
Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity


Abstract
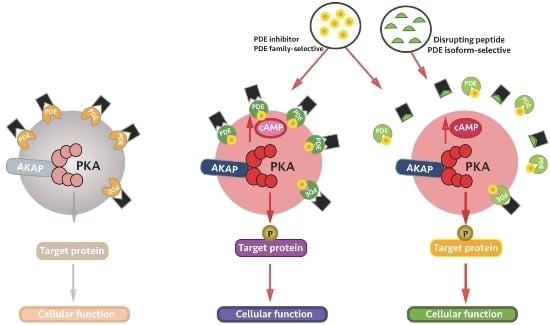
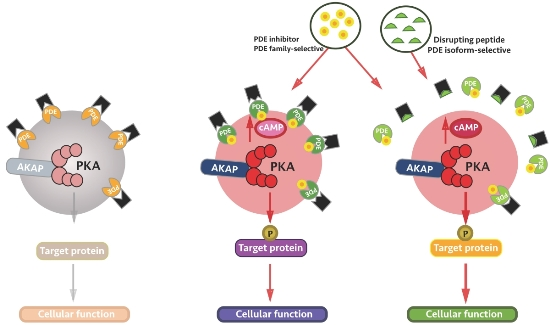
:
{kind=link}
{kind=link}
1. Introduction to Cyclic Nucleotides
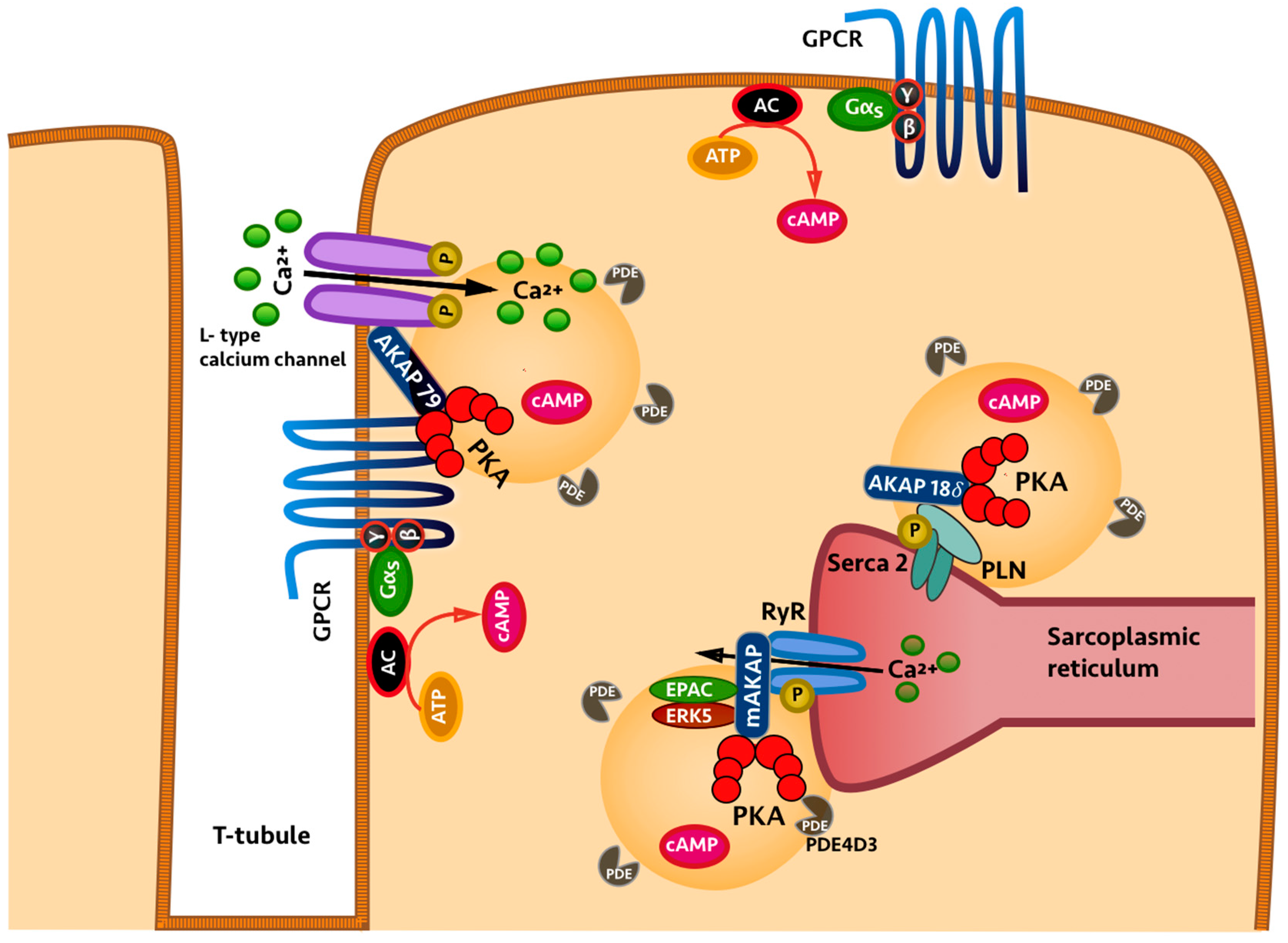
2. Compartmentalisation of Cyclic Nucleotides
3. Role of Phosphodiesterases in Cyclic Nucleotide Compartmentalisation
4. Local Inhibition of Phosphodiesterase Activity
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rall, T.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Phosphodiesterases and compartmentalised cAMP signalling in the heart. Eur. J. Cell Biol. 2006, 85, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 2011, 11, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Schneider, E.H.; Bähre, H. From canonical to non-canonical cyclic nucleotides as second messengers: Pharmacological implications. Pharmacol. Ther. 2015, 148, 154–184. [Google Scholar] [CrossRef] [PubMed]
- Beckert, U.; Grundmann, M.; Wolter, S.; Schwede, F.; Rehmann, H.; Kaever, V.; Kostenis, E.; Seifert, R. cNMP-AMs mimic and dissect bacterial nucleotidyl cyclase toxin effects. Biochem. Biophys. Res. Commun. 2014, 451, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Desch, M.; Schinner, E.; Kees, F.; Hofmann, F.; Seifert, R.; Schlossmann, J. Cyclic cytidine 3′,5′-monophosphate (cCMP) signals via cGMP kinase I. FEBS Lett. 2010, 584, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R. cCMP and cUMP: Emerging second messengers. Trends Biochem. Sci. 2015, 40, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.; Zaccolo, M. Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology 2014, 29, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Baillie, G.S.; Houslay, M.D. Mdm2 directs the ubiquitination of β-arrestin-sequestered cAMP phosphodiesterase-4D5. J. Biol. Chem. 2009, 284, 16170–16182. [Google Scholar] [CrossRef] [PubMed]
- Florio, V.A.; Sonnenburg, W.K.; Johnson, R.; Kwak, K.S.; Jensen, G.S.; Walsh, K.A.; Beavo, J.A. Phosphorylation of the 61-kDa calmodulin-stimulated cyclic nucleotide phosphodiesterase at serine 120 reduces its affinity for calmodulin. Biochemistry 1994, 33, 8948–8954. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Zaccolo, M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.; Baillie, G.S. Compartmentalisation of second messenger signalling pathways. Curr. Opin. Genet. Dev. 2014, 27, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, E.W.; Robison, G.A.A. The Role of Cyclic-3′, 5′-AMP in Responses to Catecholamines and Other Hormones. Pharmacol. Rev. 1966, 18, 145–161. [Google Scholar] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [PubMed]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yatani, A.; Dell’Acqua, M.L.; Sako, H.; Green, S.A.; Dascal, N.; Scott, J.D.; Hosey, M.M. cAMP-dependent regulation of cardiac l-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 1997, 19, 185–196. [Google Scholar] [CrossRef]
- Reuter, H. Ion channels in cardiac cell membranes. Annu. Rev. Physiol. 1984, 46, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Dodge, K.L.; Khouangsathiene, S.; Kapiloff, M.S.; Mouton, R.; Hill, E.V.; Houslay, M.D.; Langeberg, L.K.; Scott, J.D. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001, 20, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.V.; Christian, F.; Baillie, G.S. cAMP: Novel concepts in compartmentalised signalling. Semin. Cell Dev. Biol. 2012, 23, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.-S.; Hamdani, N.; Holewinski, R.; Jo, S.-H.; Danner, T.; Zhang, M.; Rainer, P.P. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kass, D.A. Phosphodiesterases and cardiac cGMP: Evolving roles and controversies. Trends Pharmacol. Sci. 2011, 32, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.K.; Gettemy, J.M.; Coghlan, V.M. Identification of cGMP-dependent protein kinase anchoring proteins (GKAPs). Biochem. Biophys. Res. Commun. 1998, 246, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; de Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2000, 2, 25–29. [Google Scholar] [PubMed]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004, 5, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Gambaryan, S.; Lohse, M.J. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat. Methods 2006, 3, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Adams, S.R.; Sawyer, C.L.; Lev-Ram, V.; Tsien, R.Y.; Dostmann, W.R. Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA 2001, 98, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hida, N.; Ozawa, T.; Umezawa, Y. Fluorescent indicators for cyclic GMP based on cyclic GMP-dependent protein kinase Iα and green fluorescent proteins. Anal. Chem. 2000, 72, 5918–5924. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jäger, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Sood, A.; McPhee, I.; Gall, I.; Perry, S.J.; Lefkowitz, R.J.; Houslay, M.D. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA 2003, 100, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Boess, F.G.; Hendrix, M.; van der Staay, F.-J.; Erb, C.; Schreiber, R.; van Staveren, W.; de Vente, J.; Prickaerts, J.; Blokland, A.; Koenig, G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 2004, 47, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T. Fluorescence resonance energy transfer–based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hatzelmann, A.; Schudt, C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J. Pharmacol. Exp. Ther. 2001, 297, 267–279. [Google Scholar] [PubMed]
- Huang, Z.; Ducharme, Y.; Macdonald, D.; Robichaud, A. The next generation of PDE4 inhibitors. Curr. Opin. Chem. Biol. 2001, 5, 432–438. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.M. Endocrine Cardiomyopathy: Response to Cyclic GMP PDE5 Inhibitors in Acromegaly Cardiomyopathy (SUM) ClinicalTrials.gov Identifier: NCT02611336. Available online: https://clinicaltrials.gov/ct2/show/NCT02611336?term=pde5&rank=40 (accessed on 30 June 2016).
- Gilead Sciences, G.S. Study of Add-on Ambrisentan Therapy to Background Phosphodiesterase Type-5 Inhibitor (PDE5i) Therapy in Pulmonary Arterial Hypertension (ATHENA-1) (ATHENA-1) ClinicalTrials.gov Identifier: NCT00617305. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00617305?term=pde5&rank=13 (accessed on 30 June 2016).
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Burgin, A.B.; Magnusson, O.T.; Stewart, L.J. Small molecule allosteric modulators of phosphodiesterase 4. In Phosphodiesterases as Drug Targets; Springer: Berlin Heidelberg, 2011; pp. 167–192. [Google Scholar]
- Pandit, J. PDE4: New structural insights into the regulatory mechanism and implications for the design of selective inhibitors. Phosphodiesterases Their Inhib. 2014, 29–44. [Google Scholar] [CrossRef]
- Sharma, V.K.; Rungta, P.; Prasad, A.K. Nucleic acid therapeutics: Basic concepts and recent developments. RSC Adv. 2014, 4, 16618–16631. [Google Scholar] [CrossRef]
- Lynch, M.J.; Baillie, G.S.; Mohamed, A.; Li, X.; Maisonneuve, C.; Klussmann, E.; van Heeke, G.; Houslay, M.D. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with β-arrestin to control the protein kinase A/AKAP79-mediated switching of the β2-adrenergic receptor to activation of ERK in HEK293B2 cells. J. Biol. Chem. 2005, 280, 33178–33189. [Google Scholar] [CrossRef] [PubMed]
- Pekkinen, M.; Ahlström, M.E.; Riehle, U.; Huttunen, M.M.; Lamberg-Allardt, C.J. Effects of phosphodiesterase 7 inhibition by RNA interference on the gene expression and differentiation of human mesenchymal stem cell-derived osteoblasts. Bone 2008, 43, 84–91. [Google Scholar] [CrossRef] [PubMed]
- McCahill, A.; McSorley, T.; Huston, E.; Hill, E.V.; Lynch, M.J.; Gall, I.; Keryer, G.; Lygren, B.; Tasken, K.; van Heeke, G. In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase A type II located in the centrosomal region. Cell. Signal. 2005, 17, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.A.; Collas, P.; Kemmner, W.A.; Witczak, O.; Conti, M.; Taskén, K. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 2001, 276, 21999–22002. [Google Scholar] [CrossRef] [PubMed]
- Sachs, B.D.; Baillie, G.S.; McCall, J.R.; Passino, M.A.; Schachtrup, C.; Wallace, D.A.; Dunlop, A.J.; MacKenzie, K.F.; Klussmann, E.; Lynch, M.J. p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J. Cell Biol. 2007, 177, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Wiesner, B.; Baillie, G.S.; Mollajew, R.; Henn, V.; Lorenz, D.; Furkert, J.; Santamaria, K.; Nedvetsky, P.; Hundsrucker, C. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 2007, 18, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.; Edwards, H.; Li, X.; Day, J.; Christian, F.; Dunlop, A.; Adams, D.; Zaccolo, M.; Houslay, M.; Baillie, G. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)–HSP20 complex attenuates the β-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E. Protein–protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell. Signal. 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.S.; Baillie, G.S.; Pritchard, L.M.; Umana, B.; Terrin, A.; Zaccolo, M.; Houslay, M.D.; Maurice, D.H. A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J. Biol. Chem. 2011, 286, 16285–16296. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brescia, M.; Zaccolo, M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. Int. J. Mol. Sci. 2016, 17, 1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101672
Brescia M, Zaccolo M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. International Journal of Molecular Sciences. 2016; 17(10):1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101672
Chicago/Turabian StyleBrescia, Marcella, and Manuela Zaccolo. 2016. "Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity" International Journal of Molecular Sciences 17, no. 10: 1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101672