
New Potential Biomarker for Methasterone Misuse in Human Urine by Liquid Chromatography Quadrupole Time of Flight Mass Spectrometry

Abstract
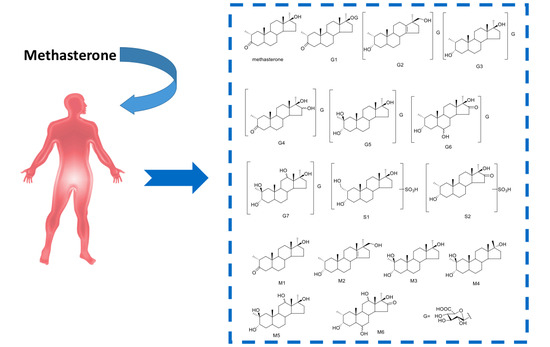
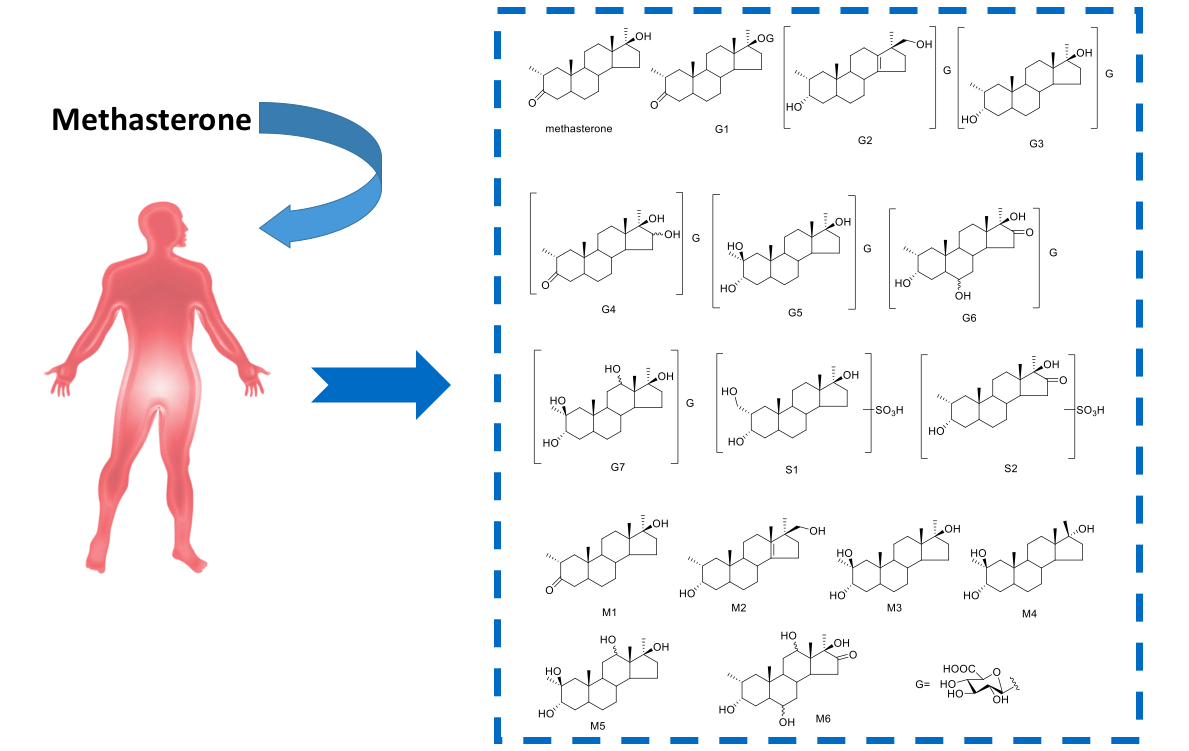
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
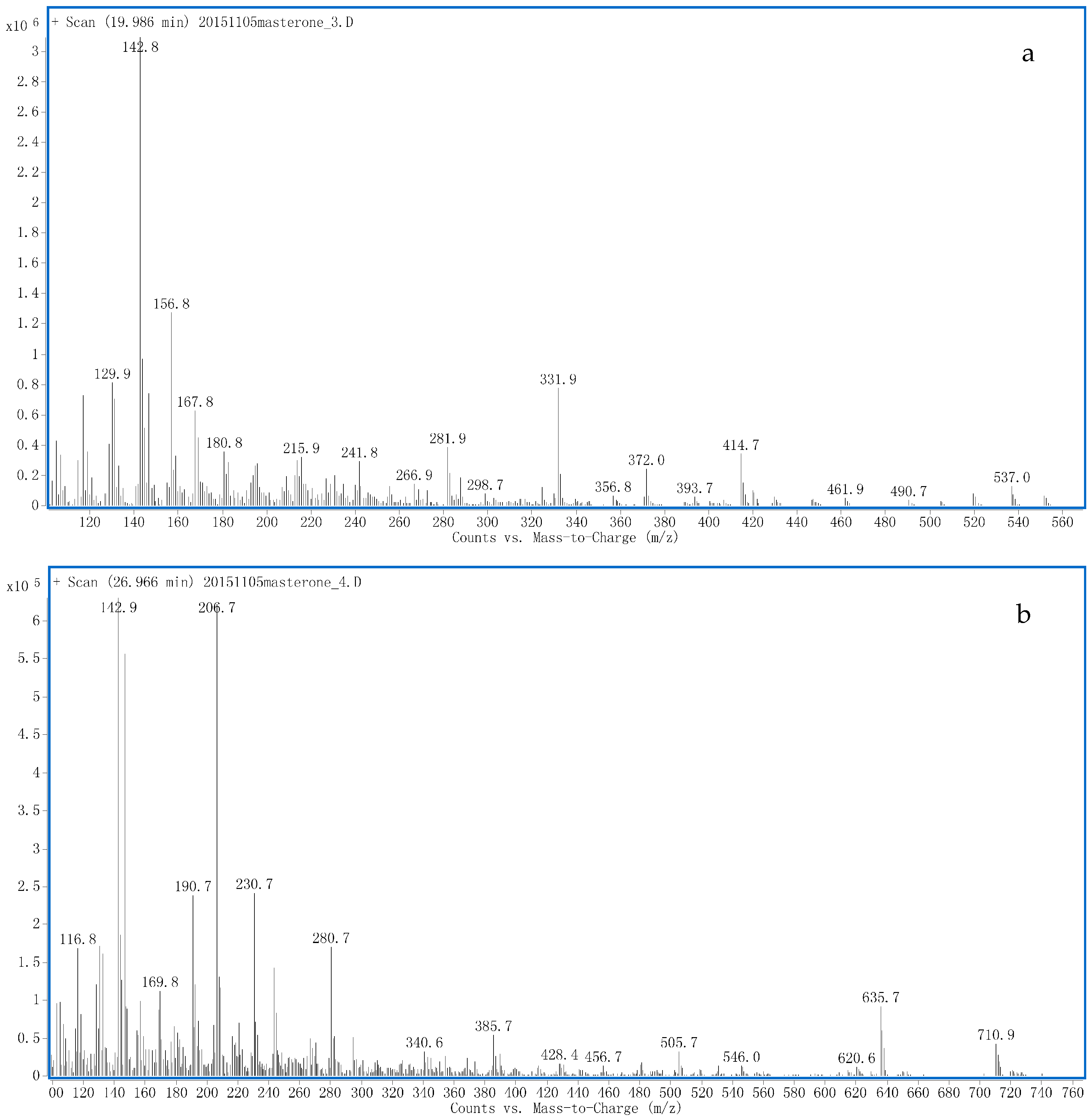
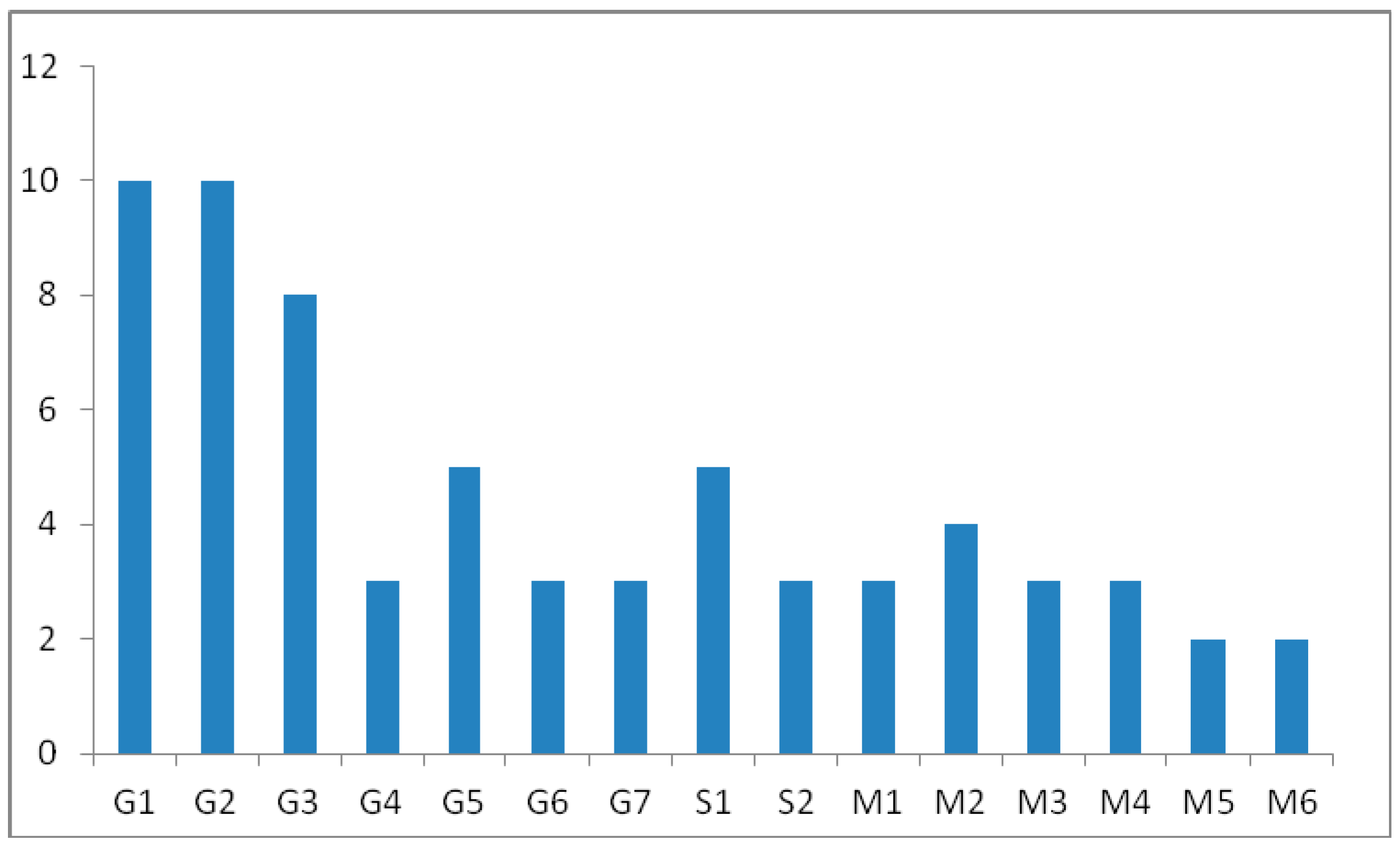
2. Results
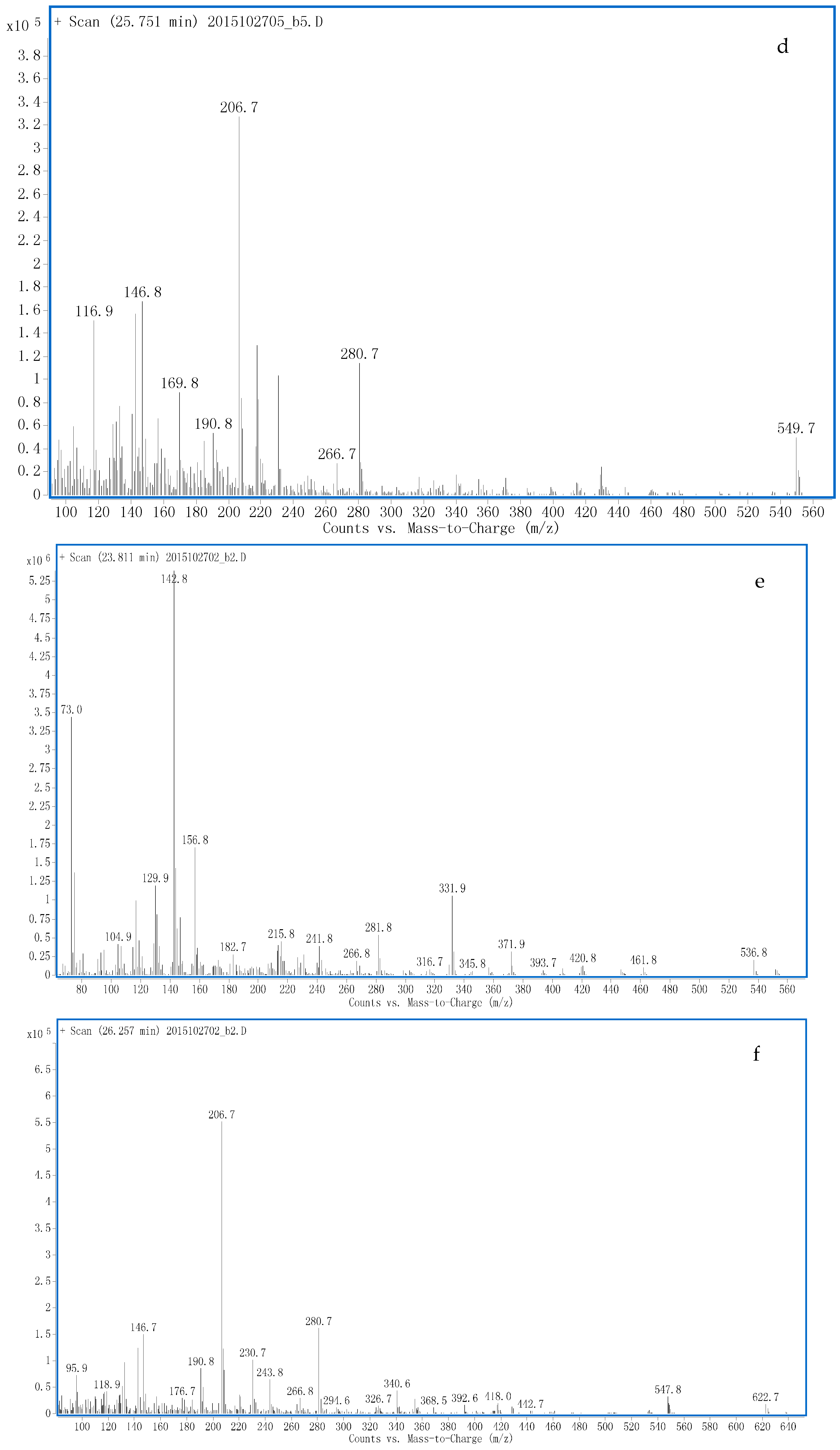
Proposed Metabolic Pathway and Application for Doping Control
3. Discussion
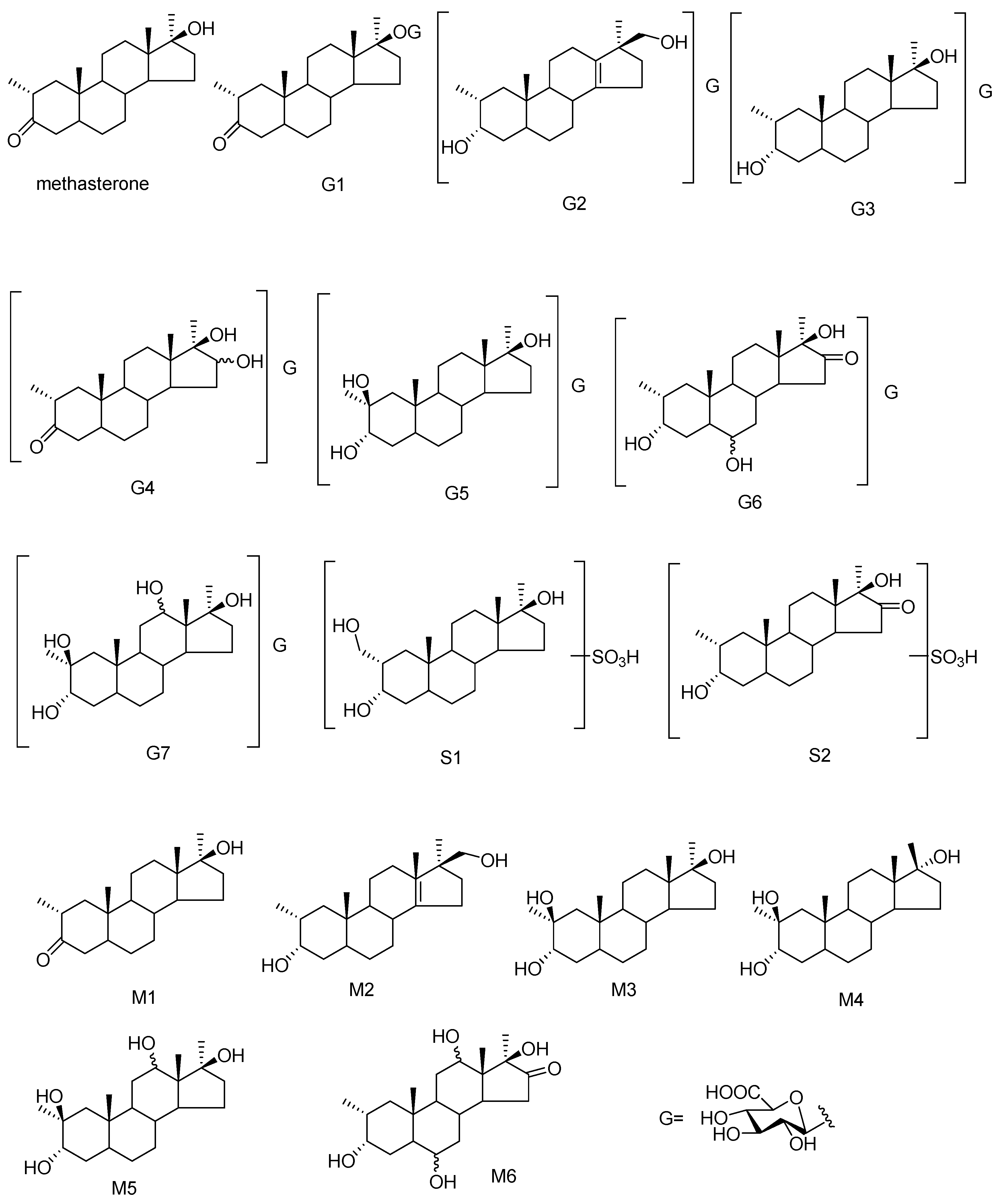
3.1. Identification of Potential Glucuronide-Conjugated Metabolites
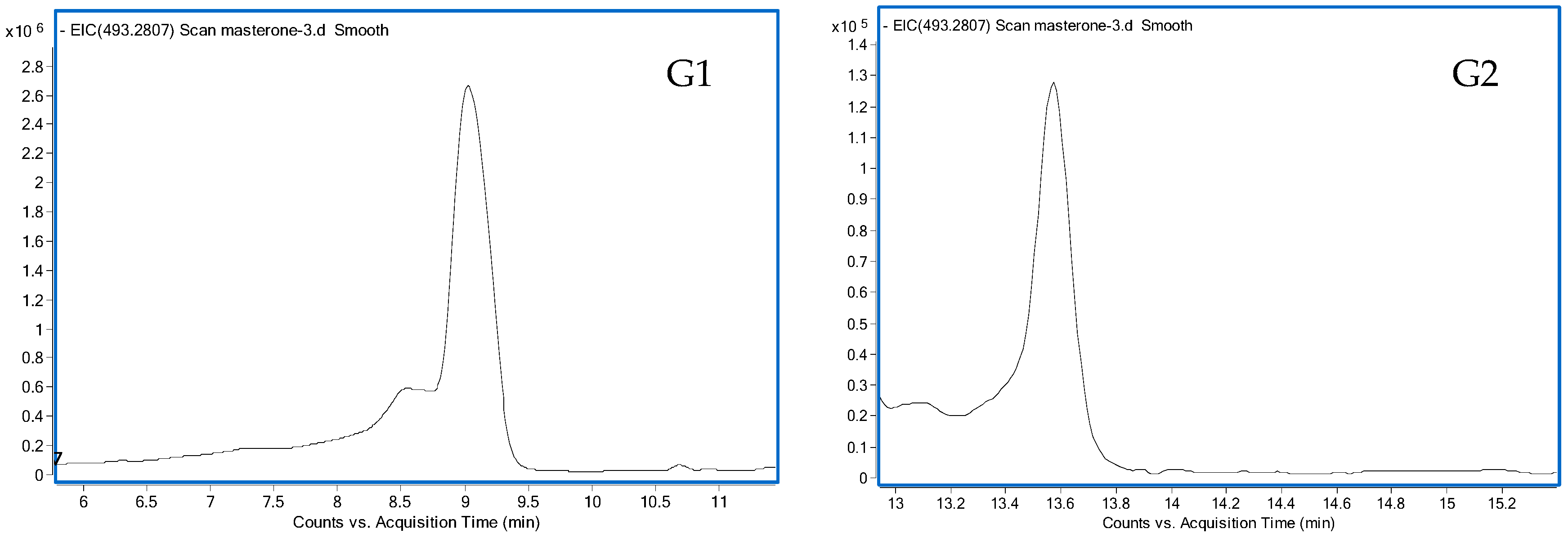
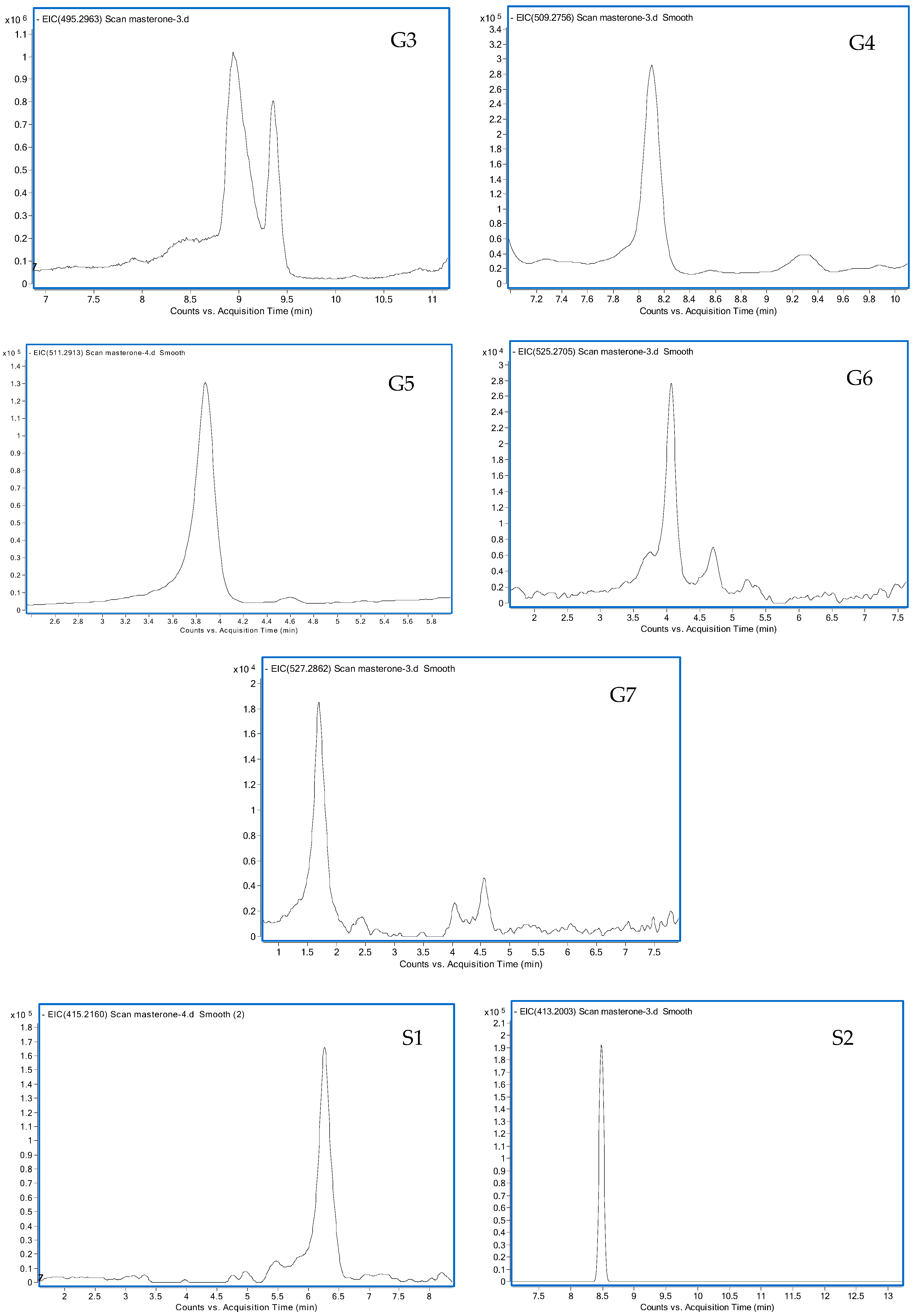
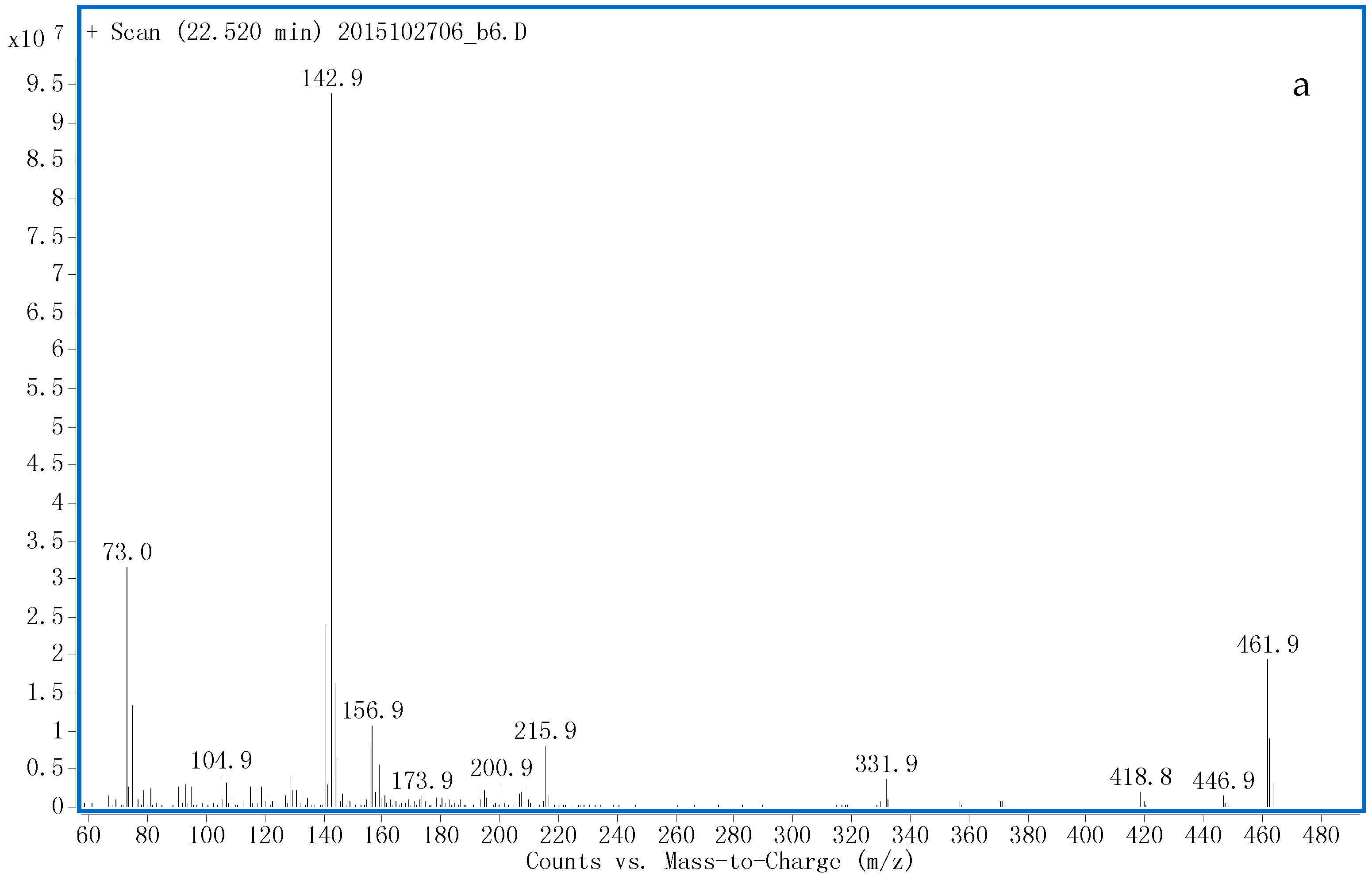
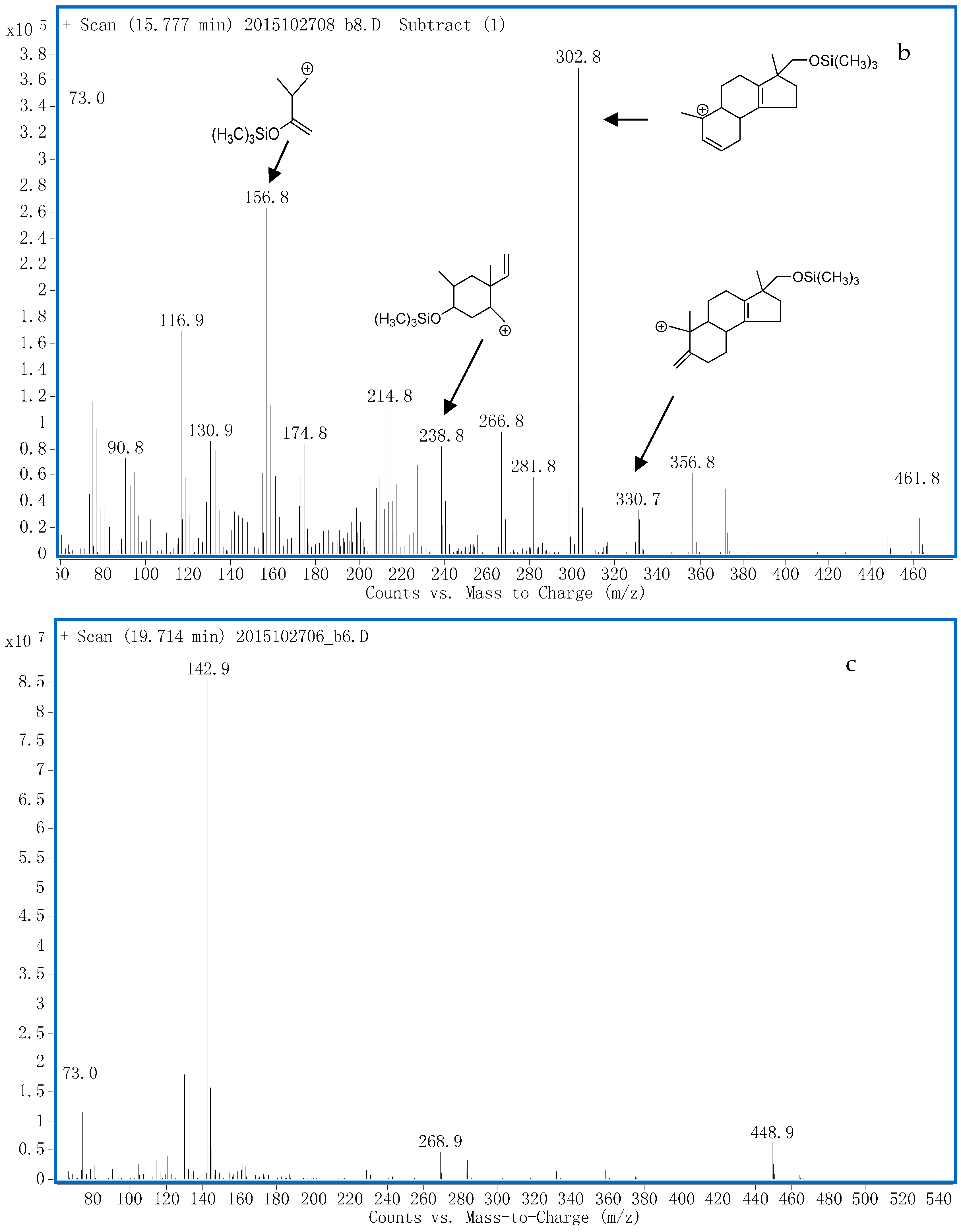
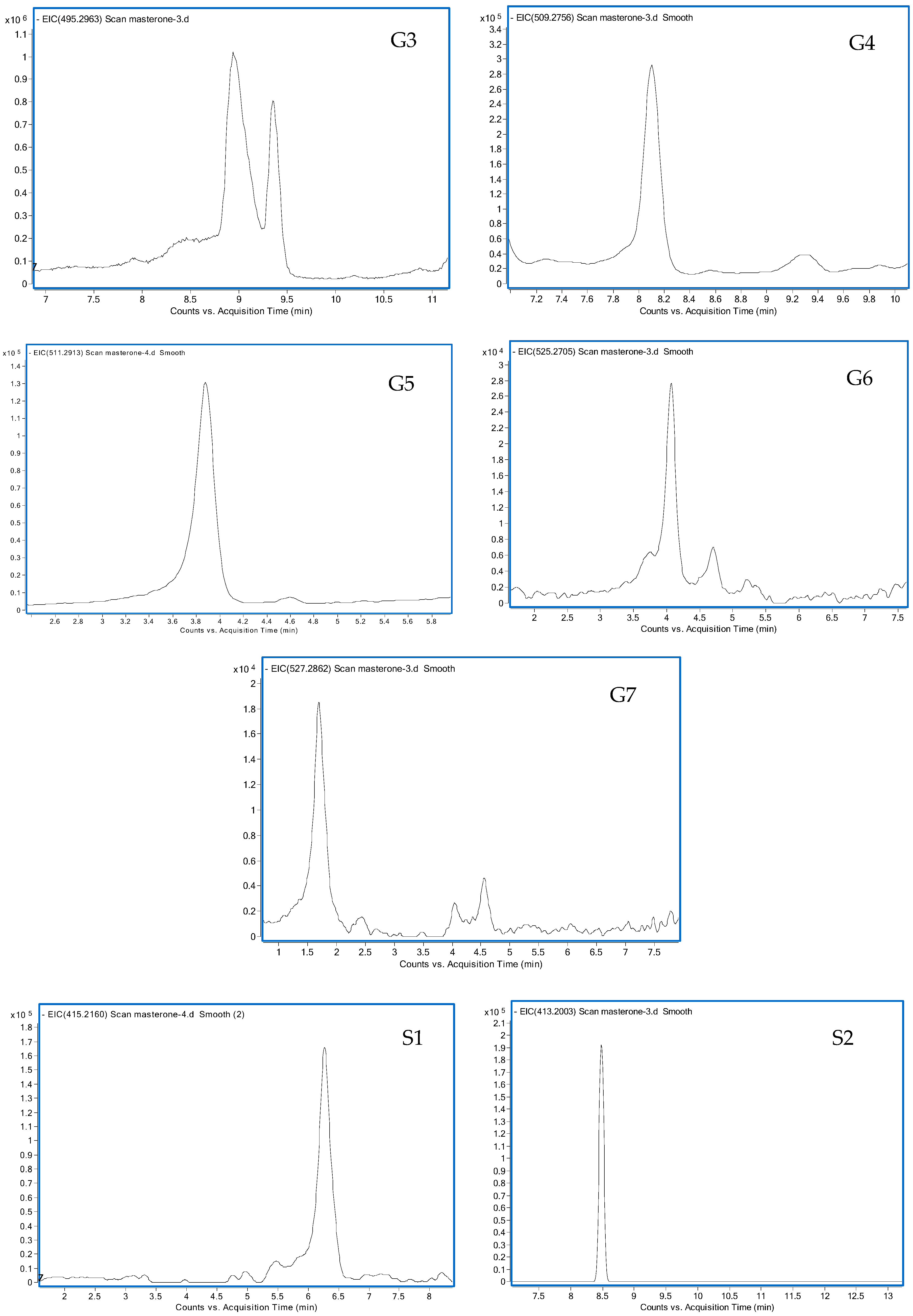
3.1.1. G1 and G2
3.1.2. G3
3.1.3. G4
3.1.4. G5
3.1.5. G6
3.1.6. G7
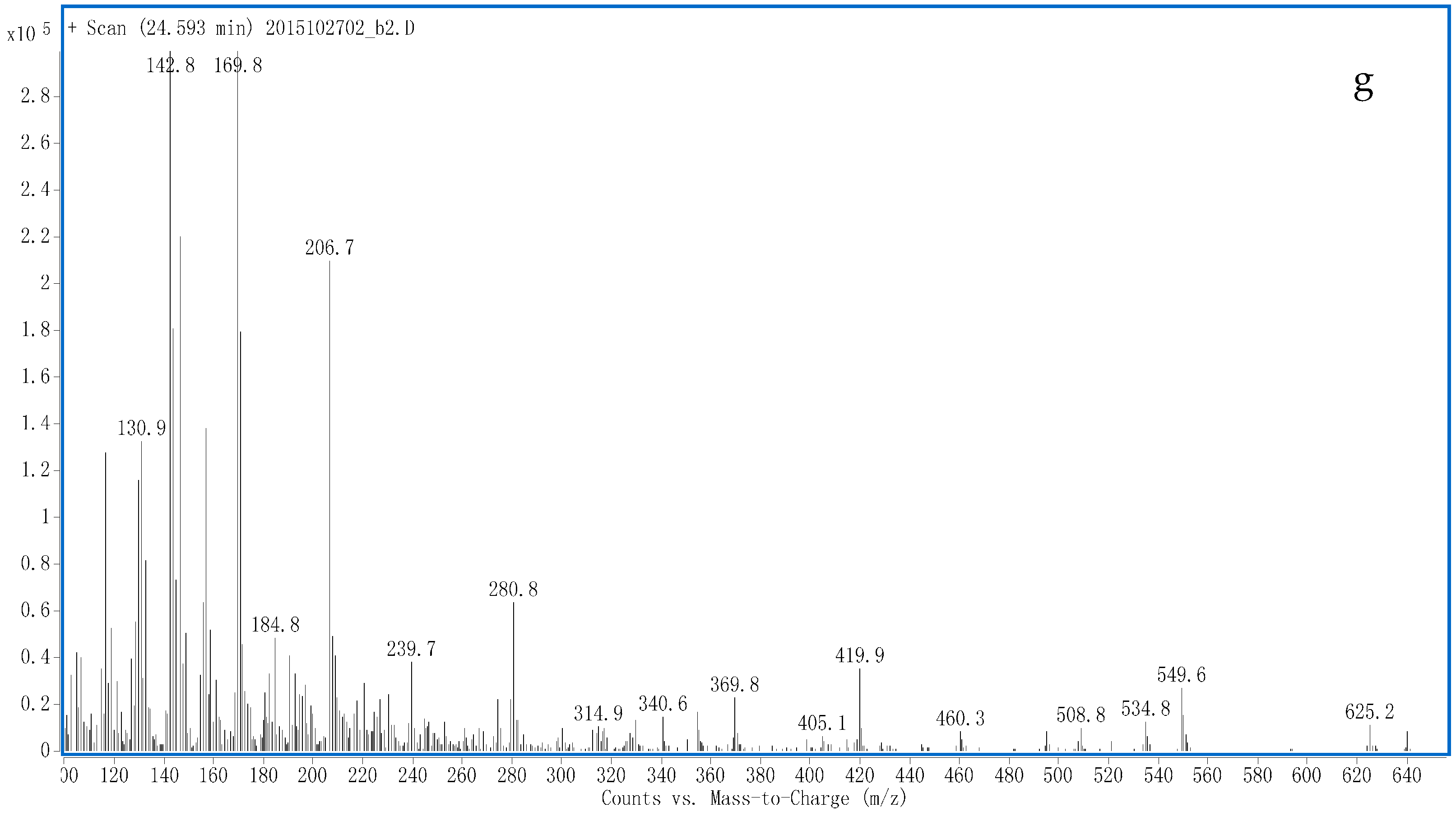
3.2. Identification of Potential Sulfate-Conjugated Metabolites
3.2.1. S1
3.2.2. S2
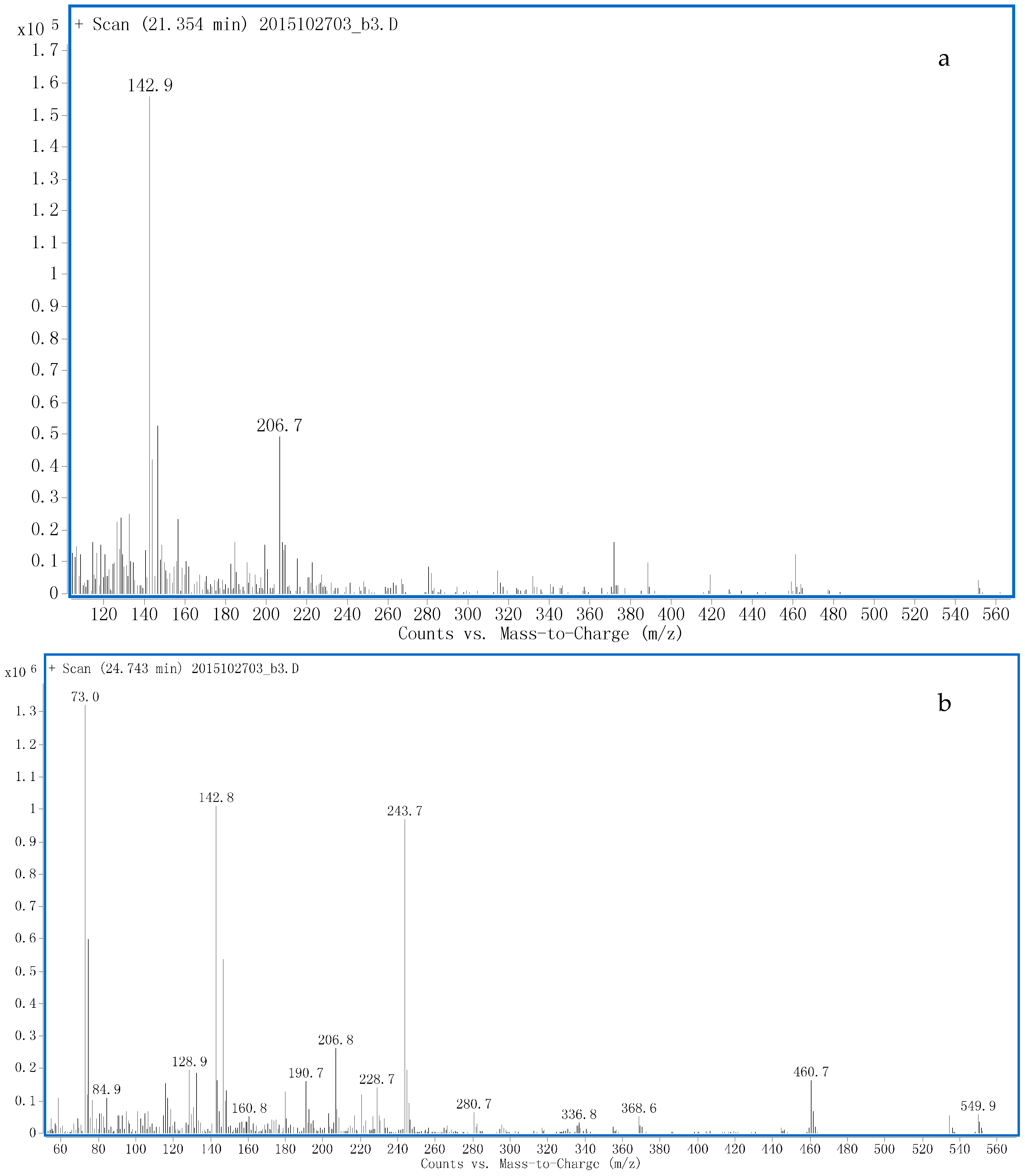
3.3. Identification of Potential Free Metabolites
3.3.1. M4
3.3.2. M6
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrumentation
4.2.1. LC/QTOF-MS Studies
4.2.2. GC-MS Analysis
4.3. Sample Preparation
4.3.1. Unconjugated Steroids
4.3.2. Glucuronide and Sulfate-Conjugated Steroids
4.4. LC Fractionation and Enzymatic Hydrolysis
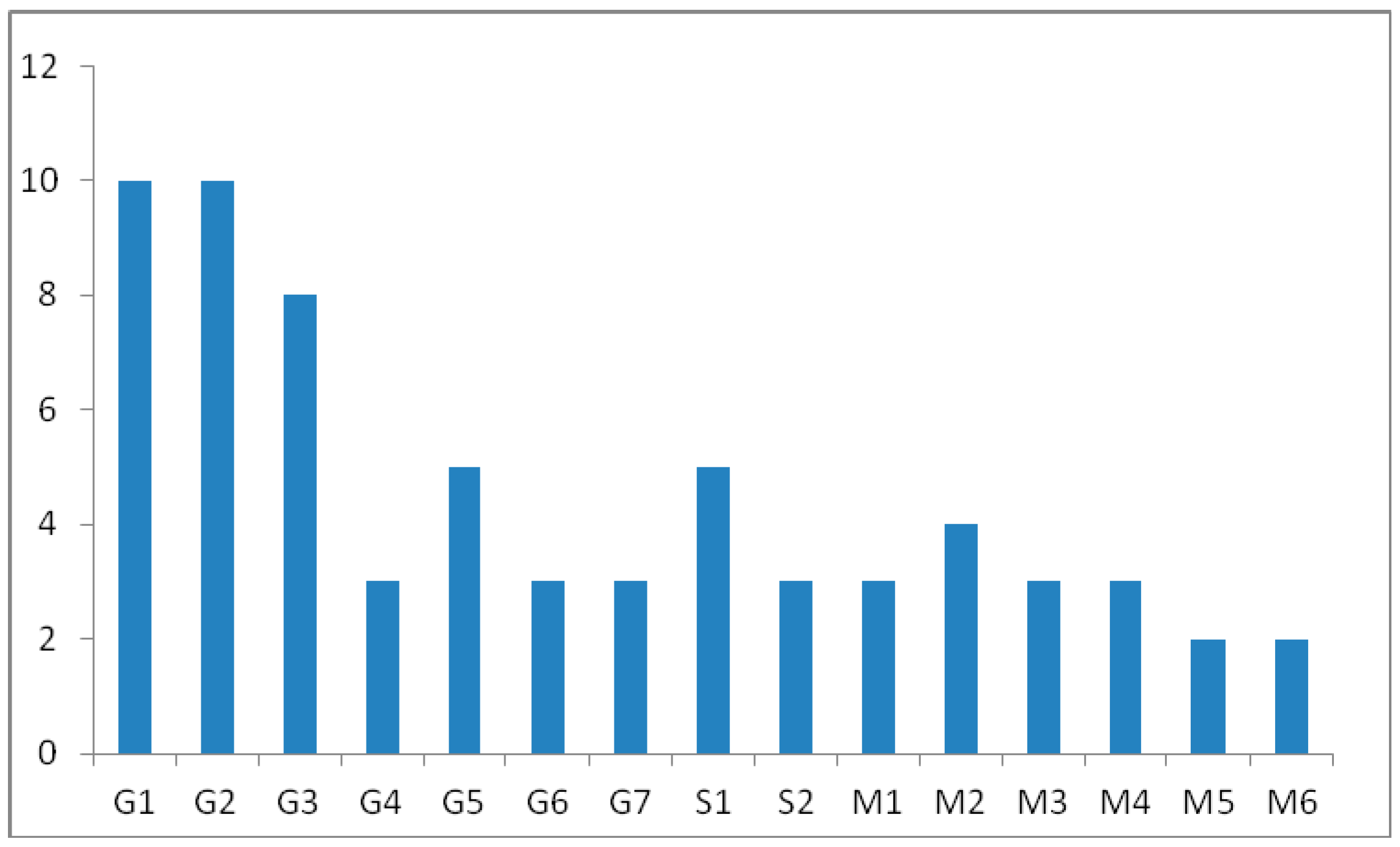
4.5. Excretion Studies
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schänzer, W. Metabolism of anabolic androgenic steroids. Clin. Chem. 1996, 42, 1001–1020. [Google Scholar] [PubMed]
- Fragkaki, A.G.; Angelis, Y.S.; Koupparis, M.; Tsantili-Kakoulidou, A. Structural characteristics of anabolic androgenic steroids contributing to binding to the androgen receptor and to their anabolic and androgenic activities applied modifications in the steroidal structure. Steroids 2009, 74, 172–197. [Google Scholar] [CrossRef] [PubMed]
- Schänzer, W.; Opfermann, G.; Donike, M. 17-Epimerization of 17α-methyl anabolic steroids in humans: Metablolism and synthesis of 17α-hydroxy-17β-methyl steroids. Steroids 1992, 57, 537–550. [Google Scholar] [CrossRef]
- Geyer, H.; Parr, M.K.; Koehler, K.; Mareck, U.; Schänzer, W.; Thevis, M. Nutritional supplements cross-contaminated and faked with doping substances. J. Mass Spectrom. 2008, 43, 892–902. [Google Scholar] [CrossRef] [PubMed]
- World Anti-Doping Agency. The World Anti-Doping Code, The 2016 Prohibited List International Standard. Available online: https://wada-main-prod.s3.amazonaws.com/resources/files/wada-2016-prohibited-list-en.pdf (accessed on 1 July 2016).
- Robles-Diaz, M.; Gonzalez-Jimenez, A.; Medina-Caliz, I.; Stephens, C.; García-Cortes, M.; García-Muñoz, B.; Ortega-Alonso, A.; Blanco-Reina, E.; Gonzalez-Grande, R.; Jimenez-Perez, M.; et al. Distinct phenotype of hepatotoxicity associated with illicit use of anabolic androgenic steroids. Aliment. Pharmacol. Ther. 2015, 41, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bylina, D.V.; Gryn, S.V.; Tkachuk, A.A.; Kruchek, Y.I. Detection of the methasterone and its metabolite in human urine by the gas chromatography/high resolution mass spectrometry (HRMS) method. Methods Objects Chem. Anal. 2012, 7, 87–93. [Google Scholar]
- Clarke, A.; Scarth, J.; Teale, P.; Pearce, C.; Hillyer, L. The use of in vitro technologies and high-resolution/accurate-mass LC-MS to screen for metabolites of “designer” steroids in the equine. Drug Test. Anal. 2011, 3, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Goudreault, D.; Poirier, D.; Ayotte, C. Identification of drostanolone and 17-methyldrostanolone metabolites produced by cryopreserved human hepatocytes. Steroids 2009, 74, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Lootens, L.; Meuleman, P.; Leroux-Roels, G.; Van Eenoo, P. Metabolic studies with promagnon, methylclostebol and methasterone in the uPA+/+-SCID chimeric mice. J. Steroid Biochem. Mol. Biol. 2011, 127, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Geldof, L.; Tudela, E.; Lootens, L.; Van Lysebeth, J.; Meuleman, P.; Leroux-Roels, G.; van Eenoo, P.; Deventer, K. In-vitro and in vivo metabolism studies of dimethazine. Biomed. Chromatogr. 2016, 30, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Geldof, L.; Lootens, L.; Polet, M.; Eichner, D.; Campbell, T.; Nair, V.; Botrè, F.; Meuleman, P.; Leroux-Roels, G.; Deventer, K.; Eenoo, P.V. Metabolism of methylstenbolone studied with human liver microsomes and the uPA+/+-SCID chimeric mouse model. Biomed. Chromatogr. 2014, 28, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Tudela, E.; Deventer, K.; Geldof, L.; Van Eenoo, P. Urinary detection of conjugated and unconjugated anabolic steroids by dilute-and-shoot liquid chromatography-high resolution mass spectrometry. Drug Test. Anal. 2015, 7, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Rodchenkov, G.; Sobolevsky, T.; Sizoi, V. New desiger anabolic steroids from internet. In Proceedings of the Manfred Donike Workshop 24th Colegne Workshop on Dope Analysis; SPORTVERLAG Strauẞ: Köln, Germany, 2006; pp. 141–150. [Google Scholar]
- Parr, M.K.; Opfermann, G.; Schänzer, W. Detection of new 17-alkylated anabolic steroids on WADA 2006 list. In Proceedings of the Manfred Donike Workshop 24th Colegne Workshop on Dope Analysis; SPORTVERLAG Strauẞ: Köln, Germany, 2006; pp. 249–258. [Google Scholar]
- Balcells, G.; Pozo, O.J.; Esquivel, A.; Kotronoulas, A.; Joglar, J.; Segura, J.; Ventura, R. Screening for anabolic steroids in sports: Analytical strategy based on the detection of phase I and phase II intact urinary metabolites by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2015, 1389, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Rzeppa, S.; Heinrich, G.; Hemmersbach, P. Analysis of anabolic androgenic steroids as sulfate conjugates using high performance liquid chromatography coupled to tandem mass spectrometry. Drug Test. Anal. 2015, 7, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Pozo, O.J.; Marcos, J.; Segura, J.; Ventura, R. Use of LC-MS/MS for the open detection of steroid metabolites conjugated with glucuronic acid. Anal. Chem. 2013, 85, 5005–5014. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.; Fabregat, A.; Pozo, O.J.; Marcos, J.; Segura, J.; Ventura, R. Analytical strategies based on mass spectrometric techniques for the study of steroid metabolism. Trends Anal. Chem. 2014, 53, 106–116. [Google Scholar] [CrossRef]
- Marcos, J.; Pozo, O.J. Current LC-MS methods and procedures applied to the identification of new steroid metabolites. J. Steroid Biochem. Mol. Biol. 2016, 162, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Masse, R.; Goudreault, D. Studies on anabolic steroids—11. 18-hydroxylated metabolites of mesterolone, methenolone and stenbolone: New steroids isolated from human urine. J. Steroid Biochem. Mol. Biol. 1992, 42, 399–410. [Google Scholar] [CrossRef]
- de Boer, D.; de Jong, E.G.; Maes, R.A.; van Rossum, J.M. The methyl-5 α-dihydrotestosterones mesterolone and drostanolone; gas chromatographic/mass spectrometric characterization of the urinary metabolites. J. Steroid Biochem. Mol. Biol. 1992, 42, 411–419. [Google Scholar] [CrossRef]
- Schänzer, W.; Geyer, H.; Fusshöller, G.; Halatcheva, N.; Kohler, M.; Parr, M.K.; Guddat, S.; Thomas, A.; Thevis, M. Mass spectrometric identification and characterization of a new long-term metabolite of metandienone in human urine. Rapid Commun. Mass Spectrom. 2006, 20, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Fragkaki, A.G.; Angelis, Y.S.; Tsantili-Kakoulidou, A.; Koupparis, M.; Georgakopoulos, C. Statistical analysis of fragmentation patterns of electron ionization mass spectra of enolized-trimethylsilylated androgenic steroids. Int. J. Mass Spectrom. 2009, 285, 58–69. [Google Scholar] [CrossRef]
- Yan, Y.; Rempel, D.L.; Holy, T.E.; Gross, M.L. Mass spectrometry combinations for structural characterization of sulfated-steroid metabolites. J. Am. Soc. Mass Spectrom. 2014, 25, 869–879. [Google Scholar] [CrossRef] [PubMed]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Lu, J.; Wu, Y.; Wang, X.; Xu, Y.; Zhang, Y.; Wang, Y. New Potential Biomarker for Methasterone Misuse in Human Urine by Liquid Chromatography Quadrupole Time of Flight Mass Spectrometry. Int. J. Mol. Sci. 2016, 17, 1628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101628
Zhang J, Lu J, Wu Y, Wang X, Xu Y, Zhang Y, Wang Y. New Potential Biomarker for Methasterone Misuse in Human Urine by Liquid Chromatography Quadrupole Time of Flight Mass Spectrometry. International Journal of Molecular Sciences. 2016; 17(10):1628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101628
Chicago/Turabian StyleZhang, Jianli, Jianghai Lu, Yun Wu, Xiaobing Wang, Youxuan Xu, Yinong Zhang, and Yan Wang. 2016. "New Potential Biomarker for Methasterone Misuse in Human Urine by Liquid Chromatography Quadrupole Time of Flight Mass Spectrometry" International Journal of Molecular Sciences 17, no. 10: 1628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101628