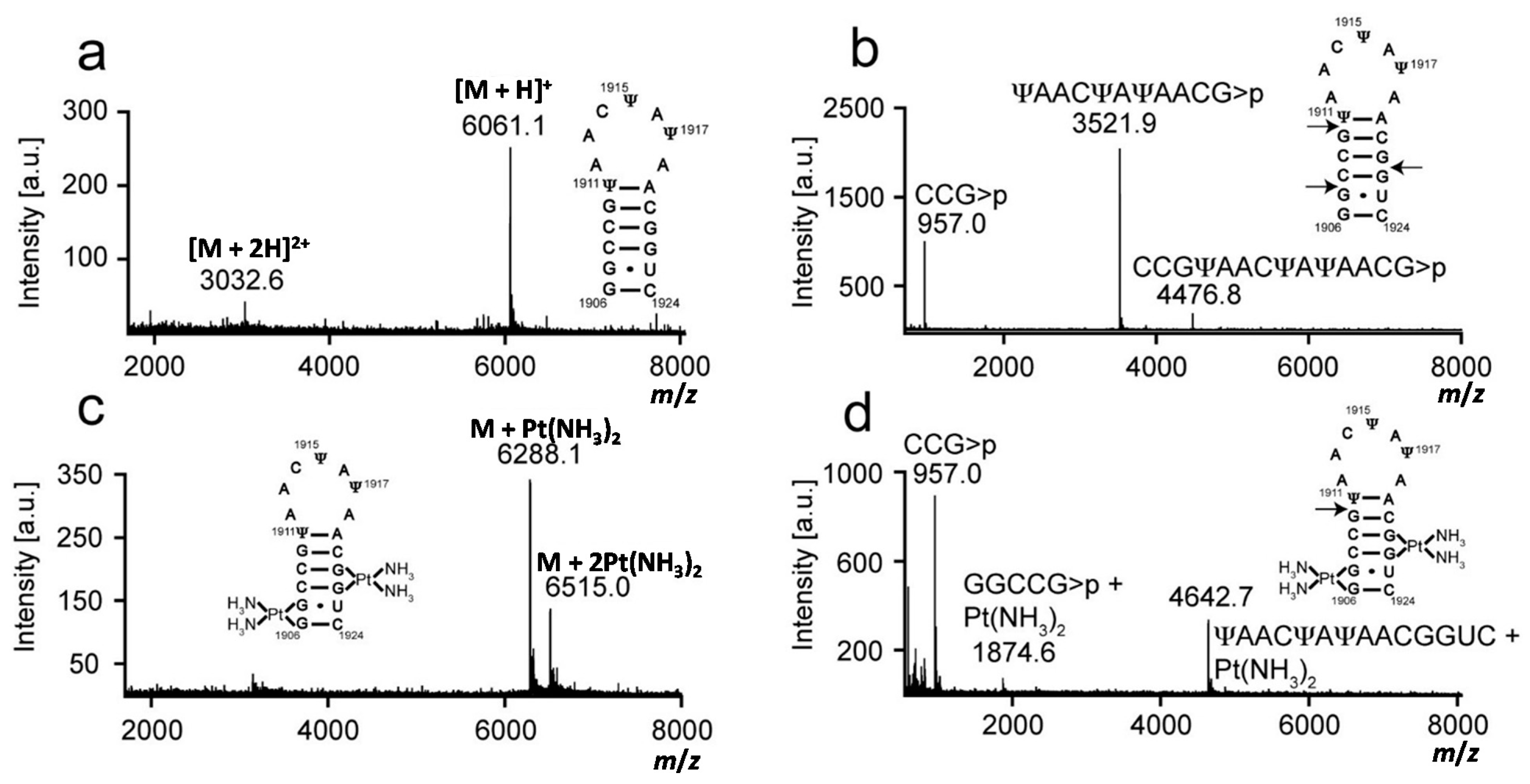
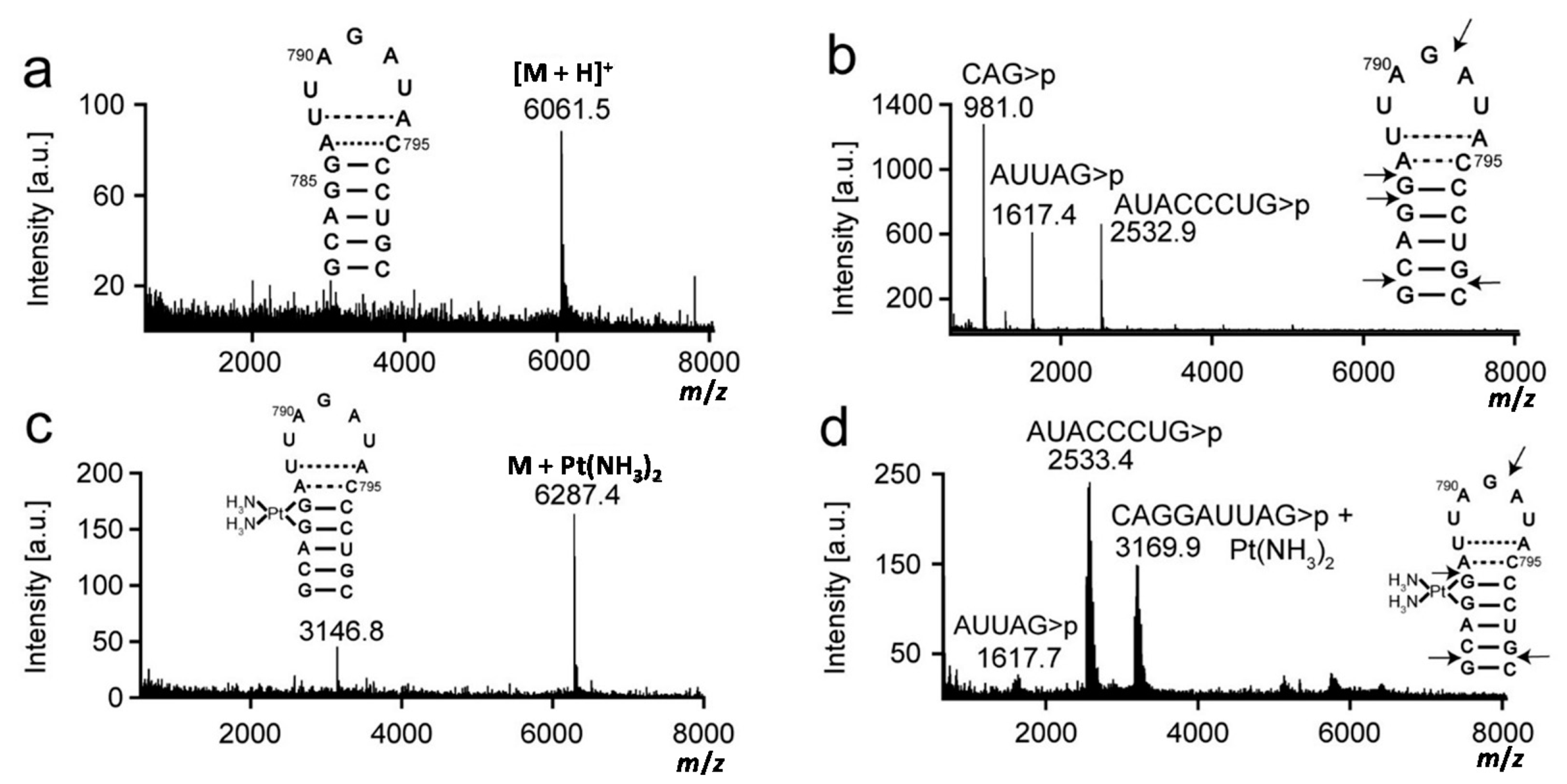
2.1. Matrix-Assisted Laser Desorption-Ionization Time-of-Flight (MALDI) Mass Spectrometry and RNase T1 Mapping Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | [RNA]+ | Predicted Mass a m/z | Experimental Mass m/z |
|---|---|---|---|
| unmodified H69 | Parent strand + H+ | 6061.7 | 6061.3 |
| Parent strand + [Pt(NH3)2]2+ − H+ | 6288.7 | 6287.9 | |
| Parent strand + 2[Pt(NH3)2]2+ − 3H+ | 6515.7 | 6514.9 | |
| 5′CCG>p3′ + H+ | 956.6 | 957.2/956.9 b | |
| 5′UAACUAUAACG>p3′ + H+ | 3521.1 | 3521.8 | |
| 5′GGCCG>p3′ + [Pt(NH3)2]2+ − H+ | 1873.9 | 1874.7 | |
| 5′UAACUAUAACGGUC3′ + [Pt(NH3)2]2+ − H+ | 4642.7 | 4642.5 | |
| modified H69 | Parent strand + H+ | 6061.7 | 6061.1 |
| Parent strand + [Pt(NH3)2]2+ − H+ | 6288.7 | 6288.1 | |
| Parent strand + 2[Pt(NH3)2]2+ − 3H+ | 6515.7 | 6515.0 | |
| 5′CCG>p3′ + H+ | 956.6 | 957.0/957.0 b | |
| 5′ΨAACΨAΨAACG>p3′ + H+ | 3521.1 | 3521.9 | |
| 5′CCGΨAACΨAΨAACG>p3′ + H+ | 4476.7 | 4476.8 | |
| 5′GGCCG>p3′ + [Pt(NH3)2]2+ − H+ | 1874.0 | 1874.6 | |
| 5′ΨAACΨAΨAACGGUC3′ + [Pt(NH3)2]2+ − H+ | 4642.7 | 4642.7 | |
| 790 loop | Parent strand + H+ Parent strand + [Pt(NH3)2]2+ − H+ | 6061.7 6288.7 | 6061.5 6287.4 |
| 5′CAG>p3′ + H+ | 980.6 | 981.0 | |
| 5′AUUAG>p3′ + H+ | 1616.9 | 1617.4/1617.7 b | |
| 5′AUACCCUG>p3′ + H+ | 2532.5 | 2532.9/2533.4 b | |
| 5′CAGGAUUAG>p3′ + [Pt(NH3)2]2+ − H+ | 3168.7 | 3169.9 |



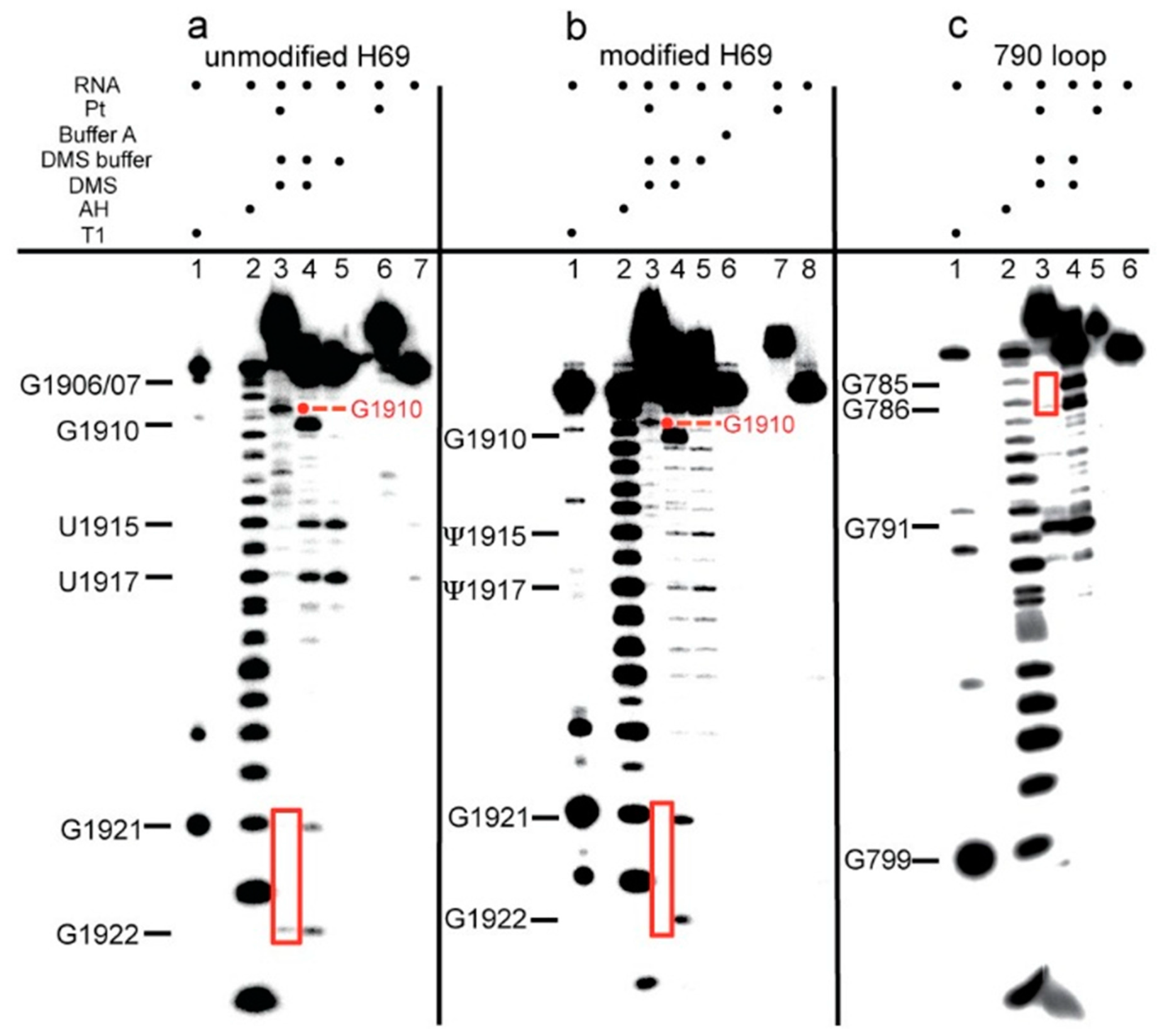
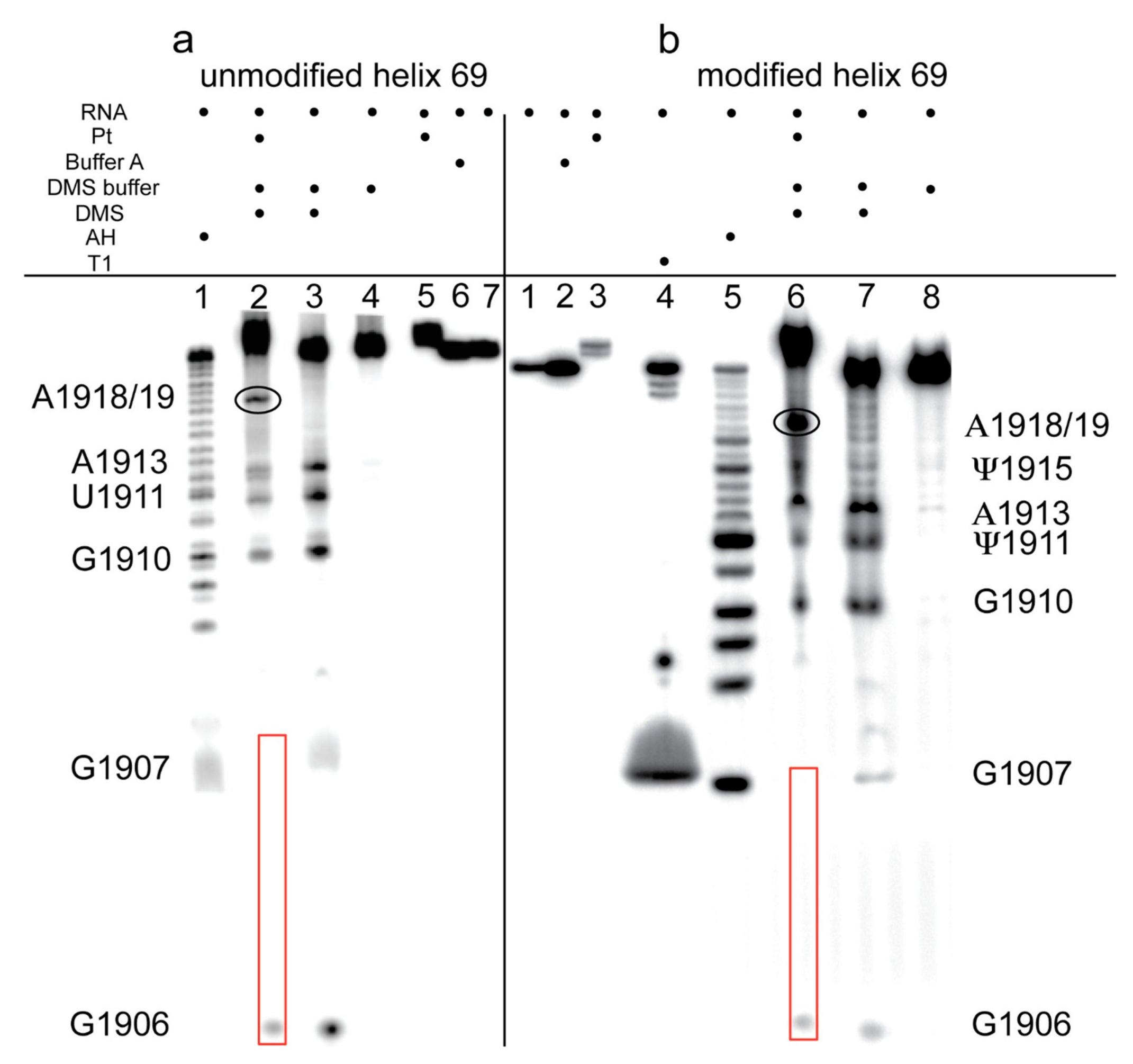
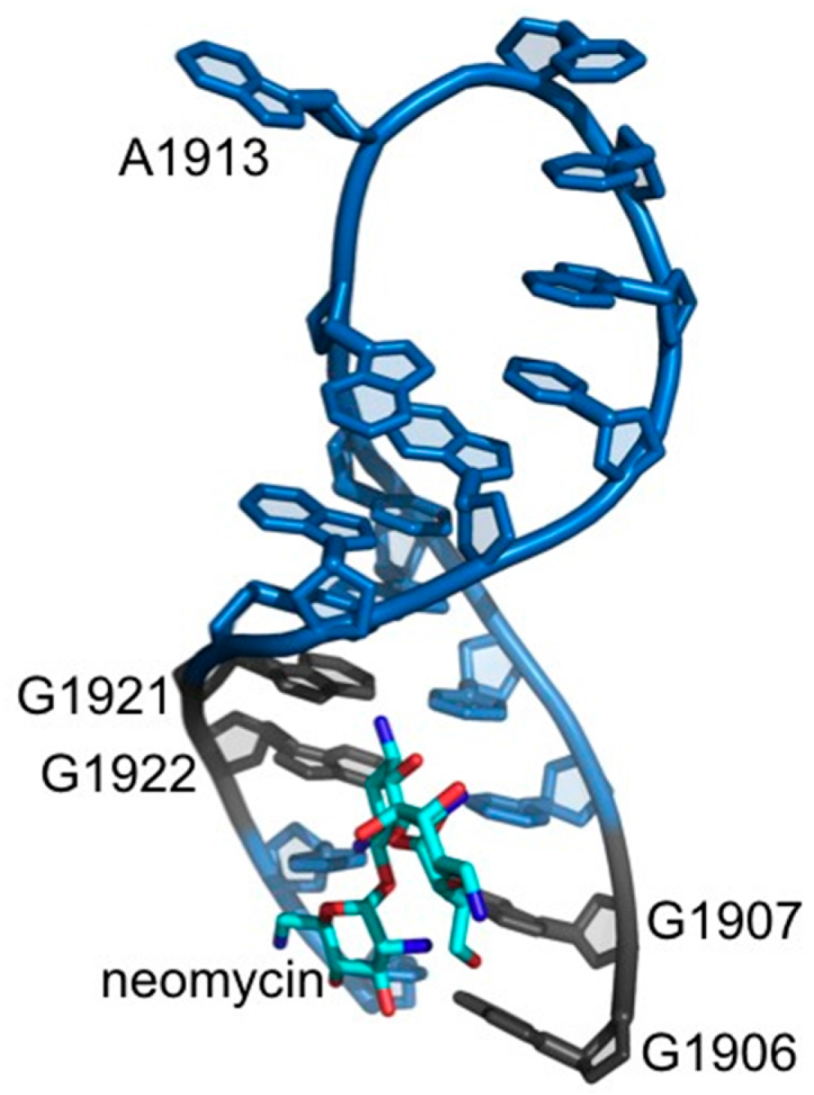
2.2. Dimethyl Sulfate Probing of Platination Sites



3. Experimental Section
3.1. Buffers
3.2. Metal Complexes
3.3. Nucleic Acids
3.4. End Labeling of Ribonucleic Acid (RNA) Constructs
3.5. Large-Scale Platination and RNase T1 Mapping
3.6. Chemical Probing of Platinated RNAs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rosenberg, B.; van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, D.P.; Lepre, C.A.; Lippard, S.J. 195Pt NMR kinetic and mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to DNA. J. Am. Chem. Soc. 1990, 112, 6860–6871. [Google Scholar] [CrossRef]
- Miller, S.E.; House, D.A. The hydrolysis products of cis-diamminedichloroplatinum(II). I. The kinetics of formation and anation of the cis-diammine(aqua)chloroplatinum(II) cation in acidic aqueous solution. Inorg. Chim. Acta 1989, 161, 131–137. [Google Scholar] [CrossRef]
- Van Hemelryck, B.; Girault, J.P.; Chottard, G.; Valadon, P.; Laoui, A.; Chottard, J.C. Sequence-dependent platinum chelation by adenylyl(3′-5′)guanosine and guanylyl(3′-5′)adenosine reacting with cis-diamminedichloroplatinum(II) and its diaqua derivative. Inorg. Chem. 1987, 26, 787–795. [Google Scholar]
- Gelasco, A.; Lippard, S.J. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry 1998, 37, 9230–9239. [Google Scholar] [CrossRef] [PubMed]
- Herder, H.C.; Rosenberg, B. Inhibitory effects of anti-tumor platinum compounds on DNA, RNA and protein syntheses in mammalian cells in vitro. Int. J. Cancer 1970, 6, 207–216. [Google Scholar] [CrossRef]
- Rosenberg, J.; Sato, P. Messenger RNA loses the ability to direct in vitro peptide synthesis following incubation with cisplatin. Mol. Pharmacol. 1988, 33, 611–616. [Google Scholar] [PubMed]
- Heminger, K.A.; Hartson, S.D.; Rogers, J.; Matts, R.L. Cisplatin inhibits protein synthesis in rabbit reticulocyte lysate by causing an arrest in elongation. Arch. Biochem. Biophys. 1997, 344, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Danenberg, P.V.; Shea, L.C.C.; Danenberg, K.D.; Horikoshi, T. Inactivation of Tetrahymena rRNA self-splicing by cis-platin proceeds through dissociable complexes. Nucleic Acids Res. 1991, 19, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.M.; Sato, P.H. Cisplatin inhibits in vitro translation by preventing the formation of complete initiation complex. Mol. Pharmacol. 1993, 43, 491–497. [Google Scholar] [PubMed]
- Hägerlöf, M.; Papsai, P.; Chow, C.S.; Elmroth, S.K. More pronounced salt dependence and higher reactivity for platination of the hairpin r(CGCGUUGUUCGCG) compared with d(CGCGTTGTTCGCG). J. Biol. Inorg. Chem. 2006, 11, 974–990. [Google Scholar] [CrossRef] [PubMed]
- Papsai, P.; Aldag, J.; Persson, T.; Elmroth, S.K.C. Kinetic preference for interaction of cisplatin with the G-C-rich wobble basepair region in both tRNAAla and MhAla. Dalton Trans. 2006, 29, 3515–3517. [Google Scholar] [CrossRef] [PubMed]
- Rijal, K.; Chow, C.S. A new role for cisplatin: Probing ribosomal RNA structure. Chem. Commun. 2009, 7, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Rijal, K.; Bao, X.; Chow, C.S. Amino acid-linked platinum(II) analogues have altered specificity for RNA compared to cisplatin. Chem. Commun. 2014, 50, 3918–3920. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, A.A.; Osborn, M.F.; DeRose, V.J. RNA-Pt adducts following cisplatin treatment of Saccharomyces cerevisiae. ACS Chem. Biol. 2011, 7, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, A.A.; Chapman, E.G.; DeRose, V.J. Rapid cross-linking of an RNA internal loop by the anticancer drug cisplatin. J. Am. Chem. Soc. 2009, 131, 9250–9257. [Google Scholar] [CrossRef] [PubMed]
- Polonyi, C.; Albertsson, I.; Damian, M.S.; Elmroth, S.K.C. Comparison of cis- and oxaliplatin-induced destabilization of 15-mer DNA- and RNA duplexes by binding to centrally located GG- and GNG sequences. Z. Anorg. Allg. Chem. 2013, 639, 1655–1660. [Google Scholar] [CrossRef]
- Sato, P.H.; Rosenberg, J.M.; Sato, R.I. Differences in the inhibition of translation by cisplatin, transplatin, and certain related compounds. Biochem. Pharmacol. 1996, 52, 1895–1902. [Google Scholar] [CrossRef]
- Hermann, T. Drugs targeting the ribosome. Curr. Opin. Struct. Biol. 2005, 15, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.R.; Gonzalez, R.L.; Puglisi, J.D. Comparison of X-ray crystal structure of the 30S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure 2003, 11, 43–53. [Google Scholar] [CrossRef]
- Schuwirth, B.S.; Borovinskaya, M.A.; Hau, C.W.; Zhang, W.; Vila-Sanjurjo, A.; Holton, J.M.; Cate, J.H.D. Structures of the bacterial ribosome at 3.5 Å resolution. Science 2005, 310, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Aduri, R.; Chow, C.S.; SantaLucia, J., Jr. Structure modulation of helix 69 from Escherichia coli 23S ribosomal RNA by pseudouridylations. Nucleic Acids Res. 2013, 42, 3971–3981. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Varma, S.; SantaLucia, J., Jr.; Cunningham, P.R. In vivo determination of RNA structure-function relationships: Analysis of the 790 loop in ribosomal RNA. J. Mol. Biol. 1997, 269, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.D.; Noller, H.F. Crystal structure of the ribosome at 5.5 Å resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Gutell, R.R.; Gray, M.W.; Schnare, M.N. A compilation of large subunit (23S and 23S-like) ribosomal RNA structures: 1993. Nucleic Acids Res. 1993, 21, 3055–3074. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N.; Schluenzen, F.; Harms, J.M.; Yoshida, T.; Ohkubo, T.; Albrecht, R.; Buerger, J.; Kobayashi, Y.; Fucini, P. X-ray crystallography study on ribosome recycling: The mechanism of binding and action of RRF on the 50S ribosomal subunit. EMBO J. 2005, 24, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.K.; Sharma, M.R.; Kiel, M.C.; Hirokawa, G.; Booth, T.M.; Spahn, C.M.T.; Grassucci, R.A.; Kaji, A.; Frank, J. Visualization of ribosome-recycling factor on the Escherichia coli 70S ribosome: Functional implications. Proc. Natl. Acad. Sci. USA 2004, 101, 8900–8905. [Google Scholar] [CrossRef] [PubMed]
- Kipper, K.; Hetényi, C.; Sild, S.; Remme, J.; Liiv, A. Ribosomal intersubunit bridge B2a is involved in factor-dependent translation initiation and translational processivity. J. Mol. Biol. 2009, 385, 405–422. [Google Scholar] [CrossRef] [PubMed]
- Abeysirigunawardena, S.C.; Chow, C.S. pH-Dependent structural changes of helix 69 from Escherichia coli 23S ribosomal RNA. RNA 2008, 14, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Chow, C.S. Role of pseudouridine in structural rearrangements of helix 69 during bacterial ribosome assembly. ACS Chem. Biol. 2012, 7, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Chow, C.S. Probing conformational states of modified helix 69 in 50S ribosomes. J. Am. Chem. Soc. 2011, 133, 8396–8399. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Magrum, L.J.; Gupta, R.; Siegel, R.B.; Stahl, D.A.; Kop, J.; Crawford, N.; Brosius, R.; Gutell, R.; Hogan, J.J.; et al. Secondary structure model for bacterial 16S ribosomal RNA: Phylogenetic, enzymatic and chemical evidence. Nucleic Acids Res. 1980, 8, 2275–2293. [Google Scholar] [CrossRef] [PubMed]
- Tapprich, W.E.; Hill, W.E. Involvement of bases 787–795 of Escherichia coli 16S ribosomal RNA in ribosomal subunit association. Proc. Natl. Acad. Sci. USA 1986, 83, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Muralikrishna, P.; Wickstrom, E. Escherichia coli initiation factor 3 protein binding to 30S ribosomal subunits alters the accessibility of nucleotides within the conserved central region of 16S rRNA. Biochemistry 1989, 28, 7505–7510. [Google Scholar] [CrossRef] [PubMed]
- Tapprich, W.E.; Goss, D.J.; Dahlberg, A.E. Mutation at position 791 in Escherichia coli 16S ribosomal RNA affects processes involved in the initiation of protein synthesis. Proc. Natl. Acad. Sci. USA 1989, 86, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pulk, A.; Wasserman, M.R.; Feldman, M.B.; Altman, R.B.; Cate, J.H.D.; Blanchard, S.C. Allosteric control of the ribosome by small-molecule antibiotics. Nat. Struct. Mol. Biol. 2012, 19, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Pioletti, M.; Schlünzen, F.; Harms, J.; Zarivach, R.; Glühmann, M.; Avila, H.; Bashan, A.; Bartels, H.; Auerbach, T.; Jacobi, C.; et al. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Saenger, W. Crystallographic study of mechanism of ribonuclease T1-catalysed specific RNA hydrolysis. J. Biomol. Struct. Dyn. 1983, 1, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Ehresmann, C.; Baudin, F.; Mougel, M.; Romby, P.; Ebel, J.P.; Ehresmann, B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987, 15, 9109–9128. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.G.; DeRose, V.J. Enzymatic processing of platinated RNAs. J. Am. Chem. Soc. 2010, 132, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Iannitti-Tito, P.; Weimann, A.; Wickham, G.; Sheil, M.M. Structural analysis of drug-DNA adducts by tandem mass spectrometry. Analyst 2000, 125, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Guittard, J.; Pacifico, C.; Blais, J.C.; Bolbach, G.; Chottard, J.C.; Spassky, A. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry of DNA–Pt(II) complexes. Rapid Commun. Mass Spectrom. 1995, 9, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.E.; Nordhoff, E.; Hillenkamp, F. Matrix-assisted UV and IR laser desorption—Ionization time-of-flight mass spectrometry of diamminoplatinum(II) oligodeoxyribonucleotide adducts and their unplatinated analogs. Int. J. Mass Spectrom. Ion Processes 1994, 132, 239–249. [Google Scholar] [CrossRef]
- Peattie, D.A. Direct chemical method for sequencing RNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Meroueh, M.; Kjellström, J.; Mårtensson, K.S.M.; Elmroth, S.K.C.; Chow, C.S. Reactions of platinum(II) complexes with a DNA hairpin, d(CGCGTTGTTCGCG): Structural characterization and kinetic studies. Inorg. Chim. Acta 2000, 297, 145–155. [Google Scholar] [CrossRef]
- Fichtinger-Schepman, A.M.J.; van der Veer, J.L.; den Hartog, J.H.J.; Lohman, P.H.M.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Takahara, P.M.; Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 1995, 377, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Abeysirigunawardena, S.C.; Duc, A.-C.E.; Dremann, D.N.; Chow, C.S. Ligand- and pH-induced conformational changes of RNA domain helix 69 revealed by 2-aminopurine fluorescence. Angew. Chem. Int. Ed. Engl. 2012, 51, 12095–12098. [Google Scholar] [CrossRef] [PubMed]
- Borovinskaya, M.A.; Pai, R.D.; Zhang, W.; Schuwirth, B.S.; Holton, J.M.; Hirokawa, G.; Kaji, H.; Kaji, A.; Cate, J.H.D. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mol. Biol. 2007, 14, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Wang, L.; Feldman, M.B.; Pulk, A.; Chen, V.B.; Kapral, G.J.; Noeske, J.; Richardson, J.S.; Blanchard, S.C.; Cate, J.H.D. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 2011, 332, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Rijal, K. Exploring Potential Drug Target Sites in the Ribosome Using Cisplatin and Its Analogues. Ph.D. Thesis, Wayne State University, Detroit, MI, USA, January 2011. [Google Scholar]
- Osborn, M.F.; White, J.D.; Haley, M.M.; DeRose, V.J. Platinum-RNA modifications following drug treatment in S. cerevisiae identified by click chemistry and enzymatic mapping. ACS Chem. Biol. 2014, 9, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 3I24, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Elmroth, S.K.C.; Lippard, S.J. Surface and electrostatic contributions to DNA-promoted reactions of platinum(II) complexes with short oligonucleotides: A kinetic study. Inorg. Chem. 1995, 34, 5234–5243. [Google Scholar] [CrossRef]
- England, T.E.; Uhlenbeck, O.C. 3′-Terminal labelling of RNA with T4 RNA ligase. Nature 1978, 275, 560–561. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedduwa-Mudalige, G.N.P.; Chow, C.S. Cisplatin Targeting of Bacterial Ribosomal RNA Hairpins. Int. J. Mol. Sci. 2015, 16, 21392-21409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921392
Dedduwa-Mudalige GNP, Chow CS. Cisplatin Targeting of Bacterial Ribosomal RNA Hairpins. International Journal of Molecular Sciences. 2015; 16(9):21392-21409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921392
Chicago/Turabian StyleDedduwa-Mudalige, Gayani N. P., and Christine S. Chow. 2015. "Cisplatin Targeting of Bacterial Ribosomal RNA Hairpins" International Journal of Molecular Sciences 16, no. 9: 21392-21409. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160921392