
Pharmacogenetics of BCR/ABL Inhibitors in Chronic Myeloid Leukemia

 and
and
Abstract
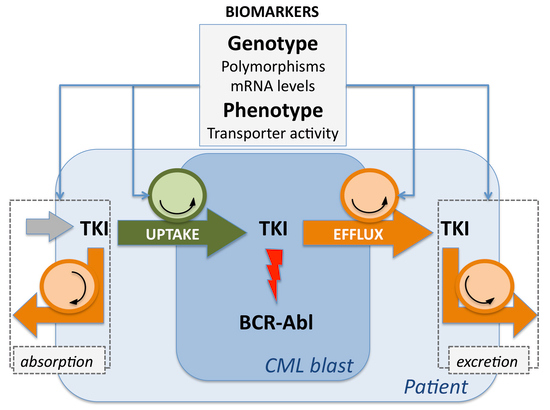
:
1. Introduction
2. Many Pieces for the Pharmacogenetic Puzzle
2.1. Liver Enzymes
2.2. Transmembrane Transporters
2.2.1. ABCB1

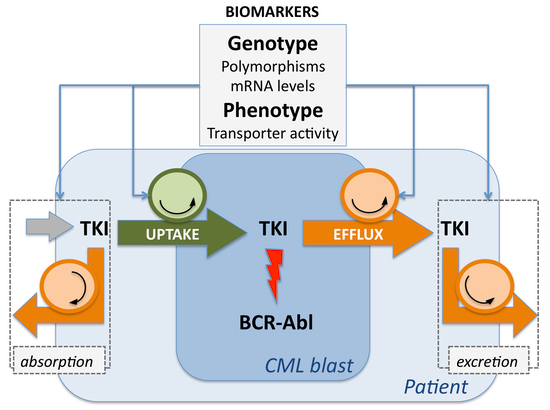{kind=link}
| Transporter | SNPs * | Patients | Main Results | Reference |
|---|---|---|---|---|
| ABCB1 | c.-129T>C | 189 | c.3435CT/TT was an adverse genotype for complete MR in Caucasians a | [15] |
| 215 | c.1236CC and CGC haplotype were associated with resistance, while c.2677TT/TA/AA were related with better CCyR | [30] | ||
| 100 | TGT haplotype was associated with worse therapeutic effect from imatinib | [31] | ||
| 90 | c.1236TT and c.2677TT/TA were associated with better major MR rate | [28] | ||
| 90 | CGC haplotype associated with less frequent major MR | [28] | ||
| 52 | c.1236TT or c.3435CT/TT were associated with higher resistance; patients with the c.2677AG/AT/AA genotype had better CCyR than those carrying c.2677TT/GT/GG | [32] | ||
| 84 | c.3435TT associated with significantly longer times to major MR compared to CC/CT genotypes | [33] | ||
| 28 | Polymorphic alleles were associated with a reduced ex vivo ABCB1 activity; the highest transporter activity was present in patients who did not achieve major MR | [29] | ||
| ABCG2 | c.34G>A | 229 | c.34GG genotype was associated with lowest rates of major MR and CCyR | [14] |
| c.34G>A, c.421C>A | 215 | c.421CC associated with resistance; AA haplotype, better response | [30] | |
| c.421C>A | 82 | c.421CC/CA associated with lower rate of major MR b | [34] |
2.2.2. ABCG2
2.2.3. SLC22A1
| Genes | Polymorphisms, Gene Expression, Functional Test | Patients | Main Results | Reference |
|---|---|---|---|---|
| SLC22A1 | c.1002C>T | 189 | c.1022CT/TT were adverse genotypes for major cytogenetic response in all patients | [15] |
| c.1260-1262delGAT, c.1222>G a | 336 | Deletion was associated with time to treatment failure, but it was restored by the c.1222G allele | [64] | |
| c.480G>C | 229 | c.480GG associated with high rate of loss of response or treatment failure | [14] | |
| c.1260-1262delGAT, c.1222A>G | 153 | c.1222AA/AG genotypes associated with longer time to MR | [65] | |
| Gene expression | 28 | Higher levels of mRNA were observed in patients who achieved major and complete MR | [29] | |
| Gene expression | 70 | Highest pre-treatment mRNA levels were associated with better CCyR rates, PFS and OS b | [61] | |
| Transporter activity | - | High TA, better major MR at 60 months; low TA, lower OS, EFS and higher kinase domain mutation rate | [66] | |
| Haplotype c | 189 | Associated with both complete and major MR | [15] | |
| OCTN1 | c.1507C>T | 189 | c.1507TT was an adverse genotype for major MR in all patients and in Caucasians | [15] |
| SLC22A1, OCTN1, OATP1A2 | various | 189 | Complete and major MR were associated with a combination of SNPs | [15] |
| CYP3A5 | g.12083G>A | 229 | g.12083AA genotype was associated with lowest rates of major MR and CCyR | [14] |
2.2.4. Other Transmembrane Transporters
3. Discussion
| Study Phase | Issues | Causes | Possible Solutions |
|---|---|---|---|
| Preclinical studies | Discrepancies in substrate affinity/role of transporters in TKIs kinetics at both the cellular and systemic level | Differences in cell lines, in in vivo models, and range of drug concentrations used | Adoption of homogenous in vitro and in vivo models Same experimental conditions |
| Clinical Trials | Contradictory results reported for the evaluation of the relationship between pharmacogenetic results and treatment outcome | Gene candidate strategy with limited number of genes/polymorphisms Definition of clinical endpoints has been changed over time Patients’ compliance and adherence have not been fully investigated | Increase the number of genes/polymorphisms investigated at one time Strict adherence to clinical guidelines for endpoints definition Check for patients’ compliance |
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Hernandez-Boluda, J.C.; Cervantes, F. Imatinib mesylate (Gleevec, Glivec): A new therapy for chronic myeloid leukemia and other malignancies. Drugs Today 2002, 38, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Piazza, R. How I treat newly diagnosed chronic myeloid leukemia in 2015. Am. J. Hematol. 2015, 90, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; O’Brien, S.G.; Guilhot, F.; Druker, B.J.; Branford, S.; Foroni, L.; Goldman, J.M.; Muller, M.C.; Radich, J.P.; Rudoltz, M.; et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Hanfstein, B.; Muller, M.C.; Hehlmann, R.; Erben, P.; Lauseker, M.; Fabarius, A.; Schnittger, S.; Haferlach, C.; Gohring, G.; Proetel, U.; et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (CML). Leukemia 2012, 26, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Hochhaus, A.; Hughes, T.P.; Clark, R.E.; Etienne, G.; Kim, D.W.; Flinn, I.W.; Kurokawa, M.; Moiraghi, B.; Yu, R.; et al. Nilotinib vs. imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia 2012, 26, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Ibrahim, A.R.; Lucas, C.; Gerrard, G.; Wang, L.; Szydlo, R.M.; Clark, R.E.; Apperley, J.F.; Milojkovic, D.; Bua, M.; et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J. Clin. Oncol. 2012, 30, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Breccia, M.; Alimena, G.; Baccarani, M.; Bocchia, M.; Di Raimondo, F.; Gambacorti-Passerini, C.; Gozzini, A.; Morra, E.; Pane, F.; Pregno, P.; et al. Current management of CML patients: Summary of the Italian Consensus Meeting held in Rome, April 11–12, 2013. Crit. Rev. Oncol. Hematol. 2014, 90, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Hayes, M.; Resta, D.; Racine-Poon, A.; Druker, B.J.; Talpaz, M.; Sawyers, C.L.; Rosamilia, M.; Ford, J.; Lloyd, P.; et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J. Clin. Oncol. 2004, 22, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P.; Faber, E.; Kosztyu, P. Interactions of N-desmethyl imatinib, an active metabolite of imatinib, with P-glycoprotein in human leukemia cells. Ann. Hematol. 2011, 90, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Bolton, A.E.; Peng, B.; Hubert, M.; Krebs-Brown, A.; Capdeville, R.; Keller, U.; Seiberling, M. Effect of rifampicin on the pharmacokinetics of imatinib mesylate (Gleevec, STI571) in healthy subjects. Cancer Chemother. Pharmacol. 2004, 53, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Dutreix, C.; Peng, B.; Mehring, G.; Hayes, M.; Capdeville, R.; Pokorny, R.; Seiberling, M. Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother. Pharmacol. 2004, 54, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Skoglund, K.; Rommel, F.; Mirghani, R.A.; Lotfi, K. CYP3A activity influences imatinib response in patients with chronic myeloid leukemia: A pilot study on in vivo CYP3A activity. Eur. J. Clin. Pharmacol. 2010, 66, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sriharsha, L.; Xu, W.; Kamel-Reid, S.; Liu, X.; Siminovitch, K.; Messner, H.A.; Lipton, J.H. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin. Cancer Res. 2009, 15, 4750–4758. [Google Scholar] [CrossRef] [PubMed]
- Angelini, S.; Soverini, S.; Ravegnini, G.; Barnett, M.; Turrini, E.; Thornquist, M.; Pane, F.; Hughes, T.P.; White, D.L.; Radich, J.; et al. Association between imatinib transporters and metabolizing enzymes genotype and response in newly diagnosed chronic myeloid leukemia patients receiving imatinib therapy. Haematologica 2013, 98, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Cameron, M.D. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1175–1193. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, N.I.; Dorer, D.J.; Niland, K.; Haluska, F.; Sonnichsen, D. Effects of ketoconazole on the pharmacokinetics of ponatinib in healthy subjects. J. Clin. Pharmacol. 2013, 53, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Abbas, R.; Hug, B.A.; Leister, C.; Burns, J.; Sonnichsen, D. Effect of ketoconazole on the pharmacokinetics of oral bosutinib in healthy subjects. J. Clin. Pharmacol. 2011, 51, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Yin, O.Q.; Smith, T.; Sethuraman, V.; Grouss, K.; Galitz, L.; Harrell, R.; Schran, H. Effects of rifampin and ketoconazole on the pharmacokinetics of nilotinib in healthy participants. J. Clin. Pharmacol. 2011, 51, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Abbas, R.; Boni, J.; Sonnichsen, D. Effect of rifampin on the pharmacokinetics of bosutinib, a dual Src/Abl tyrosine kinase inhibitor, when administered concomitantly to healthy subjects. Drug Metabol. Personal. Ther. 2015, 30, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saunders, V.A.; Leclercq, T.M.; Hughes, T.P.; White, D.L. Ponatinib is not transported by ABCB1, ABCG2 or OCT-1 in CML cells. Leukemia 2015, 29, 1792–1794. [Google Scholar] [CrossRef] [PubMed]
- Illmer, T.; Schaich, M.; Platzbecker, U.; Freiberg-Richter, J.; Oelschlagel, U.; von Bonin, M.; Pursche, S.; Bergemann, T.; Ehninger, G.; Schleyer, E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 2004, 18, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S.V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, S.; Dulucq, S.; Pasquet, J.M.; Lagarde, V.; Molimard, M.; Mahon, F.X. From in vitro to in vivo: Intracellular determination of imatinib and nilotinib may be related with clinical outcome. Leukemia 2013, 27, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Saunders, V.A.; Hughes, T.P.; White, D.L. Degree of kinase inhibition achieved in vitro by imatinib and nilotinib is decreased by high levels of ABCB1 but not ABCG2. Leuk. Lymphoma 2013, 54, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Shugarts, S.; Benet, L.Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009, 26, 2039–2054. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Bouchet, S.; Turcq, B.; Lippert, E.; Etienne, G.; Reiffers, J.; Molimard, M.; Krajinovic, M.; Mahon, F.X. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 2008, 112, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Vivona, D.; Lima, L.T.; Rodrigues, A.C.; Bueno, C.T.; Alcantara, G.K.; Barros, L.S.R.; Hungria, V.T.D.M.; Chiattone, C.S.; Chauffaille, M.L.L.F.; Guerra-Shinohara, E.M. ABCB1 haplotypes are associated with P-gp activity and affect a major molecular response in chronic myeloid leukemia patients treated with a standard dose of imatinib. Oncol. Lett. 2014, 7, 1313–1319. [Google Scholar] [PubMed]
- Au, A.; Aziz Baba, A.; Goh, A.S.; Wahid Fadilah, S.A.; Teh, A.; Rosline, H.; Ankathil, R. Association of genotypes and haplotypes of multi-drug transporter genes ABCB1 and ABCG2 with clinical response to imatinib mesylate in chronic myeloid leukemia patients. Biomed. Pharmacother. 2014, 68, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Elsalakawy, W.A. ABCB1 haplotypes but not individual SNPs predict for optimal response/failure in Egyptian patients with chronic-phase chronic myeloid leukemia receiving imatinib mesylate. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.N.; Li, J.Y.; Miao, K.R.; Qiao, C.; Zhang, S.J.; Qiu, H.R.; Qian, S.X. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Med. Oncol. 2011, 28, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Vine, J.; Cohen, S.B.; Ruchlemer, R.; Goldschmidt, N.; Levin, M.; Libster, D.; Gural, A.; Gatt, M.E.; Lavie, D.; Ben-Yehuda, D.; et al. Polymorphisms in the human organic cation transporter and the multidrug resistance gene: Correlation with imatinib levels and clinical course in patients with chronic myeloid leukemia. Leuk. Lymphoma 2014, 55, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.J.; Lim, M.; Sohn, S.K.; Moon, J.H.; Oh, S.J.; Kim, B.S.; Ryoo, H.M.; Chung, J.S.; Joo, Y.D.; Bang, S.M.; et al. Influence of enzyme and transporter polymorphisms on trough imatinib concentration and clinical response in chronic myeloid leukemia patients. Ann. Oncol. 2013, 24, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, K.; Moreno, S.B.; Baytar, M.; Jonsson, J.I.; Green, H. ABCB1 haplotypes do not influence transport or efficacy of tyrosine kinase inhibitors in vitro. Pharmagenom. Pers. Med. 2013, 6, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Menon-Andersen, D.; Mondick, J.T.; Jayaraman, B.; Thompson, P.A.; Blaney, S.M.; Bernstein, M.; Bond, M.; Champagne, M.; Fossler, M.J.; Barrett, J.S. Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother. Pharmacol. 2009, 63, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Petain, A.; Kattygnarath, D.; Azard, J.; Chatelut, E.; Delbaldo, C.; Geoerger, B.; Barrois, M.; Seronie-Vivien, S.; LeCesne, A.; Vassal, G.; et al. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin. Cancer Res. 2008, 14, 7102–7109. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, A.; Polillo, M.; Capecchi, M.; Cervetti, G.; Barate, C.; Angelini, S.; Guerrini, F.; Fontanelli, G.; Arici, R.; Ciabatti, E.; et al. The c.480C>G polymorphism of hOCT1 influences imatinib clearance in patients affected by chronic myeloid leukemia. Pharmacogenom. J. 2014, 14, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.; Chan, J.Y.; Lin, K.; Heng, C.C.; Chowbay, B. SLC22A1-ABCB1 haplotype profiles predict imatinib pharmacokinetics in Asian patients with chronic myeloid leukemia. PLoS ONE 2012, 7, e51771. [Google Scholar] [CrossRef] [PubMed]
- Gurney, H.; Wong, M.; Balleine, R.L.; Rivory, L.P.; McLachlan, A.J.; Hoskins, J.M.; Wilcken, N.; Clarke, C.L.; Mann, G.J.; Collins, M.; et al. Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clin. Pharmacol. Ther. 2007, 82, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Sodani, K.; Wang, S.R.; Kuang, Y.H.; Ashby, C.R., Jr.; Chen, X.; Chen, Z.S. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem. Pharmacol. 2009, 78, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.K.; Shi, C.J.; Zhang, H.; Hu, Y.P.; Chen, Y.F.; Fu, L.W. Nilotinib enhances the efficacy of conventional chemotherapeutic drugs in CD34(+)CD38(−) stem cells and ABC transporter overexpressing leukemia cells. Molecules 2014, 19, 3356–3375. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Abuznait, A.H.; Singh, S.; Xiao, Z.J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Dolezel, P.; Mlejnek, P. Can P-glycoprotein mediate resistance to nilotinib in human leukaemia cells? Pharmacol. Res. 2013, 67, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Galeotti, L.; Ceccherini, F.; Polillo, M.; Baratè, C.; Fontanelli, G.; Guerrini, F.; Arici, R.; Barsotti, S.; Ricci, F.; et al. Transmembrane transporters hOCT1 and SLCO1B3 polymorphisms could have a role in patients affected by chronic myeloid leukemia treated with nilotinib. Blood 2014, 124, 3608. [Google Scholar]
- Hiwase, D.K.; Saunders, V.A.; Nievergall, E.; Ross, D.D.; White, D.L.; Hughes, T.P. Dasatinib targets chronic myeloid leukemia-CD34+ progenitors as effectively as it targets mature cells. Haematologica 2013, 98, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van Waterschoot, R.A.; van Tilburg, V.A.; Hillebrand, M.J.; Lankheet, N.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin. Cancer Res. 2009, 15, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Perini, P.; Ceccon, M.; Piazza, R.; Rigolio, R.; Mauri, M.; Boschelli, F.; Giannoudis, A.; Gambacorti-Passerini, C. In vitro and in vivo identification of ABCB1 as an efflux transporter of bosutinib. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Scharenberg, C.; Dohse, M.; Robey, R.W.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; Wang, Y.; Wennemuth, G.; Burchert, A.; et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007, 21, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Germain, G.S.; Harwood, F.C.; Schuetz, J.D.; Stewart, C.F.; Buchdunger, E.; Traxler, P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004, 64, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Nooter, K. Pharmacokinetic resistance to imatinib mesylate: Role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinib. Cell Cycle 2004, 3, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Miura, M.; Scott, S.A.; Kagaya, H.; Kameoka, Y.; Tagawa, H.; Saitoh, H.; Fujishima, N.; Yoshioka, T.; Hirokawa, M.; et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J. Hum. Genet. 2010, 55, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Takahashi, N.; Nishiwaki, K.; Hino, M.; Kashimura, M.; Wakita, H.; Hatano, Y.; Hirasawa, A.; Nakagawa, Y.; Itoh, K.; et al. A multicenter clinical study evaluating the confirmed complete molecular response rate in imatinib-treated patients with chronic phase chronic myeloid leukemia by using the international scale of real-time quantitative polymerase chain reaction. Haematologica 2013, 98, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Delord, M.; Rousselot, P.; Cayuela, J.M.; Sigaux, F.; Guilhot, J.; Preudhomme, C.; Guilhot, F.; Loiseau, P.; Raffoux, E.; Geromin, D.; et al. High imatinib dose overcomes insufficient response associated with ABCG2 haplotype in chronic myelogenous leukemia patients. Oncotarget 2013, 4, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Sun, J.; Howell, C.E.; Zhou, Q.Y.; He, Z.X.; Yang, T.; Chew, H.; Duan, W.; Zhou, Z.W.; Kanwar, J.R.; et al. Prediction of the likelihood of drug interactions with kinase inhibitors based on in vitro and computational studies. Fundam. Clin. Pharmacol. 2014, 28, 551–582. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Giannoudis, A.; Lane, S.; Williamson, P.; Pirmohamed, M.; Clark, R.E. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin. Pharmacol. Ther. 2008, 83, 258–264. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Zannettino, A.C.; Cambareri, A.C.; Quinn, S.R.; Manley, P.W.; Hughes, T.P. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): Reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 2006, 108, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Kerb, R.; Brinkmann, U.; Chatskaia, N.; Gorbunov, D.; Gorboulev, V.; Mornhinweg, E.; Keil, A.; Eichelbaum, M.; Koepsell, H. Identification of genetic variations of the human organic cation transporter hOCT1 and their functional consequences. Pharmacogenetics 2002, 12, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, A.; Wang, L.; Jorgensen, A.L.; Xinarianos, G.; Davies, A.; Pushpakom, S.; Liloglou, T.; Zhang, J.E.; Austin, G.; Holyoake, T.L.; et al. The hOCT1 SNPs M420del and M408V alter imatinib uptake and M420del modifies clinical outcome in imatinib-treated chronic myeloid leukemia. Blood 2013, 121, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Gerrard, G.; Alikian, M.; Alonso-Dominguez, J.; Ale, S.; Valganon, M.; Nteliopoulos, G.; White, D.; Marin, D.; Hedgley, C.; et al. A common novel splice variant of SLC22A1 (OCT1) is associated with impaired responses to imatinib in patients with chronic myeloid leukaemia. Br. J. Haematol. 2013, 163, 631–639. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Dang, P.; Engler, J.; Frede, A.; Zrim, S.; Osborn, M.; Saunders, V.A.; Manley, P.W.; Hughes, T.P. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J. Clin. Oncol. 2010, 28, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, M.V.; Seitz, T.; Bokelmann, K.; Mueller, T.; Brockmoller, J.; Koepsell, H. Does the haplotype Met408-Del420, which was apparently predictive for imatinib efficacy, really exist and how strongly may it affect OCT1 activity? Blood 2014, 123, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Crossman, L.C.; Druker, B.J.; Deininger, M.W.; Pirmohamed, M.; Wang, L.; Clark, R.E. hOCT 1 and resistance to imatinib. Blood 2005, 106, 1133–1134. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Bazeos, A.; Mahon, F.X.; Eliasson, L.; Milojkovic, D.; Bua, M.; Apperley, J.F.; Szydlo, R.; Desai, R.; Kozlowski, K.; et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J. Clin. Oncol. 2010, 28, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Cortes, J.E.; Yao, H.; Zhang, L.; Reddy, N.G.; Jabbour, E.; Kantarjian, H.M.; Jones, D. Predictors of primary imatinib resistance in chronic myelogenous leukemia are distinct from those in secondary imatinib resistance. J. Clin. Oncol. 2009, 27, 3642–3649. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Wagenaar, E.; Mol, C.A.; Buitelaar, M.; Koepsell, H.; Smit, J.W.; Schinkel, A.H. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 (Slc22a1)) gene. Mol. Cell. Biol. 2001, 21, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Wagenaar, E.; van Eijl, S.; Schinkel, A.H. Deficiency in the organic cation transporters 1 and 2 (Oct1/Oct2 (Slc22a1/Slc22a2)) in mice abolishes renal secretion of organic cations. Mol. Cell. Biol. 2003, 23, 7902–7908. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Franke, R.M.; Filipski, K.K.; Hu, C.; Orwick, S.J.; de Bruijn, E.A.; Burger, H.; Baker, S.D.; Sparreboom, A. Interaction of imatinib with human organic ion carriers. Clin. Cancer Res. 2008, 14, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Venables, A.; Zrim, S.; Zannettino, A.; Lynch, K.; Manley, P.W.; Hughes, T. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: Higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood 2007, 110, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Hughes, T.P. OCT-1 activity measurement provides a superior imatinib response predictor than screening for single-nucleotide polymorphisms of OCT-1. Leukemia 2010, 24, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Schaeffeler, E.; van der Kuip, H.; Cascorbi, I.; Bruhn, O.; Kneba, M.; Pott, C.; Hofmann, U.; Volk, C.; Hu, S.; et al. Cellular uptake of imatinib into leukemic cells is independent of human organic cation transporter 1 (OCT1). Clin. Cancer Res. 2014, 20, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Jordanides, N.E.; Giannoudis, A.; Lucas, C.M.; Hatziieremia, S.; Harris, R.J.; Jorgensen, H.G.; Holyoake, T.L.; Pirmohamed, M.; Clark, R.E.; et al. Nilotinib concentration in cell lines and primary CD34(+) chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia 2009, 23, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, A.; Davies, A.; Lucas, C.M.; Harris, R.J.; Pirmohamed, M.; Clark, R.E. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): Implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood 2008, 112, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Fava, C.; Rege-Cambrin, G.; Saglio, G. The choice of first-line chronic myelogenous leukemia treatment. Ann. Hematol. 2015, 94 (Suppl. 2), S123–S131. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Gui, X.; He, X.; Chen, Y.; Pan, J.; Qiu, H.; Wu, D.; Chen, S.; Guo, L.; Cen, J. Deep molecular responses achieved in chronic myeloid leukemia in chronic phase patients with BCR-ABL1 >10% at 3 months who are early switched to nilotinib. Hematology 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I. Functional significance of genetic polymorphisms in P-glycoprotein (MDR1, ABCB1) and breast cancer resistance protein (BCRP, ABCG2). Drug Metab. Pharmacokinet. 2012, 27, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.J.; Bachtiar, M.; Wang, J.; Sim, T.S.; Chong, S.S.; Lee, C.G. An update on ABCB1 pharmacogenetics: Insights from a 3D model into the location and evolutionary conservation of residues corresponding to SNPs associated with drug pharmacokinetics. Pharmacogenom. J. 2011, 11, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Zhang, H.; Zhang, Y.; Wei, D.Y. ABCB1 (1199G>A) polymorphism regulates the efficacy of docetaxel and imatinib mesylate in HEK293 recombinant cell lines. Cancer Chemother. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, P.; Beijnen, J.H.; Schellens, J.H. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 2006, 27, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hatziieremia, S.; Jordanides, N.E.; Holyoake, T.L.; Mountford, J.C.; Jorgensen, H.G. Inhibition of MDR1 does not sensitize primitive chronic myeloid leukemia CD34+ cells to imatinib. Exp. Hematol. 2009, 37, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, T.; Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Picard, S.; Titier, K.; Etienne, G.; Teilhet, E.; Ducint, D.; Bernard, M.A.; Lassalle, R.; Marit, G.; Reiffers, J.; Begaud, B.; et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 2007, 109, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Di Paolo, A.; Liu, H.H.; Polillo, M.; Clermont, P.L.; Guerrini, F.; Ciabatti, E.; Ricci, F.; Baratè, C.; Fontanelli, G.; et al. Polycomb genes are associated with response to imatinib in chronic myeloid leukemia. Epigenomics 2015, in press. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polillo, M.; Galimberti, S.; Baratè, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Pharmacogenetics of BCR/ABL Inhibitors in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2015, 16, 22811-22829. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922811
Polillo M, Galimberti S, Baratè C, Petrini M, Danesi R, Di Paolo A. Pharmacogenetics of BCR/ABL Inhibitors in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2015; 16(9):22811-22829. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922811
Chicago/Turabian StylePolillo, Marialuisa, Sara Galimberti, Claudia Baratè, Mario Petrini, Romano Danesi, and Antonello Di Paolo. 2015. "Pharmacogenetics of BCR/ABL Inhibitors in Chronic Myeloid Leukemia" International Journal of Molecular Sciences 16, no. 9: 22811-22829. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160922811