
In Vivo Analysis of Protein–Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects

Abstract
:1. Introduction
2. Principles of Bioluminescence Resonance Energy Transfer (BRET) and Comparison with Related Methods
2.1. Principles of BRET
2.2. FRET and Protein-Fragment Complementation Assays
2.3. Compare BRET with FRET and Mainstream Protein-Fragment Complementation Assays (PCAs)
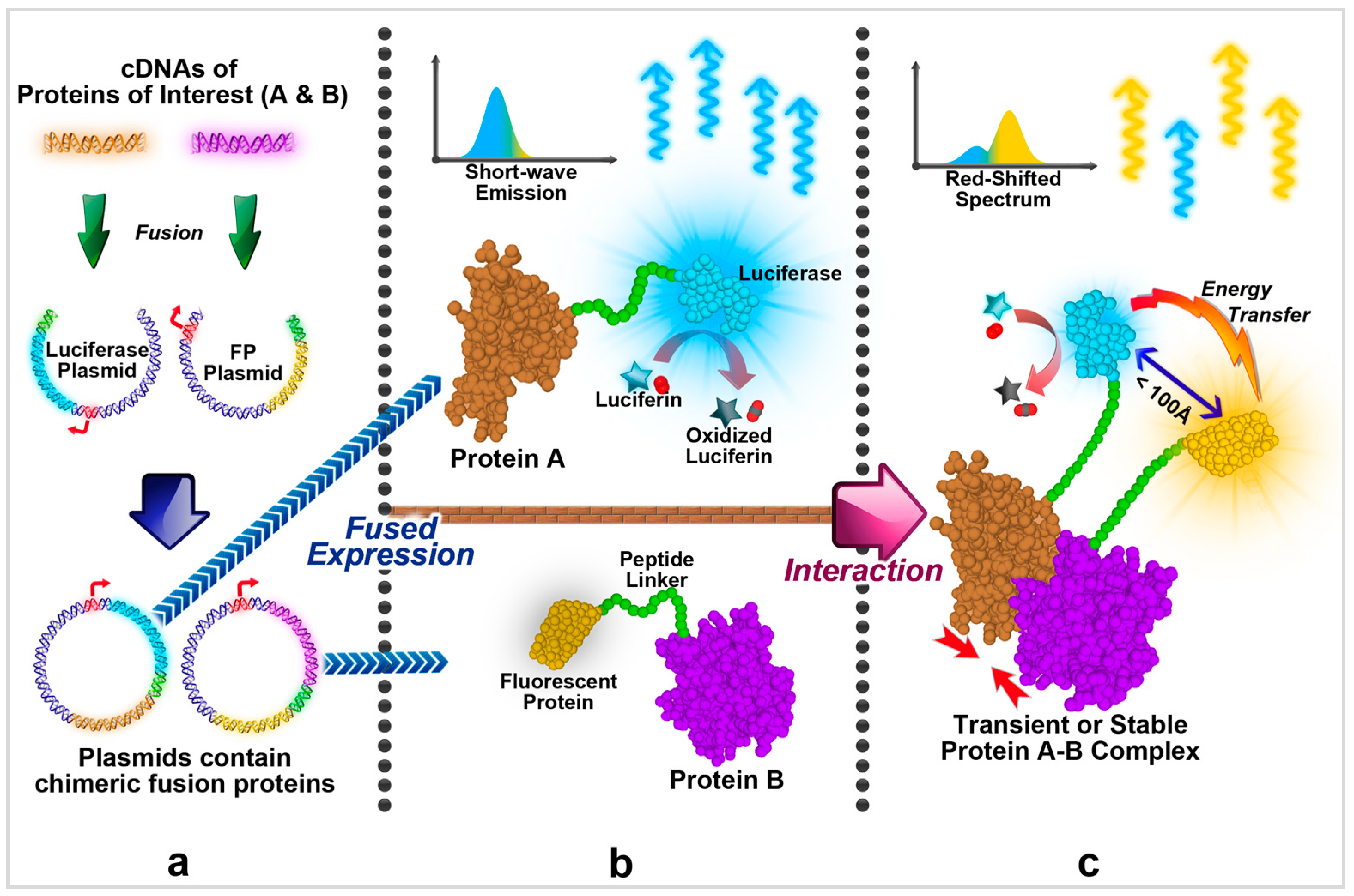
3. General Approach of Conducting BRET Assays
3.1. Vector Design and Acquiring
3.2. Fusion Expression and Signal Observation
3.3. Signal Analysis
3.4. BRET Imaging
4. Developed BRET Systems and Their Applications
4.1. Overview
4.2. Renilla Luciferase Based BRET Systems
4.3. BRET with Firefly and Gaussia Luciferases
4.4. NanoBRET
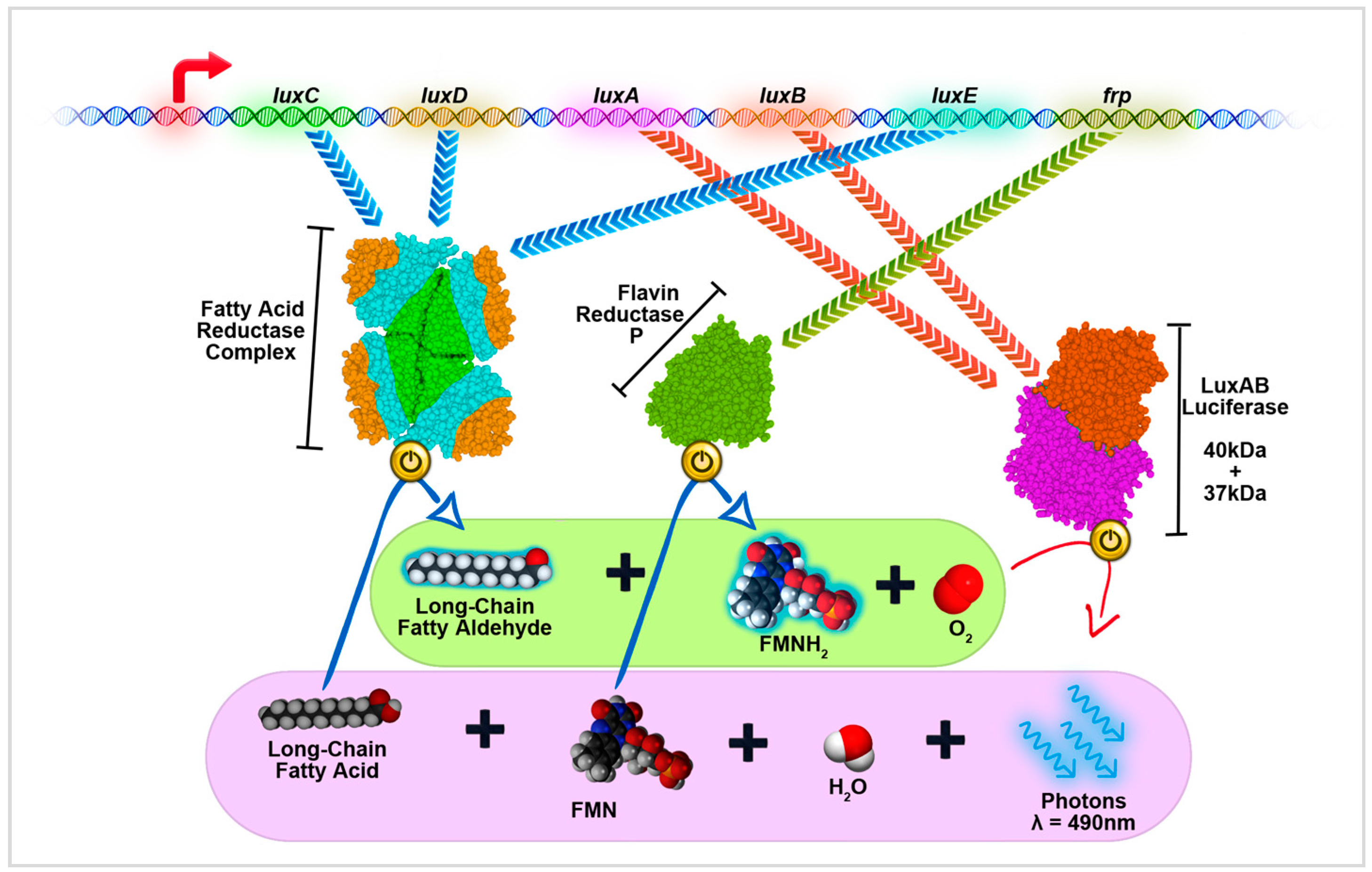
4.5. LuxBRET in Bacteria
5. Prospects of BRET
5.1. The Broader Selections of Fluorescent Proteins
5.2. Vast Development Potentials of Luciferases
5.3. Novel BRET Configurations and Applications
6. Recapitulation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Hunke, S.; Müller, V.S. Approaches to analyze protein–protein interactions of membrane proteins. In Protein Interactions; Cai, J., Ed.; InTech: Rijeka, Croatia, 2012; pp. 327–348. [Google Scholar]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef] [PubMed]
- Berggård, T.; Linse, S.; James, P. Methods for the detection and analysis of protein–protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Ramachandran, N. Protein microarrays as tools for functional proteomics. Curr. Opin. Chem. Biol. 2005, 9, 14–19. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Gamsjaeger, R.; Mackay, J.P. The structural analysis of protein–protein interactions by NMR spectroscopy. Proteomics 2009, 9, 5224–5232. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein–protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.; Dikici, E.; Daunert, S. Engineering bioluminescent proteins: Expanding their analytical potential. Anal. Chem. 2009, 81, 8662–8668. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Woo, J.; Cai, X.; Xu, X.; Servick, S.; Johnson, C.H.; Nebenführ, A.; von Arnim, A.G. A suite of tools and application notes for in vivo protein interaction assays using bioluminescence resonance energy transfer (BRET). Plant J. 2006, 48, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Houle, B.; Dionne, P.; Taylor, S.; Ménard, L. Bioluminescence Resonance Energy Transfer (BRET2) Principles, Applications, and Products. Available online: http://web4.cbm.uam.es/joomla-rl/images/Servicios/070.Microscopia-optica-cfocal/documentos/manuales/bret.pdf (accessed on 10 June 2016).
- Helms, V. Fluorescence resonance energy transfer. In Principles of Computational Cell Biology; Wiley-Blackwell: Hoboken, NJ, USA, 2008; p. 202. [Google Scholar]
- Scholes, G.D. Long-range resonance energy transfer in molecular systems. Annu. Rev. Phys. Chem. 2003, 54, 57–87. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.E.; Berti, L.; Medintz, I.L. Materials for fluorescence resonance energy transfer analysis: Beyond traditional donor-acceptor combinations. Angew. Chem. Int. Ed. 2006, 45, 4562–4589. [Google Scholar] [CrossRef] [PubMed]
- Boute, N.; Jockers, R.; Issad, T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 2002, 23, 351–354. [Google Scholar] [CrossRef]
- Morell, M.; Ventura, S.; Avilés, F.X. Protein complementation assays: Approaches for the in vivo analysis of protein interactions. FEBS Lett. 2009, 583, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008, 37, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Luker, K.E. Luciferase protein complementation assays for bioluminescence imaging of cells and mice. Methods Mol. Biol. 2011, 680, 29–43. [Google Scholar] [PubMed]
- Li, Y.C.; Rodewald, L.W.; Hoppmann, C.; Wong, E.T.; Lebreton, S.; Safar, P.; Patek, M.; Wang, L.; Wertman, K.F.; Wahl, G.M. A versatile platform to analyze low-affinity and transient protein–protein interactions in living cells in real time. Cell Rep. 2014, 9, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Wehr, M.C.; Laage, R.; Bolz, U.; Fischer, T.M.; Grünewald, S.; Scheek, S.; Bach, A.; Nave, K.A.; Rossner, M.J. Monitoring regulated protein–protein interactions using split TEV. Nat. Methods 2006, 3, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nagamune, T.; Kawahara, M. Split focal adhesion kinase for probing protein–protein interactions. Biochem. Eng. J. 2014, 90, 272–278. [Google Scholar] [CrossRef]
- Dünkler, A.; Müller, J.; Johnsson, N. Detecting protein-protein interactions with the split-ubiquitin sensor. Methods Mol. Biol. 2012, 786, 115–130. [Google Scholar] [PubMed]
- Remy, I.; Campbell-Valois, F.X.; Michnick, S.W. Detection of protein–protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2007, 2, 2120–2125. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Soutto, M.; Xu, X.; Zhang, Y.; Johnson, C.H. Bioluminescence resonance energy transfer (BRET) imaging in plant seedlings and mammalian cells. Methods Mol. Biol. 2011, 680, 3–28. [Google Scholar] [PubMed]
- Pfleger, K.D.; Seeber, R.M.; Eidne, K.A. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat. Protoc. 2006, 1, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Wang, Y.; Song, Y.; Wang, T.; Li, C.; Wei, Y.; Luo, Z.Q.; Shen, X. Bioluminescence resonance energy transfer system for measuring dynamic protein–protein interactions in Bacteria. mBio 2014. [Google Scholar] [CrossRef] [PubMed]
- Marullo, S.; Bouvier, M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol. Sci. 2007, 28, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Deriziotis, P.; Graham, S.A.; Estruch, S.B.; Fisher, S.E. Investigating protein-protein interactions in live cells using bioluminescence resonance energy transfer. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mandić, M.; Drinovec, L.; Glisic, S.; Veljkovic, N.; Nøhr, J.; Vrecl, M. Demonstration of a direct interaction between β2-adrenergic receptor and insulin receptor by BRET and bioinformatics. PLoS ONE 2014, 9, e112664. [Google Scholar] [CrossRef] [PubMed]
- Szalai, B.; Hoffmann, P.; Prokop, S.; Erdélyi, L.; Várnai, P.; Hunyady, L. Improved methodical approach for quantitative BRET analysis of G protein coupled receptor dimerization. PLoS ONE 2014, 9, e109503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eidne, K.A.; Kroeger, K.M.; Hanyaloglu, A.C. Applications of novel resonance energy transfer techniques to study dynamic hormone receptor interactions in living cells. Trends Endocrinol. Metab. 2002, 13, 415–421. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Flajolet, M.; Agnati, L.F.; Greengard, P.; Fuxe, K. Bioluminescence resonance energy transfer methods to study G protein-coupled receptor-receptor tyrosine kinase heteroreceptor complexes. Methods Cell Biol. 2013, 117, 141–164. [Google Scholar] [PubMed]
- Promega Corporation. NanoLuc® Luciferase: Redefining Reporter Assays. Available online: https://www.promega.com/products/reporter-assays-and-transfection/reporter-assays/nanoluc-luciferase-redefining-reporter-assays (accessed on 7 July 2016).
- De, A.; Gambhir, S.S. Noninvasive imaging of protein–protein interactions from live cells and living subjects using bioluminescence resonance energy transfer. FASEB J. 2005, 19, 2017–2019. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Soutto, M.; Xie, Q.; Servick, S.; Subramanian, C.; von Arnim, A.G.; Johnson, C.H. Imaging protein interactions with bioluminescence resonance energy transfer (BRET) in plant and mammalian cells and tissues. Proc. Natl. Acad. Sci. USA 2007, 104, 10264–10269. [Google Scholar] [CrossRef] [PubMed]
- Goyet, E.; Bouquier, N.; Ollendorff, V.; Perroy, J. Fast and high resolution single-cell BRET imaging. Sci. Rep. 2016, 6, 28231. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Piston, D.W.; Johnson, C.H. A bioluminescence resonance energy transfer (BRET) system: Application to interacting circadian clock proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Parent, S.; Caron, M.; Legault, M.; Joly, E.; Angers, S.; Bouvier, M.; Brown, M.; Houle, B.; Ménard, L. The BRET2/arrestin assay in stable recombinant cells: A platform to screen for compounds that interact with G protein-coupled receptors (GPCRS). J. Recept. Signal Transduct. Res. 2002, 22, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Kocan, M.; See, H.B.; Seeber, R.M.; Eidne, K.A.; Pfleger, K.D. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J. Biomol. Screen. 2008, 13, 888–898. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Ray, P.; Loening, A.M.; Gambhir, S.S. BRET3: A red-shifted bioluminescence resonance energy transfer (BRET)-based integrated platform for imaging protein-protein interactions from single live cells and living animals. FASEB J. 2009, 23, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Gammon, S.T.; Villalobos, V.M.; Roshal, M.; Samrakandi, M.; Piwnica-Worms, D. Rational design of novel red-shifted BRET pairs: Platforms for real-time single-chain protease biosensors. Biotechnol. Prog. 2009, 25, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, K.D.; Dromey, J.R.; Dalrymple, M.B.; Lim, E.M.; Thomas, W.G.; Eidne, K.A. Extended bioluminescence resonance energy transfer (eBRET) for monitoring prolonged protein-protein interactions in live cells. Cell Signal. 2006, 18, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, H.; Nakajima, Y.; Ohmiya, Y. Luciferase-YFP fusion tag with enhanced emission for single-cell luminescence imaging. Nat. Methods 2007, 4, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu-Andrasi, A.; Chan, C.T.; De, A.; Massoud, T.F.; Gambhir, S.S. Bioluminescence resonance energy transfer (BRET) imaging of protein–protein interactions within deep tissues of living subjects. Proc. Natl. Acad. Sci. USA 2011, 108, 12060–12065. [Google Scholar] [CrossRef] [PubMed]
- Dimri, S.; Basu, S.; De, A. Use of BRET to study protein-protein interactions in vitro and in vivo. Methods Mol. Biol. 2016, 1443, 57–78. [Google Scholar] [PubMed]
- Li, F.; Yu, J.; Zhang, Z.; Cui, Z.; Wang, D.; Wei, H.; Zhang, X.E. Use of hGluc/tdTomato pair for sensitive BRET sensing of protease with high solution media tolerance. Talanta 2013, 109, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Machleidt, T.; Woodroofe, C.C.; Schwinn, M.K.; Méndez, J.; Robers, M.B.; Zimmerman, K.; Otto, P.; Daniels, D.L.; Kirkland, T.A.; Wood, K.V. NanoBRET—A novel BRET platform for the analysis of protein–protein interactions. ACS Chem. Biol. 2015, 10, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, T.; Okuda-Ashitaka, E.; Kojima, S.; Akiyama, H.; Ito, S.; Ohmiya, Y. Monitoring for dynamic biological processing by intramolecular bioluminescence resonance energy transfer system using secreted luciferase. Anal. Chem. 2004, 329, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Rathnayaka, T.; Kuroda, Y. Bacterial expression and re-engineering of Gaussia princeps luciferase and its use as a reporter protein. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Loening, A.M.; Wu, A.M.; Gambhir, S.S. Red-shifted Renilla reniformis luciferase variants for imaging in living subjects. Nat. Methods 2007, 4, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs-theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Salahpour, A.; Espinoza, S.; Masri, B.; Lam, V.; Barak, L.; Gainetdinov, R.R. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kocan, M.; Pfleger, K.D. Study of GPCR-protein interactions by BRET. Methods Mol. Biol. 2011, 746, 357–371. [Google Scholar] [PubMed]
- Stoddart, L.A.; Johnstone, E.K.; Wheal, A.J.; Goulding, J.; Robers, M.B.; Machleidt, T.; Wood, K.V.; Hill, S.J.; Pfleger, K.D. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. Applications of bioluminescence-and fluorescence resonance energy transfer to drug discovery at G protein-coupled receptors. Eur. J. Pharm. Sci. 2004, 21, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Rebois, R.V.; Maki, K.; Meeks, J.A.; Fishman, P.H.; Hébert, T.E.; Northup, J.K. D2-like dopamine and β-adrenergic receptors form a signaling complex that integrates Gs-and Gi-mediated regulation of adenylyl cyclase. Cell Signal. 2012, 24, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Biotium Inc. Physical Properties of Coelenterazine Analogs. Available online: http://www.bioscience.co.uk/userfiles/pdf/Physical-Properties-of-Coelenterazine-Analogs.pdf (accessed on 15 July 2016).
- Arai, R.; Nakagawa, H.; Kitayama, A.; Ueda, H.; Nagamune, T. Detection of protein-protein interaction by bioluminescence resonance energy transfer from firefly luciferase to red fluorescent protein. J. Biosci. Bioeng. 2002, 94, 362–364. [Google Scholar] [CrossRef]
- Thorne, N.; Inglese, J.; Auld, D.S. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 2010, 17, 646–657. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Jasani, A.; Arora, R.; Gambhir, S.S. Evolution of BRET biosensors from live cell to tissue-scale in vivo imaging. Front. Endocrinol. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.T.; Case, J.F. Bioluminescence in the mesopelagic copepod, Gaussia princeps (T. Scott). J. Exp. Mar. Biol. Ecol. 1972, 8, 53–71. [Google Scholar] [CrossRef]
- Verhaegen, M.; Christopoulos, T.K. Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal. Chem. 2002, 74, 4378–4385. [Google Scholar] [CrossRef]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yu, J.; Zhang, Z.; Cui, Z.; Wang, D.; Wei, H.; Zhang, X.E. Buffer enhanced bioluminescence resonance energy transfer sensor based on Gaussia luciferase for in vitro detection of protease. Anal. Chim. Acta 2012, 724, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hiraoka, K.; Kasahara, N.; Logg, C.R. Optimization of enzyme-substrate pairing for bioluminescence imaging of gene transfer using Renilla and Gaussia luciferases. J. Gene Med. 2010, 12, 538–537. [Google Scholar] [CrossRef] [PubMed]
- Inouye, S.; Shimomura, O. The use of Renilla luciferase, Oplophorus luciferase, and apoaequorin as bioluminescent reporter protein in the presence of coelenterazine analogues as substrate. Biochem. Biophys. Res. Commun. 1997, 233, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Inouye, S.; Sasaki, S. Overexpression, purification and characterization of the catalytic component of Oplophorus luciferase in the deep-sea shrimp, Oplophorus gracilirostris. Protein Expr. Purif. 2007, 56, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L. NanoLuc®: A Smaller, Brighter, and More Versatile Luciferase Reporter. Available online: http://www.promega.com/~/media/files/promega%20worldwide/europe/promega%20uk/webinars%20and%20events/cell%20analysis%20seminar%20tour/terry-riss-02.pdf (accessed on 3 July 2016).
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A small luciferase is brightening up the field of bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Promega Corporation. Coelenterazines. Available online: https://www.promega.com/products/biochemicals-and-labware/biochemical-buffers-and-reagents/coelenterazines (accessed on 5 July 2016).
- Promega Corporation. EnduRen™ Live Cell Substrate. Available online: http://www.promega.com/products/reporter-assays-and-transfection/reporter-assays/enduren-live-cell-substrate (accessed on 5 July 2016).
- Biotium. Coelenterazine 400a (DeepBlueC). Available online: https://biotium.com/product/coelenterazine-400a-also-known-as-deepblue-ctm (accessed on 5 July 2016).
- Promega Corporation. Nano-Glo® Luciferase Assay System. Available online: http://www.promega.com/products/reporter-assays-and-transfection/reporter-assays/nano_glo-luciferase-assay-system (accessed on 20 July 2016).
- Sigma-Aldrich Co. d-Luciferin Synthetic. Available online: http://www.sigmaaldrich.com/catalog/product/sigma/l9504 (accessed on 20 July 2016).
- Bhaumik, S.; Gambhir, S.S. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc. Natl. Acad. Sci. USA 2002, 99, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ripp, S.; Sayler, G.S.; Close, D.M. Expression of a humanized viral 2A-mediated lux operon efficiently generates autonomous bioluminescence in human cells. PLoS ONE 2014, 9, e96347. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.L.; Hastings, J.; Krause, K.L. Luciferases and light-emitting accessory proteins: Structural biology. In eLS Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Andreu, N.; Zelmer, A.; Fletcher, T.; Elkington, P.T.; Ward, T.H.; Ripoll, J.; Parish, T.; Bancroft, G.J.; Schaible, U.; Robertson, B.D.; et al. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS ONE 2010, 5, e10777. [Google Scholar] [CrossRef] [PubMed]
- Close, D.; Xu, T.; Smartt, A.; Rogers, A.; Crossley, R.; Price, S.; Ripp, S.; Sayler, G. The evolution of the bacterial luciferase gene cassette (lux) as a real-time bioreporter. Sensors 2012, 12, 732–752. [Google Scholar] [CrossRef] [PubMed]
- Sigma-Aldrich Co. Decanal. Available online: http://www.sigmaaldrich.com/catalog/product/sigma/d7384 (accessed on 22 July 2016).
- Velten, J.; Pogson, B.; Cazzonelli, C.I. Luciferase as a reporter of gene activity in plants. Transgenic Plant J. 2008, 2, 1–13. [Google Scholar]
- Close, D.M.; Xu, T.; Sayler, G.S.; Ripp, S. In vivo bioluminescent imaging (BLI): Noninvasive visualization and interrogation of biological processes in living animals. Sensors 2010, 11, 180–206. [Google Scholar] [CrossRef] [PubMed]
- Welham, P.A.; Stekel, D.J. Mathematical model of the Lux luminescence system in the terrestrial bacterium Photorhabdus luminescens. Mol. Biosyst. 2009, 5, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Loening, A.M.; Fenn, T.D.; Wu, A.M.; Gambhir, S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 2006, 19, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Oba, Y.; Kato, S.I.; Ojika, M.; Inouye, S. Biosynthesis of coelenterazine in the deep-sea copepod, Metridia Pacifica. Biochem. Biophys. Res. Commun. 2009, 390, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Oba, Y.; Yoshida, N.; Kanie, S.; Ojika, M.; Inouye, S. Biosynthesis of firefly luciferin in adult lantern: Decarboxylation of l-Cysteine is a key step for benzothiazole ring formation in firefly luciferin Synthesis. PLoS ONE 2013, 8, e84023. [Google Scholar] [CrossRef] [PubMed]
- Low, J.C.; Tu, S.C. Energy transfer evidence for in vitro and in vivo complexes of Vibrio harveyi flavin reductase P and luciferase. J. Photochem. Photobiol. 2003, 77, 446–452. [Google Scholar] [CrossRef]
- Cui, B.; Zhang, L.; Song, Y.; Wei, J.; Li, C.; Wang, T.; Wang, Y.; Zhao, T.; Shen, X. Engineering an enhanced, thermostable, monomeric bacterial luciferase gene as a reporter in plant protoplasts. PLoS ONE 2014, 9, e107885. [Google Scholar] [CrossRef] [PubMed]
- Close, D.M.; Patterson, S.S.; Ripp, S.; Baek, S.J.; Sanseverino, J.; Sayler, G.S. Autonomous bioluminescent expression of the bacterial luciferase gene cassette (lux) in a mammalian cell line. PLoS ONE 2010, 5, e12441. [Google Scholar] [CrossRef] [PubMed]
- McNamara, G.; Boswell, C.A. A thousand proteins of light: 15 years of advances in fluorescent proteins. In Modern Research and Educational Topics in Microscopy; Méndez-Vilas, A., Díaz, J., Eds.; Formatex: Badajoz, Spain, 2007; pp. 287–296. [Google Scholar]
- Lambert, T.; Thorn, K. Fluorescent Protein Properties. Available online: http://nic.ucsf.edu/FPvisualization (accessed on 30 July 2016).
- Kremers, G.J.; Gilbert, S.G.; Cranfill, P.J.; Davidson, M.W.; Piston, D.W. Fluorescent proteins at a glance. J. Cell Sci. 2011, 124, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Dedecker, P.; De-Schryver, F.C.; Hofkens, J. Fluorescent proteins: Shine on, you crazy diamond. J. Am. Chem. Soc. 2013, 135, 2387–2402. [Google Scholar] [CrossRef] [PubMed]
- Shemiakina, I.I.; Ermakova, G.V.; Cranfill, P.J.; Baird, M.A.; Evans, R.A.; Souslova, E.A.; Staroverov, D.B.; Gorokhovatsky, A.Y.; Putintseva, E.V.; Gorodnicheva, T.V.; et al. A monomeric red fluorescent protein with low cytotoxicity. Nat. Commun. 2012, 3, 1204:1–1204:7. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.A.; Tran, G.N.; Gross, L.A.; Crisp, J.L.; Shu, X.; Lin, J.Y.; Tsien, R.Y. A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein. Nat. Methods 2016, 13, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 2011, 29, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Kim, Y.P. Analysis of protease activity using quantum dots and resonance energy transfer. Theranostics 2012, 2, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Matsuzaki, K. Tag–probe labeling methods for live-cell imaging of membrane proteins. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2124–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Niwa, K.; Yamada, N.; Enomoto, T.; Irie, T.; Kubota, H.; Ohmiya, Y.; Akiyama, H. Firefly bioluminescence quantum yield and colour change by pH-sensitive green emission. Nat. Photonics 2008, 2, 44–47. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of α 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.; Dey, D.; Roy, A.D.; Bhattacharjee, D.; Hussain, S.A. Multi step FRET among three laser dyes Pyrene, Acriflavine and Rhodamine B. J. Lumin. 2016, 172, 168–174. [Google Scholar] [CrossRef]
- Aneja, A.; Mathur, N.; Bhatnagar, P.K.; Mathur, P.C. Triple-FRET technique for energy transfer between conjugated polymer and TAMRA dye with possible applications in medical diagnostics. J. Biol. Phys. 2008, 34, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Zylstra, J.; Fontaine, D.M.; Branchini, B.R.; Maye, M.M. Novel multistep BRET-FRET energy transfer using nanoconjugates of firefly proteins, quantum dots, and red fluorescent proteins. Nanoscale 2013, 5, 5303–5306. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Susha, A.S.; Lutich, A.A.; Stefani, F.D.; Feldmann, J.; Rogach, A.L. Cascaded FRET in conjugated polymer/quantum dot/dye-labeled DNA complexes for DNA hybridization detection. ACS Nano 2009, 3, 4127–4131. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Shuhendler, A.J.; Rao, J. Self-luminescing BRET-FRET near-infrared dots for in vivo lymph-node mapping and tumour imaging. Nat. Commun. 2012. [Google Scholar] [CrossRef] [PubMed]
- So, M.K.; Xu, C.; Loening, A.M.; Gambhir, S.S.; Rao, J. Self-illuminating quantum dot conjugates for in vivo imaging. Nat. Biotechnol. 2006, 24, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Mitsunaga, M.; Bhattacharyya, S.; Miller, S.C.; Choyke, P.L.; Kobayashi, H. Self-illuminating in vivo lymphatic imaging using a bioluminescence resonance energy transfer quantum dot nano-particle. Contrast Media Mol. Imaging 2011, 6, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, J.; Wu, B.; Feng, S.; Cai, G.; Tuluc, F.; Peränen, J.; Guo, W. Activation of Rab8 guanine nucleotide exchange factor Rabin8 by ERK1/2 in response to EGF signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Sleno, R.; Pétrin, D.; Devost, D.; Goupil, E.; Zhang, A.; Hébert, T.E. Designing BRET-based conformational biosensors for G protein-coupled receptors. Methods 2016, 92, 11–18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Advantages | Disadvantages | Applications |
|---|---|---|---|
| BRET | Real-time PPI monitoring In vivo More sensitive than FRET | Requires expensive luciferin, but this might be solvable in the future with LuxBRET | PPI regulatory study Binary PPI detection |
| FRET | Real-time PPI monitoring In vivo | Requires external excitation Higher background noise Potential detriments to sample | PPI regulatory study Binary PPI detection |
| Y2H | Easy to use Low cost | Only suitable for proteins that are nucleus co-localizable Non-reversible detection In vivo only | Binary PPI detection |
| BiFC | Suitable for wider range of proteins than Y2H | Requires external excitation Non-reversible detection | Binary PPI detection |
| Co-IP | Detect multiple PPIs | Lower sensitivity Higher error rate In vitro only | Initial PPI screening Protein complex detection |
| Name § | Luciferase | Luciferase Emission * | FP | FP Emission * | Luciferin (Substrate) | Reference |
|---|---|---|---|---|---|---|
| BRET1 | Rluc/Rluc8 | 480 | eYFP | 530 | Coelenterazine-h | Xu et al. [39] |
| BRET2 | Rluc | 395 | GFP2 | 510 | DeepBlueC | Bertrand et al. [40] |
| eBRET2 | Rluc8 | 395 | GFP2 | 510 | DeepBlueC | Kocan et al. [41] |
| BRET3 (1) | Rluc8 | 480 | mOrange | 564 | Coelenterazine-h | De et al. [42] |
| BRET3 (2) | Fluc | 565 | DsRed | 583 | d-Luciferin | Gammon et al. [43] |
| eBRET | Rluc | 480 | eYFP | 530 | EnduRen | Pfleger et al. [44] |
| BAF-Y | Rluc | 480 | eYFP | 530 | ViviRen | Hoshino et al. [45] |
| BRET3.1 | Rluc8 | 515 | mOrange | 564 | Coelenterazine-v | Dragulescu et al. [46] |
| BRET4 | Rluc8 | 480 | TagRFP | 584 | Coelenterazine-h | Dimri et al. [47] |
| BRET4.1 | Rluc8 | 515 | TagRFP | 584 | Coelenterazine-v | Dragulescu et al. [46] |
| BRET5 | Rluc8.6 | 535 | TagRFP | 584 | Coelenterazine-h | Dragulescu et al. [46] |
| BRET6 | Rluc8.6 | 535 | TurboFP | 635 | Coelenterazine-h | Dragulescu et al. [46] |
| BRET6.1 | Rluc8.6 | 570 | TurboFP | 635 | Coelenterazine-v | Dragulescu et al. [46] |
| BRET7 | Rluc8 | 515 | TurboFP | 635 | Coelenterazine-v | Dimri et al. [47] |
| BRET8 | Rluc8.6 | 535 | TurboFP | 635 | Coelenterazine-h | Dimri et al. [47] |
| GlucBRET | hGluc | 470 | tdTomato | 580 | Coelenterazine | Li et al. [48] |
| NanoBRET | NLuc | 460 | HaloTag | 618 | Furimazine | Machleidt et al. [49] |
| LuxBRET | LuxAB | 490 | eYFP | 530 | FMNH2 + Long-chain fatty aldehyde | Cui et al. [28] |
| VlucBRET | Vluc | 460 | eYFP | 530 | Vargulin | Otsuji et al. [50] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Yang, X.; Wang, Y.; Shen, X. In Vivo Analysis of Protein–Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects. Int. J. Mol. Sci. 2016, 17, 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101704
Sun S, Yang X, Wang Y, Shen X. In Vivo Analysis of Protein–Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects. International Journal of Molecular Sciences. 2016; 17(10):1704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101704
Chicago/Turabian StyleSun, Sihuai, Xiaobing Yang, Yao Wang, and Xihui Shen. 2016. "In Vivo Analysis of Protein–Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects" International Journal of Molecular Sciences 17, no. 10: 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101704