
Extracellular Matrix, a Hard Player in Angiogenesis



Abstract
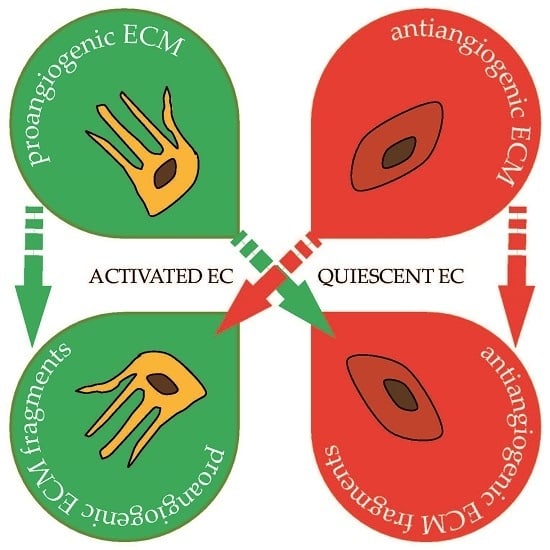
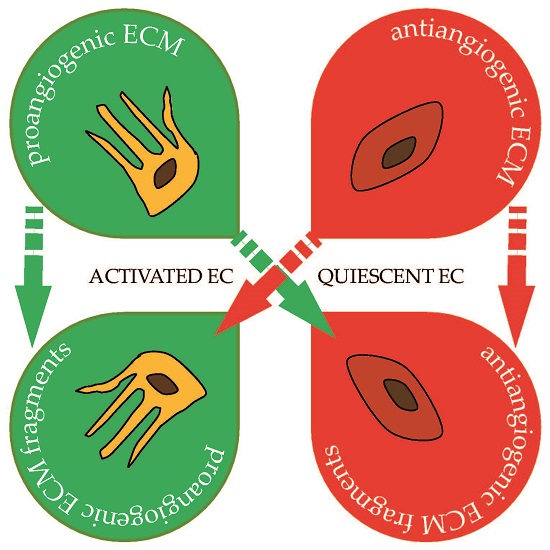
:
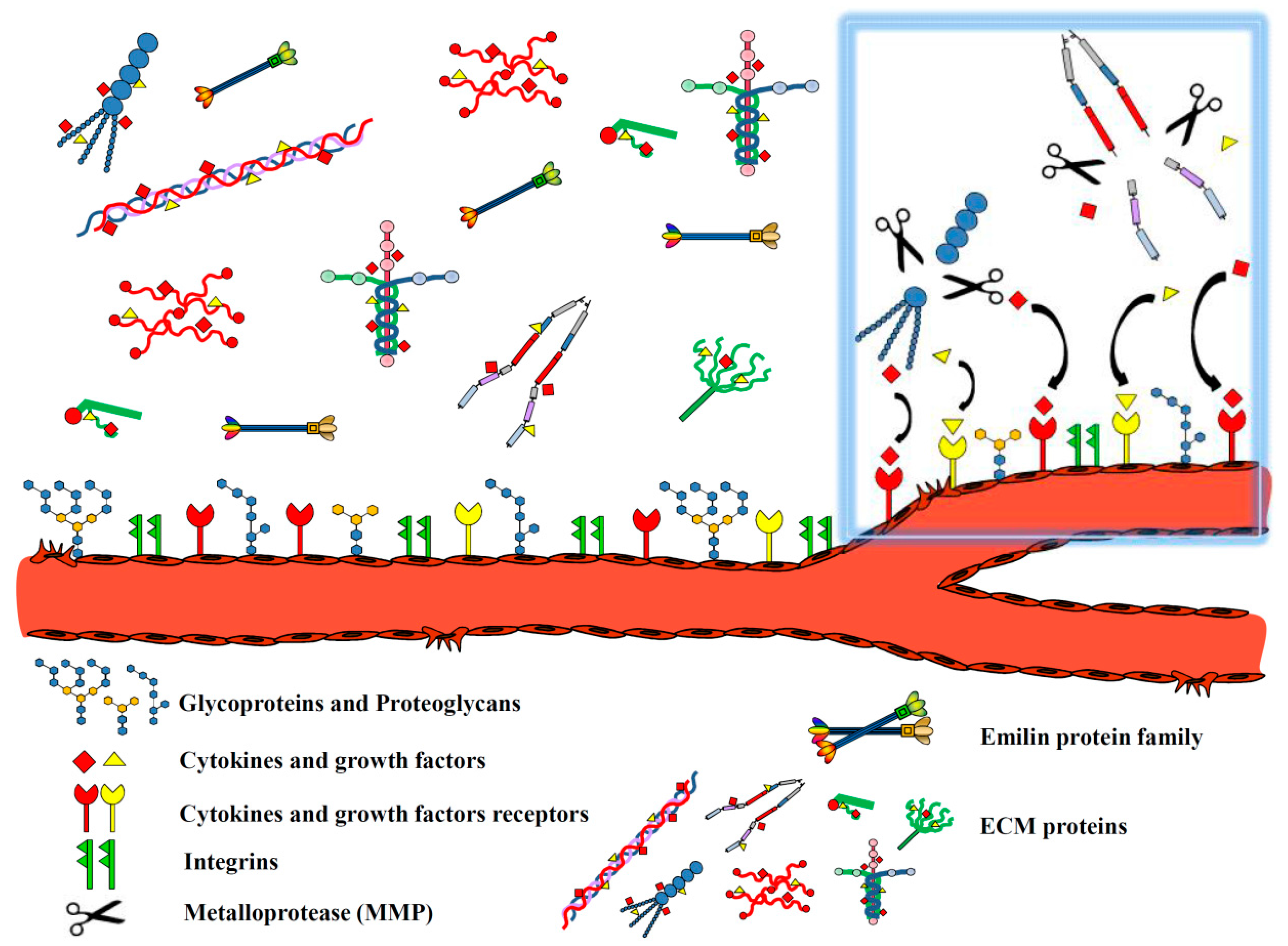
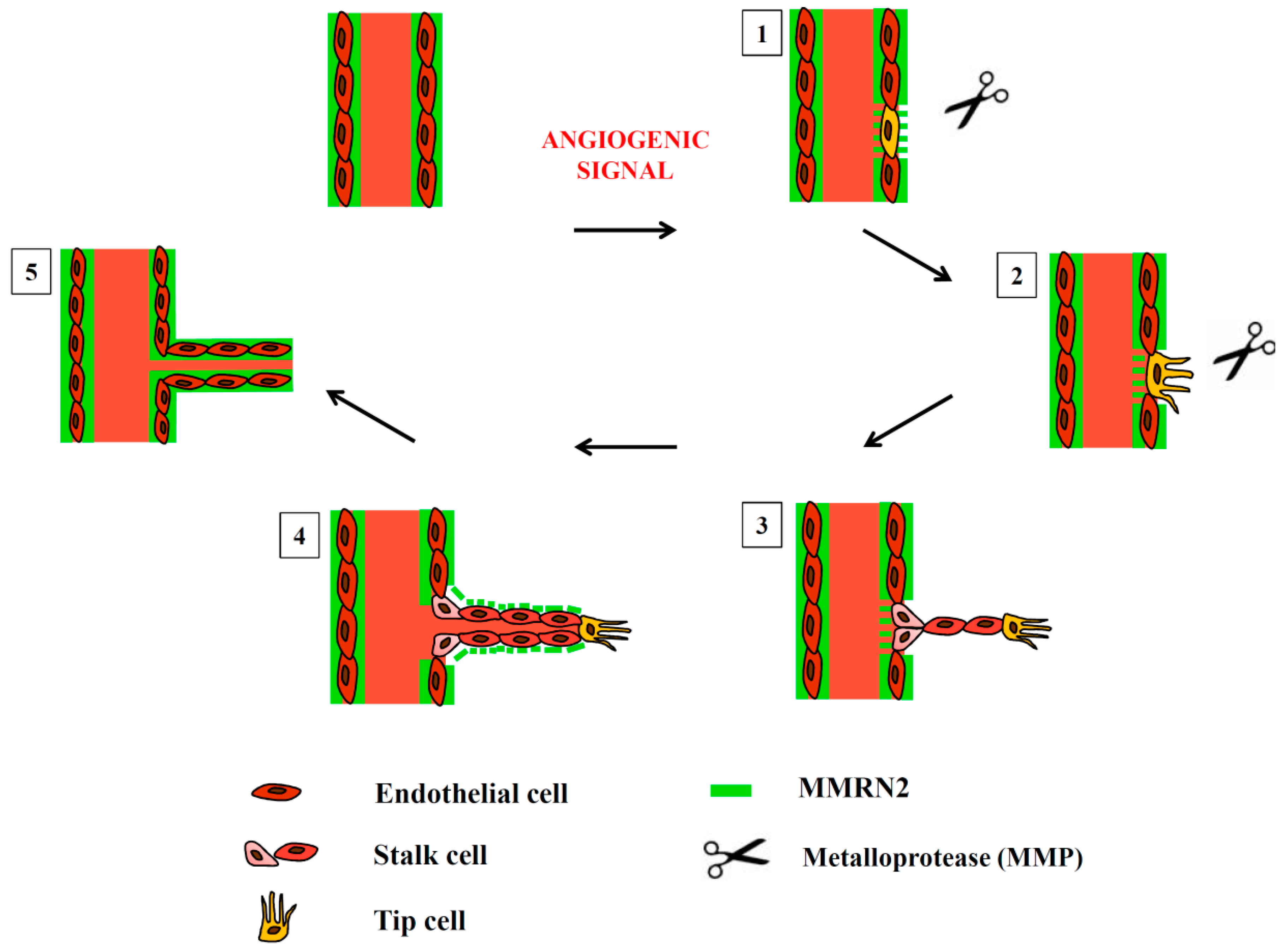
1. Introduction
2. Thrombospondins: Endogenous Angiogenesis Inhibitors
3. Fibronectin: Key Function in Pathological Angiogenesis
4. Collagens: A Major Source of Anti-Angiogenic Fragments
5. Laminins: Multiple Chains for Multiple Functions
6. Proteoglycans: Complex Functions from the Protein Core and Carbohydrate Chains
7. Hyaluronan: Not Only a Mere Glue
8. The EDEN Family: Two Members with Opposite Functions
9. The CCN Family of Proteins as Regulators of Vascular Development and Pathological Angiogenesis
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECM Proteins | Fragments | Anti-Angiogenic Activity | Pro-Angiogenic Activity | References |
|---|---|---|---|---|
| Trombospondins | ||||
| TSP-1 | √ | [27,28,30,32,33,34,35,36,37,38,39,40,41,42,43,44] | ||
| TSP-2 | √ | [29,31,44] | ||
| Fibronectin | ||||
| Fibronectin | √ | [51,52,53,54,55,56] | ||
| Collagens | ||||
| Type I | √ | [58,59,60,61] | ||
| Type IV | √ | [67] | ||
| arresten | √ | [65,71] | ||
| canstatin | √ | [72,73,74,75] | ||
| tumstatin | √ | [76,77] | ||
| Type XV | restin | √ | [80,81,82] | |
| Type XVIII | endostatin | √ | [81,83,84,85,86,87,92] | |
| Laminins | ||||
| Laminin 411 and 421 | √ | [101,102,103,105] | ||
| Laminin 511 | √ | [104,106] | ||
| Proteoglicans | ||||
| Perlecan | √ | [112,133,134,135,136,137,138,139,140,141,142] | ||
| endorepellin | √ | [148,149,150,151,152,153,154,155,156,157] | ||
| Decorin | √ | [162,163,164,165,166,167,168,169,170,171,172,173,174,175] | ||
| √ | [159,160,161,163,164,165,169,170,171,172,173,174,175] | |||
| Biglycan | √ | [176,177,178] | ||
| Syndecan 1 | √ | [181,182,183,185] | ||
| Syndecan 2 | √ | [184,186] | ||
| Syndecan 4 | √ | [187,188] | ||
| Glypicans | √ | [189,190,191,192] | ||
| Lumican | √ | [196,197,198] | ||
| Hyaluronan | ||||
| LMW-HA | √ | [200,201,202,203,204,205,206] | ||
| √ | [210] | |||
| HMW-HA | √ | [207,208] | ||
| √ | [211] | |||
| EDEN Family | ||||
| Multimerin 2 | √ | [224,225] | ||
| √ | [226,227] | |||
| Δ2 fragment | √ | [225] | ||
| Emilin 2 | √ | [230,231] | ||
| CCN Family | ||||
| CCN1 | √ | [235,236,237] | ||
| CCN2 | √ | [239,240] | ||
| CCN3 | √ | [241,242,243,244] | ||
| CCN4 | √ | [245,246] | ||
| CCN5 | √ | [248] | ||
References
- Ozbek, S.; Balasubramanian, P.G.; Chiquet-Ehrismann, R.; Tucker, R.P.; Adams, J.C. The evolution of extracellular matrix. Mol. Biol. Cell 2010, 21, 4300–4305. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, S.; Ligresti, G.; Lorenzon, E.; Colombatti, A.; Mongiat, M. Extracellular matrix: A matter of life and death. Connect. Tissue Res. 2008, 49, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Weeraratna, A.T.; Rangel, L.B.; Pizer, E.S.; Cho, K.R.; Schwartz, D.R.; Shock, T.; Morin, P.J. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell 2003, 3, 377–386. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, S.; Andreuzzi, E.; Paulitti, A.; Colladel, R.; Pellicani, R.; Todaro, F.; Schiavinato, A.; Bonaldo, P.; Colombatti, A.; Mongiat, M. EMILIN2 down-modulates the Wnt signalling pathway and suppresses breast cancer cell growth and migration. J. Pathol. 2014, 232, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.T.; Tang, M.J. SPARC in cancer biology: Its role in cancer progression and potential for therapy. Drug Resist. Updates 2008, 11, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Ray, A. The CCN family proteins in carcinogenesis. Exp. Oncol. 2010, 32, 2–9. [Google Scholar] [PubMed]
- Reed, C.C.; Waterhouse, A.; Kirby, S.; Kay, P.; Owens, R.T.; McQuillan, D.J.; Iozzo, R.V. Decorin prevents metastatic spreading of breast cancer. Oncogene 2005, 24, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Ligresti, G.; Marastoni, S.; Lorenzon, E.; Doliana, R.; Colombatti, A. Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Mol. Cell. Biol. 2007, 27, 7176–7187. [Google Scholar] [CrossRef] [PubMed]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-Action Combination Therapy Enhances Angiogenesis while Reducing Tumor Growth and Spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, C.M.; Miller, J.W. Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Curr. Opin. Ophthalmol. 2007, 18, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Dejana, E.; Languino, L.R.; Polentarutti, N.; Balconi, G.; Ryckewaert, J.J.; Larrieu, M.J.; Donati, M.B.; Mantovani, A.; Marguerie, G. Interaction between fibrinogen and cultured endothelial cells. Induction of migration and specific binding. J. Clin. Investig. 1985, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Streit, M.; Koteliansky, V.E.; de Fougerolles, A.R.; Detmar, M. The α1β1 and α2β1 integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am. J. Pathol. 2002, 160, 195–204. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Salza, R. Matricryptins and matrikines: Biologically active fragments of the extracellular matrix. Exp. Dermatol. 2014, 23, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C.; Lawler, J. The thrombospondins. Cold Spring Harb. Perspect. Biol. 2011, 3, a009712. [Google Scholar] [CrossRef] [PubMed]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef] [PubMed]
- Tolsma, S.S.; Volpert, O.V.; Good, D.J.; Frazier, W.A.; Polverini, P.J.; Bouck, N. Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. J. Cell Biol. 1993, 122, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, T.R.; Zhu, Y.H.; Smith, L.T.; Bain, S.D.; Yang, Z.; Lin, M.T.; Danielson, K.G.; Iozzo, R.V.; LaMarca, M.; McKinney, C.E.; et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J. Cell Biol. 1998, 140, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Bleuel, K.; Popp, S.; Fusenig, N.E.; Stanbridge, E.J.; Boukamp, P. Tumor suppression in human skin carcinoma cells by chromosome 15 transfer or thrombospondin-1 overexpression through halted tumor vascularization. Proc. Natl. Acad. Sci. USA 1999, 96, 2065–2070. [Google Scholar] [CrossRef] [PubMed]
- Streit, M.; Riccardi, L.; Velasco, P.; Brown, L.F.; Hawighorst, T.; Bornstein, P.; Detmar, M. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 14888–14893. [Google Scholar] [CrossRef] [PubMed]
- Streit, M.; Velasco, P.; Brown, L.F.; Skobe, M.; Richard, L.; Riccardi, L.; Lawler, J.; Detmar, M. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am. J. Pathol. 1999, 155, 441–452. [Google Scholar] [CrossRef]
- Weinstat-Saslow, D.L.; Zabrenetzky, V.S.; VanHoutte, K.; Frazier, W.A.; Roberts, D.D.; Steeg, P.S. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994, 54, 6504–6511. [Google Scholar] [PubMed]
- Dawson, D.W.; Pearce, S.F.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Volpert, O.V.; Pearce, S.F.; Schneider, A.J.; Silverstein, R.L.; Henkin, J.; Bouck, N.P. Three distinct D-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Mol. Pharmacol. 1999, 55, 332–338. [Google Scholar] [PubMed]
- Yee, K.O.; Connolly, C.M.; Duquette, M.; Kazerounian, S.; Washington, R.; Lawler, J. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res. Treat. 2009, 114, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Asch, A.S.; Silbiger, S.; Heimer, E.; Nachman, R.L. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem. Biophys. Res. Commun. 1992, 182, 1208–1217. [Google Scholar] [CrossRef]
- Jimenez, B.; Volpert, O.V.; Reiher, F.; Chang, L.; Munoz, A.; Karin, M.; Bouck, N. C-Jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene 2001, 20, 3443–3448. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Foglieni, C.; Resovi, A.; Giavazzi, R.; Taraboletti, G. Targeting angiogenesis with compounds from the extracellular matrix. Int. J. Biochem. Cell Biol. 2011, 43, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; Rusnati, M.; Ragona, L.; Colombo, G. Targeting tumor angiogenesis with TSP-1-based compounds: Rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010, 1, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lawler, J. Thrombospondin-based antiangiogenic therapy. Microvasc. Res. 2007, 74, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.; Detmar, M. Tumor progression: The effects of thrombospondin-1 and -2. Int. J. Biochem. Cell Biol. 2004, 36, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Haviv, F.; Bradley, M.F.; Kalvin, D.M.; Schneider, A.J.; Davidson, D.J.; Majest, S.M.; McKay, L.M.; Haskell, C.J.; Bell, R.L.; Nguyen, B.; et al. Thrombospondin-1 Mimetic Peptide Inhibitors of Angiogenesis and Tumor Growth: Design, Synthesis, and Optimization of Pharmacokinetics and Biological Activities. J. Med. Chem. 2005, 48, 2838–2846. [Google Scholar] [CrossRef] [PubMed]
- Uronis, H.E.; Cushman, S.M.; Bendell, J.C.; Blobe, G.C.; Morse, M.A.; Nixon, A.B.; Dellinger, A.; Starr, M.D.; Li, H.; Meadows, K.; et al. A phase I study of ABT-510 plus bevacizumab in advanced solid tumors. Cancer Med. 2013, 2, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.H.; Chen, G.E.; Hynes, R.O. Fibronectin isoform distribution in the mouse. II. Differential distribution of the alternatively spliced EIIIB, EIIIA, and V segments in the adult mouse. Cell Adhes. Commun. 1996, 4, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Astrof, S.; Hynes, R.O. Fibronectins in vascular morphogenesis. Angiogenesis 2009, 12, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, M.; Soltermann, A.; Arni, S.; Weder, W.; Neri, D.; Hillinger, S. Comparative immunohistochemistry of L19 and F16 in non-small cell lung cancer and mesothelioma: Two human antibodies investigated in clinical trials in patients with cancer. Lung Cancer 2009, 64, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of Fibronectin Extracellular Matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 2007, 5, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Hielscher, A.; Ellis, K.; Qiu, C.; Porterfield, J.; Gerecht, S. Fibronectin Deposition Participates in Extracellular Matrix Assembly and Vascular Morphogenesis. PLoS ONE 2016, 11, e0147600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Rowe, R.G.; Hiraoka, N.; George, J.P.; Wirtz, D.; Mosher, D.F.; Virtanen, I.; Chernousov, M.A.; Weiss, S.J. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 2008, 22, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Sun, Z.; Opitz, S.L.; Schmidt, T.E.; Peters, J.H.; George, E.L. Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood 2004, 104, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Yoshida, N.; Kataoka, Y.; Manabe, R.; Mizuno-Horikawa, Y.; Sato, M.; Kuriyama, K.; Yasui, N.; Sekiguchi, K. Mice lacking the EDB segment of fibronectin develop normally but exhibit reduced cell growth and fibronectin matrix assembly in vitro. Cancer Res. 2002, 62, 5603–5610. [Google Scholar] [PubMed]
- Astrof, S.; Crowley, D.; Hynes, R.O. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev. Biol. 2007, 311, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Perruzzi, C.A.; de Fougerolles, A.R.; Koteliansky, V.E.; Whelan, M.C.; Westlin, W.F.; Senger, D.R. Functional overlap and cooperativity among αv and β1 integrin subfamilies during skin angiogenesis. J. Investig. Dermatol. 2003, 120, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.M.; DiLullo, G.; Slater, S.J.; Martinez, J.; Iozzo, R.V.; Lauer-Fields, J.L.; Fields, G.B.; San Antonio, J.D. Angiogenesis in collagen I requires α2β1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. J. Biol. Chem. 2003, 278, 30516–30524. [Google Scholar] [CrossRef] [PubMed]
- Whelan, M.C.; Senger, D.R. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. J. Biol. Chem. 2003, 278, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kamei, M.; Saunders, W.B.; Bayless, K.J.; Dye, L.; Davis, G.E.; Weinstein, B.M. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 2006, 442, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Stratman, A.N.; Saunders, W.B.; Sacharidou, A.; Koh, W.; Fisher, K.E.; Zawieja, D.C.; Davis, M.J.; Davis, G.E. Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood 2009, 114, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Segura, I.; De, B.K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, A.; Nyberg, P.; Keshamouni, V.G.; Mannam, A.P.; Li, J.; Sugimoto, H.; Cosgrove, D.; Kalluri, R. Human α1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α1β1 integrin. J. Clin. Investig. 2005, 115, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. Discovery of type IV collagen non-collagenous domains as novel integrin ligands and endogenous inhibitors of angiogenesis. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Rodriguez, D.; Petitclerc, E.; Kim, J.J.; Hangai, M.; Moon, Y.S.; Davis, G.E.; Brooks, P.C. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol. 2001, 154, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jang, J.W.; Lee, O.H.; Yeon, J.; Choi, E.Y.; Kim, K.W.; Lee, S.T.; Kwon, Y.G. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res. 2000, 60, 5410–5413. [Google Scholar] [PubMed]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a Novel Matrix-derived Inhibitor of Angiogenesis and Tumor Growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Petitclerc, E.; Boutaud, A.; Prestayko, A.; Xu, J.; Sado, Y.; Ninomiya, Y.; Sarras, M.P., Jr.; Hudson, B.G.; Brooks, P.C. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000, 275, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Aikio, M.; Alahuhta, I.; Nurmenniemi, S.; Suojanen, J.; Palovuori, R.; Teppo, S.; Sorsa, T.; Lopez-Otin, C.; Pihlajaniemi, T.; Salo, T.; et al. Arresten, a collagen-derived angiogenesis inhibitor, suppresses invasion of squamous cell carcinoma. PLoS ONE 2012, 7, e51044. [Google Scholar] [CrossRef] [PubMed]
- He, G.A.; Luo, J.X.; Zhang, T.Y.; Wang, F.Y.; Li, R.F. Canstatin-N fragment inhibits in vitro endothelial cell proliferation and suppresses in vivo tumor growth. Biochem. Biophys. Res. Commun. 2003, 312, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Bo, J.; Yoo, K.H.; Park, J.H.; Jeong, H.S.; Chung, I.S. Recombinant canstatin inhibits angiopoietin-1-induced angiogenesis and lymphangiogenesis. Int. J. Cancer 2012, 131, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bouquet, C.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with αvβ3 and αvβ5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar] [CrossRef] [PubMed]
- Panka, D.J.; Mier, J.W. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J. Biol. Chem. 2003, 278, 37632–37636. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, Y.; Sudhakar, A.; Lively, J.C.; Ueki, K.; Kharbanda, S.; Kahn, C.R.; Sonenberg, N.; Hynes, R.O.; Kalluri, R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science 2002, 295, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Duncan, M.B.; Pahler, J.; Sugimoto, H.; Martino, M.; Lively, J.; Mundel, T.; Soubasakos, M.; Rubin, K.; Takeda, T.; et al. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc. Natl. Acad. Sci. USA 2011, 108, 9939–9944. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.C.; Dion, A.S.; Abraham, V.; Amenta, P.S. Type XV collagen exhibits a widespread distribution in human tissues but a distinct localization in basement membrane zones. Cell Tissue Res. 1996, 286, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Muragaki, Y.; Abe, N.; Ninomiya, Y.; Olsen, B.R.; Ooshima, A. The human α 1(XV) collagen chain contains a large amino-terminal non-triple helical domain with a tandem repeat structure and homology to α 1(XVIII) collagen. J. Biol. Chem. 1994, 269, 4042–4046. [Google Scholar] [PubMed]
- Ramchandran, R.; Dhanabal, M.; Volk, R.; Waterman, M.J.; Segal, M.; Lu, H.; Knebelmann, B.; Sukhatme, V.P. Antiangiogenic activity of restin, NC10 domain of human collagen XV: Comparison to endostatin. Biochem. Biophys. Res. Commun. 1999, 255, 735–739. [Google Scholar] [CrossRef] [PubMed]
- John, H.; Radtke, K.; Standker, L.; Forssmann, W.G. Identification and characterization of novel endogenous proteolytic forms of the human angiogenesis inhibitors restin and endostatin. Biochim. Biophys. Acta 2005, 1747, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Piuhola, J.; Komulainen, J.; Sormunen, R.; Ongvarrasopone, C.; Fassler, R.; Muona, A.; Ilves, M.; Ruskoaho, H.; Takala, T.E.; et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Ferreras, M.; Felbor, U.; Lenhard, T.; Olsen, B.R.; Delaisse, J. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett. 2000, 486, 247–251. [Google Scholar] [CrossRef]
- Lee, S.J.; Jang, J.W.; Kim, Y.M.; Lee, H.I.; Jeon, J.Y.; Kwon, Y.G.; Lee, S.T. Endostatin binds to the catalytic domain of matrix metalloproteinase-2. FEBS Lett. 2002, 519, 147–152. [Google Scholar] [CrossRef]
- Chang, J.H.; Gabison, E.E.; Kato, T.; Azar, D.T. Corneal neovascularization. Curr. Opin. Ophthalmol. 2001, 12, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Yang, Y.; Lu, N.; You, Q.D.; Wang, S.; Gao, Y.; Chen, Y.; Guo, Q.L. Endostar, a novel recombinant human endostatin, exerts antiangiogenic effect via blocking VEGF-induced tyrosine phosphorylation of KDR/Flk-1 of endothelial cells. Biochem. Biophys. Res. Commun. 2007, 361, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lim, S.J.; Park, Y.K. Anti-angiogenic factor endostatin in osteosarcoma. APMIS 2009, 117, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, H.; Yao, C.; Su, F.; Guan, W.; Yan, S.; Ni, Z. Antitumor activity of combined endostatin and thymidine kinase gene therapy in C6 glioma models. Cancer Med. 2016, 5, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Ferician, O.; Cimpean, A.M.; Avram, S.; Raica, M. Endostatin Effects on Tumor Cells and Vascular Network of Human Renal Cell Carcinoma Implanted on Chick Embryo Chorioallantoic Membrane. Anticancer Res. 2015, 35, 6521–6528. [Google Scholar] [PubMed]
- Guan, Y.; Li, A.; Xiao, W.; Liu, S.; Chen, B.; Lu, T.; Zhao, C.; Han, F. The efficacy and safety of Endostar combined with chemoradiotherapy for patients with advanced, locally recurrent nasopharyngeal carcinoma. Oncotarget 2015, 6, 33926–33934. [Google Scholar] [PubMed]
- Li, W.; Zhao, X.; Du, B.; Li, X.; Liu, S.; Yang, X.Y.; Ding, H.; Yang, W.; Pan, F.; Wu, X.; et al. Gold Nanoparticle-Mediated Targeted Delivery of Recombinant Human Endostatin Normalizes Tumour Vasculature and Improves Cancer Therapy. Sci. Rep. 2016, 6, 30619. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Deng, Q.H.; Yu, X.M.; Ji, Y.L.; Zheng, Y.D.; Jiang, H.; Xu, Y.P.; Ma, S.L. A phase II study of Endostatin in combination with paclitaxel, carboplatin, and radiotherapy in patients with unresectable locally advanced non-small cell lung cancer. BMC Cancer 2016, 16, 266. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Bergsland, E.K.; Ryan, D.P.; Enzinger, P.C.; Lynch, T.J.; Zhu, A.X.; Meyerhardt, J.A.; Heymach, J.V.; Fogler, W.E.; Sidor, C.; et al. Phase II study of recombinant human endostatin in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2006, 24, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Mao, L.; Chi, Z.; Si, L.; Sheng, X.; Kong, Y.; Li, S.; Lian, B.; Gu, K.; Tao, M.; et al. A phase II, randomized, double-blind, placebo-controlled multicenter trial of Endostar in patients with metastatic melanoma. Mol. Ther. 2013, 21, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M. Laminins. Cell Tissue Res. 2010, 339, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Boroujerdi, A.; Welser-Alves, J.V.; Milner, R. Matrix metalloproteinase-9 mediates post-hypoxic vascular pruning of cerebral blood vessels by degrading laminin and claudin-5. Angiogenesis 2015, 18, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Falcone, D.J. Role of laminin in matrix induction of macrophage urokinase-type plasminogen activator and 92-kDa metalloproteinase expression. J. Biol. Chem. 1997, 272, 8270–8275. [Google Scholar] [CrossRef] [PubMed]
- Yousif, L.F.; di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 2001, 153, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.M.; Gonzales, M.; Herron, G.S.; Nagavarapu, U.; Hopkinson, S.B.; Tsuruta, D.; Jones, J.C. Complex interactions between the laminin α4 subunit and integrins regulate endothelial cell behavior in vitro and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 16075–16080. [Google Scholar] [CrossRef] [PubMed]
- Wragg, J.W.; Finnity, J.P.; Anderson, J.A.; Ferguson, H.J.; Porfiri, E.; Bhatt, R.I.; Murray, P.G.; Heath, V.L.; Bicknell, R. MCAM and LAMA4 Are Highly Enriched in Tumor Blood Vessels of Renal Cell Carcinoma and Predict Patient Outcome. Cancer Res. 2016, 76, 2314–2326. [Google Scholar] [CrossRef] [PubMed]
- Estrach, S.; Cailleteau, L.; Franco, C.A.; Gerhardt, H.; Stefani, C.; Lemichez, E.; Gagnoux-Palacios, L.; Meneguzzi, G.; Mettouchi, A. Laminin-binding integrins induce Dll4 expression and Notch signaling in endothelial cells. Circ. Res. 2011, 109, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Shibuya, M.; Hoffman, M.P.; Engbring, J.A.; Hossain, R.; Mochizuki, M.; Kudoh, S.; Nomizu, M.; Kleinman, H.K. Laminin α5 chain metastasis- and angiogenesis-inhibiting peptide blocks fibroblast growth factor 2 activity by binding to the heparan sulfate chains of CD44. Cancer Res. 2005, 65, 10494–10501. [Google Scholar] [CrossRef] [PubMed]
- Thyboll, J.; Kortesmaa, J.; Cao, R.; Soininen, R.; Wang, L.; Iivanainen, A.; Sorokin, L.; Risling, M.; Cao, Y.; Tryggvason, K. Deletion of the laminin α4 chain leads to impaired microvessel maturation. Mol. Cell. Biol. 2002, 22, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Di Russo, J.; Hannocks, M.J.; Luik, A.L.; Song, J.; Zhang, X.; Yousif, L.; Aspite, G.; Hallmann, R.; Sorokin, L. Vascular laminins in physiology and pathology. Matrix Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Ehling, M.; Kato, K.; Kanai, K.; van Lessen, M.; Frye, M.; Zeuschner, D.; Nakayama, M.; Vestweber, D.; Adams, R.H. Integrin β1 controls VE-cadherin localization and blood vessel stability. Nat. Commun. 2015, 6, 6429. [Google Scholar] [CrossRef] [PubMed]
- Stamati, K.; Priestley, J.V.; Mudera, V.; Cheema, U. Laminin promotes vascular network formation in 3D in vitro collagen scaffolds by regulating VEGF uptake. Exp. Cell Res. 2014, 327, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Chiaverina, G.; Gagliardi, P.A.; di Blasio, L.; Puliafito, A.; Bouvard, C.; Sessa, R.; Tarone, G.; Sorokin, L.; Helley, D.; et al. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat. Cell Biol. 2014, 16, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Tzanakakis, G.N.; Karamanos, N.K. Proteoglycans in health and disease: Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010, 277, 3904–3923. [Google Scholar] [CrossRef] [PubMed]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavao, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Bugatti, A.; Urbinati, C.; Rusnati, M. Heparin/Heparan sulfate proteoglycans glycomic interactome in angiogenesis: Biological implications and therapeutical use. Molecules 2015, 20, 6342–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Neill, T.; Multhaupt, H.A.; Hubo, M.; Frey, H.; Gopal, S.; Gomes, A.; Afratis, N.; Lim, H.C.; et al. Insights into the key roles of proteoglycans in breast cancer biology and translational medicine. Biochim. Biophys. Acta 2015, 1855, 276–300. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Yang, Y.; Suva, L.J.; Kelly, T. Heparan sulfate proteoglycans and heparanase—Partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004, 23, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Suhovskih, A.V.; Aidagulova, S.V.; Kashuba, V.I.; Grigorieva, E.V. Proteoglycans as potential microenvironmental biomarkers for colon cancer. Cell Tissue Res. 2015, 361, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Baghy, K.; Tatrai, P.; Regos, E.; Kovalszky, I. Proteoglycans in liver cancer. World J. Gastroenterol. 2016, 22, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, A.D.; Dodge, G.R.; Cohen, I.; Tuan, R.S.; Iozzo, R.V. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 1992, 267, 8544–8557. [Google Scholar] [PubMed]
- Iozzo, R.V.; Cohen, I.R.; Grassel, S.; Murdoch, A.D. The biology of perlecan: The multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem. J. 1994, 302, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Farach-Carson, M.C.; Carson, D.D. Perlecan-a multifunctional extracellular proteoglycan scaffold. Glycobiology 2007, 17, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, A.D.; Liu, B.; Schwarting, R.; Tuan, R.S.; Iozzo, R.V. Widespread expression of perlecan proteoglycan in basement membranes and extracellular matrices of human tissues as detected by a novel monoclonal antibody against domain III and by in situ hybridization. J. Histochem. Cytochem. 1994, 42, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Handler, M.; Yurchenco, P.D.; Iozzo, R.V. Developmental expression of perlecan during murine embryogenesis. Dev. Dyn. 1997, 210, 130–145. [Google Scholar] [CrossRef]
- Whitelock, J.M.; Melrose, J.; Iozzo, R.V. Diverse cell signaling events modulated by perlecan. Biochemistry 2008, 47, 11174–11183. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Taylor, K.; Otto, J.; Aho, S.; Uitto, J.; Whitelock, J.M.; Iozzo, R.V. The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J. Biol. Chem. 2000, 275, 7095–7100. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Otto, J.; Oldershaw, R.; Ferrer, F.; Sato, J.D.; Iozzo, R.V. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J. Biol. Chem. 2001, 276, 10263–10271. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Fu, J.; Oldershaw, R.; Greenhalgh, R.; Gown, A.M.; Iozzo, R.V. Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis. J. Biol. Chem. 2003, 278, 17491–17499. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.M.; Mongiat, M.; Slater, S.J.; Baffa, R.; Iozzo, R.V. A novel interaction between perlecan protein core and progranulin: Potential effects on tumor growth. J. Biol. Chem. 2003, 278, 38113–38116. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shimono, C.; Norioka, N.; Nakano, I.; Okubo, T.; Yagi, Y.; Hayashi, M.; Sato, Y.; Fujisaki, H.; Hattori, S.; et al. Activin A binds to perlecan through its pro-region that has heparin/heparan sulfate binding activity. J. Biol. Chem. 2010, 285, 36645–36655. [Google Scholar] [CrossRef] [PubMed]
- Poluzzi, C.; Iozzo, R.V.; Schaefer, L. Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers. Adv. Drug Deliv. Rev. 2016, 97, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Handler, M.; Eichstetter, I.; Whitelock, J.M.; Nugent, M.A.; Iozzo, R.V. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Investig. 1998, 102, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; San Antonio, J.D. Heparan sulfate proteoglycans: Heavy hitters in the angiogenesis arena. J. Clin. Investig. 2001, 108, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004, 64, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Aviezer, D.; Hecht, D.; Safran, M.; Eisinger, M.; David, G.; Yayon, A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 1994, 79, 1005–1013. [Google Scholar] [CrossRef]
- Ghiselli, G.; Eichstetter, I.; Iozzo, R.V. A role for the perlecan protein core in the activation of the keratinocyte growth factor receptor. Biochem. J. 2001, 359, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Simons, M. Fibroblast growth factor regulation of neovascularization. Curr. Opin. Hematol. 2008, 15, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, J.J.; Whitelock, J.M.; Iozzo, R.V. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009, 28, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Lord, M.S.; Melrose, J.; Rees, M.D.; Knox, S.M.; Freeman, C.; Iozzo, R.V.; Whitelock, J.M. Heparan sulfate-dependent signaling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry 2010, 49, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, A.; Cooper, C.R.; Gomes, R.R., Jr. Soluble perlecan domain I enhances vascular endothelial growth factor-165 activity and receptor phosphorylation in human bone marrow endothelial cells. BMC Biochem. 2010, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Chuang, C.Y.; Melrose, J.; Davies, M.J.; Iozzo, R.V.; Whitelock, J.M. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014, 35, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Whitelock, J.M.; Murdoch, A.D.; Iozzo, R.V.; Underwood, P.A. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J. Biol. Chem. 1996, 271, 10079–10086. [Google Scholar] [PubMed]
- Marchisone, C.; Del Grosso, F.; Masiello, L.; Prat, M.; Santi, L.; Noonan, D.M. Phenotypic alterations in Kaposi's sarcoma cells by antisense reduction of perlecan. Pathol. Oncol. Res. 2000, 6, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Kang, U.B.; Kim, D.H.; Yi, J.K.; Lee, J.W.; Noh, D.Y.; Lee, C.; Yu, M.H. Identification of circulating endorepellin LG3 fragment: Potential use as a serological biomarker for breast cancer. Proteom. Clin. Appl. 2008, 2, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Granato, D.C.; Carnielli, C.M.; Cervigne, N.K.; Oliveria, C.E.; Rivera, C.; Yokoo, S.; Fonseca, F.P.; Lopes, M.; Santos-Silva, A.R.; et al. Agrin and perlecan mediate tumorigenic processes in oral squamous cell carcinoma. PLoS ONE 2014, 9, e115004. [Google Scholar] [CrossRef] [PubMed]
- Grindel, B.; Li, Q.; Arnold, R.; Petros, J.; Zayzafoon, M.; Muldoon, M.; Stave, J.; Chung, L.W.; Farach-Carson, M.C. Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: Tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget 2016, 7, 10433–10447. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Bix, G.; Fu, J.; Gonzalez, E.M.; Macro, L.; Barker, A.; Campbell, S.; Zutter, M.M.; Santoro, S.A.; Kim, J.K.; Hook, M.; et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through α2β1 integrin. J. Cell Biol. 2004, 166, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Bix, G.; Castello, R.; Burrows, M.; Zoeller, J.J.; Weech, M.; Iozzo, R.A.; Cardi, C.; Thakur, M.L.; Barker, C.A.; Camphausen, K.; et al. Endorepellin in vivo: Targeting the tumor vasculature and retarding cancer growth and metabolism. J. Natl. Cancer Inst. 2006, 98, 1634–1646. [Google Scholar] [CrossRef] [PubMed]
- Woodall, B.P.; Nystrom, A.; Iozzo, R.A.; Eble, J.A.; Niland, S.; Krieg, T.; Eckes, B.; Pozzi, A.; Iozzo, R.V. Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 2008, 283, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, A.; Shaik, Z.P.; Gullberg, D.; Krieg, T.; Eckes, B.; Zent, R.; Pozzi, A.; Iozzo, R.V. Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity. Blood 2009, 114, 4897–4906. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Poluzzi, C.; Willis, C.D.; Smythies, J.; Shellard, A.; Neill, T.; Iozzo, R.V. Endorepellin affects angiogenesis by antagonizing diverse vascular endothelial growth factor receptor 2 (VEGFR2)-evoked signaling pathways: Transcriptional repression of hypoxia-inducible factor 1α and VEGFA and concurrent inhibition of nuclear factor of activated T cell 1 (NFAT1) activation. J. Biol. Chem. 2012, 287, 43543–43556. [Google Scholar] [PubMed]
- Goyal, A.; Pal, N.; Concannon, M.; Paul, M.; Doran, M.; Poluzzi, C.; Sekiguchi, K.; Whitelock, J.M.; Neill, T.; Iozzo, R.V. Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2): A dual receptor antagonism. J. Biol. Chem. 2011, 286, 25947–25962. [Google Scholar] [CrossRef] [PubMed]
- Douglass, S.; Goyal, A.; Iozzo, R.V. The role of perlecan and endorepellin in the control of tumor angiogenesis and endothelial cell autophagy. Connect. Tissue Res. 2015, 56, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Poluzzi, C.; Casulli, J.; Goyal, A.; Mercer, T.J.; Neill, T.; Iozzo, R.V. Endorepellin evokes autophagy in endothelial cells. J. Biol. Chem. 2014, 289, 16114–16128. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Gubbiotti, M.A.; Chery, D.R.; Han, L.; Iozzo, R.V. Endorepellin-evoked autophagy contributes to angiostasis. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sofeu Feugaing, D.D.; Gotte, M.; Viola, M. More than matrix: The multifaceted role of decorin in cancer. Eur. J. Cell Biol. 2013, 92, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jarvelainen, H.; Vernon, R.B.; Gooden, M.D.; Francki, A.; Lara, S.; Johnson, P.Y.; Kinsella, M.G.; Sage, E.H.; Wight, T.N. Overexpression of decorin by rat arterial smooth muscle cells enhances contraction of type I collagen in vitro. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kalamajski, S.; Oldberg, A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010, 29, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Reese, S.P.; Underwood, C.J.; Weiss, J.A. Effects of decorin proteoglycan on fibrillogenesis, ultrastructure, and mechanics of type I collagen gels. Matrix Biol. 2013, 32, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Buraschi, S.; Xu, S.Q.; Belfiore, A.; Schaefer, L.; Iozzo, R.V.; Morrione, A. Decorin differentially modulates the activity of insulin receptor isoform A ligands. Matrix Biol. 2014, 35, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Tovey, J.C.; Sharma, A.; Schultz, G.S.; Cowden, J.W.; Tandon, A. Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS ONE 2011, 6, e26432. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Panitch, A. Decorin mimic regulates platelet-derived growth factor and interferon-gamma stimulation of vascular smooth muscle cells. Biomacromolecules 2014, 15, 2090–2103. [Google Scholar] [CrossRef] [PubMed]
- Jarvelainen, H.; Sainio, A.; Wight, T.N. Pivotal role for decorin in angiogenesis. Matrix Biol. 2015, 43, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-β1 release. Biochem. J. 1997, 322, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Boivin, W.A.; Shackleford, M.; Vanden Hoek, A.; Zhao, H.; Hackett, T.L.; Knight, D.A.; Granville, D.J. Granzyme B cleaves decorin, biglycan and soluble βglycan, releasing active transforming growth factor-β1. PLoS ONE 2012, 7, e33163. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decorin as a multivalent therapeutic agent against cancer. Adv. Drug Deliv. Rev. 2016, 97, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Painter, H.; Buraschi, S.; Owens, R.T.; Lisanti, M.P.; Schaefer, L.; Iozzo, R.V. Decorin antagonizes the angiogenic network: Concurrent inhibition of Met, hypoxia inducible factor 1α, vascular endothelial growth factor A, and induction of thrombospondin-1 and TIMP3. J. Biol. Chem. 2012, 287, 5492–5506. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Neill, T.; Owens, R.T.; Schaefer, L.; Iozzo, R.V. Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol. 2014, 35, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.A.; Iozzo, R.V. Proteoglycans regulate autophagy via outside-in signaling: An emerging new concept. Matrix Biol. 2015, 48, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, E.; Sunderkotter, C.; Schaefer, L.; Thanos, S.; Grassel, S.; Oldberg, A.; Iozzo, R.V.; Young, M.F.; Kresse, H. Decorin deficiency leads to impaired angiogenesis in injured mouse cornea. J. Vasc. Res. 2004, 41, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Jarvelainen, H.; Puolakkainen, P.; Pakkanen, S.; Brown, E.L.; Hook, M.; Iozzo, R.V.; Sage, E.H.; Wight, T.N. A role for decorin in cutaneous wound healing and angiogenesis. Wound Repair Regen. 2006, 14, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.S.; Yenisey, C.; Rose, R.W.; Tootell, M.; Santra, M.; Iozzo, R.V. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene 2002, 21, 4765–4777. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Pinnow, E.L.; Maeda, A.; Brown, A.C.; McCartney-Francis, N.; Kram, V.; Owens, R.T.; Robey, P.G.; Holmbeck, K.; de Castro, L.F.; et al. Biglycan modulates angiogenesis and bone formation during fracture healing. Matrix Biol. 2014, 35, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Myren, M.; Kirby, D.J.; Noonan, M.L.; Maeda, A.; Owens, R.T.; Ricard-Blum, S.; Kram, V.; Kilts, T.M.; Young, M.F. Biglycan potentially regulates angiogenesis during fracture repair by altering expression and function of endostatin. Matrix Biol. 2016, 52–54, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Gu, X.; Ma, T.; Ye, H. Biglycan up-regulated vascular endothelial growth factor (VEGF) expression and promoted angiogenesis in colon cancer. Tumour Biol. 2015, 36, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Barbouri, D.; Afratis, N.; Gialeli, C.; Vynios, D.H.; Theocharis, A.D.; Karamanos, N.K. Syndecans as modulators and potential pharmacological targets in cancer progression. Front. Oncol. 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Rapraeger, A.C.; Ell, B.J.; Roy, M.; Li, X.; Morrison, O.R.; Thomas, G.M.; Beauvais, D.M. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between αvβ3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J. 2013, 280, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, S.; Ferrero, S.; Aschero, S.; Monitillo, L.; Bussolati, B.; Omede, P.; Ladetto, M.; Camussi, G. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia 2012, 26, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Wei-Jie, Y.; Yi-Feng, Z.G.; Jing, H. Downregulation of Syndecan-1 induce glomerular endothelial cell dysfunction through modulating internalization of VEGFR-2. Cell Signal. 2016, 28, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Desouky, J.; Friedl, A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene 2006, 25, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Noguer, O.; Villena, J.; Lorita, J.; Vilaro, S.; Reina, M. Syndecan-2 downregulation impairs angiogenesis in human microvascular endothelial cells. Exp. Cell Res. 2009, 315, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Uyama, T.; Kobayashi, F.; Yamada, S.; Sugahara, K.; Rapraeger, A.C.; Sanderson, R.D. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood 2010, 115, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, G.; Evans, A.R.; Kay, E.; Woodfin, A.; McKay, T.R.; Nourshargh, S.; Whiteford, J.R. Shed syndecan-2 inhibits angiogenesis. J. Cell Sci. 2014, 127, 4788–4799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Manzaneque, J.C.; Carpizo, D.; Plaza-Calonge, M.C.; Torres-Collado, A.X.; Thai, S.N.; Simons, M.; Horowitz, A.; Iruela-Arispe, M.L. Cleavage of syndecan-4 by ADAMTS1 provokes defects in adhesion. Int. J. Biochem. Cell Biol. 2009, 41, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xie, J.; Wu, H.; Li, G.; Chen, J.; Chen, Q.; Wang, L.; Xu, B. Syndecan-4 shedding impairs macrovascular angiogenesis in diabetes mellitus. Biochem. Biophys. Res. Commun. 2016, 474, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, T.; Whipple, C.A.; Lopez, M.E.; Gunn, J.; Young, A.; Lander, A.D.; Korc, M. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J. Clin. Investig. 2008, 118, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Meyer, K.; Mundhenke, C.; Drew, S.A.; Friedl, A. Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J. Biol. Chem. 2003, 278, 16045–16053. [Google Scholar] [CrossRef] [PubMed]
- Fico, A.; Maina, F.; Dono, R. Fine-tuning of cell signaling by glypicans. Cell. Mol. Life Sci. 2011, 68, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Monteforte, A.J.; Lam, B.; Das, S.; Mukhopadhyay, S.; Wright, C.S.; Martin, P.E.; Dunn, A.K.; Baker, A.B. Glypican-1 nanoliposomes for potentiating growth factor activity in therapeutic angiogenesis. Biomaterials 2016, 94, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Naito, Z. Role of the small leucine-rich proteoglycan (SLRP) family in pathological lesions and cancer cell growth. J. Nippon Med. Sch. 2005, 72, 137–145. [Google Scholar] [CrossRef] [PubMed]
- D'Onofrio, M.F.; Brezillon, S.; Baranek, T.; Perreau, C.; Roughley, P.J.; Maquart, F.X.; Wegrowski, Y. Identification of β1 integrin as mediator of melanoma cell adhesion to lumican. Biochem. Biophys. Res. Commun. 2008, 365, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Albig, A.R.; Roy, T.G.; Becenti, D.J.; Schiemann, W.P. Transcriptome analysis of endothelial cell gene expression induced by growth on matrigel matrices: Identification and characterization of MAGP-2 and lumican as novel regulators of angiogenesis. Angiogenesis 2007, 10, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Niewiarowska, J.; Brezillon, S.; Sacewicz-Hofman, I.; Bednarek, R.; Maquart, F.X.; Malinowski, M.; Wiktorska, M.; Wegrowski, Y.; Cierniewski, C.S. Lumican inhibits angiogenesis by interfering with α2β1 receptor activity and downregulating MMP-14 expression. Thromb. Res. 2011, 128, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Papoutsidakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican affects tumor cell functions, tumor-ECM interactions, angiogenesis and inflammatory response. Matrix Biol. 2014, 35, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Fulford, L.A.; Albig, A.R. Lumican reduces tumor growth via induction of fas-mediated endothelial cell apoptosis. Cancer Microenviron. 2010, 4, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Kumar, S.; Gaffney, J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J. Biol. Chem. 2002, 277, 41046–41059. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Savani, R.C.; Cao, G.; Pooler, P.M.; Zaman, A.; Zhou, Z.; DeLisser, H.M. Differential involvement of the hyaluronan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J. Biol. Chem. 2001, 276, 36770–36778. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Gaffney, J.; Matou, S.; West, D.; Delisser, H.; Savani, R.C.; Kumar, S. Hyaluronan-mediated angiogenesis in vascular disease: Uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007, 26, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, C.X.; Mo, W.; Liu, Y.W.; He, Y.Q. Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing. Clin. Investig. Med. 2008, 31, E106–E116. [Google Scholar]
- Wang, Y.; Han, G.; Guo, B.; Huang, J. Hyaluronan oligosaccharides promote diabetic wound healing by increasing angiogenesis. Pharmacol. Rep. 2016, 68, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.N.; Alaniz, L.D.; Menger, M.D.; Baranao, R.I.; Laschke, M.W.; Meresman, G.F. Inhibition of Hyaluronic Acid Synthesis Suppresses Angiogenesis in Developing Endometriotic Lesions. PLoS ONE 2016, 11, e0152302. [Google Scholar] [CrossRef] [PubMed]
- Litwiniuk, M.; Krejner, A.; Speyrer, M.S.; Gauto, A.R.; Grzela, T. Hyaluronic Acid in Inflammation and Tissue Regeneration. Wounds 2016, 28, 78–88. [Google Scholar] [PubMed]
- West, D.C.; Kumar, S. The effect of hyaluronate and its oligosaccharides on endothelial cell proliferation and monolayer integrity. Exp. Cell Res. 1989, 183, 179–196. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Lord, J.D.; Masewicz, S.A.; Evanko, S.P.; Buckner, J.H.; Wight, T.N.; Nepom, G.T. Cutting edge: High molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J. Immunol. 2007, 179, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, K.; Hippe, A.; Schmaus, A.; Homey, B.; Sleeman, J.P.; Orian-Rousseau, V. Opposing effects of high- and low-molecular weight hyaluronan on CXCL12-induced CXCR4 signaling depend on CD44. Cell Death Dis. 2013, 4, e819. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Hibi, T.; Isogai, Z.; Yoneda, M.; Fujimori, M.; Amano, J.; Kawakubo, M.; Kannagi, R.; Kimata, K.; Taniguchi, S.; et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: Possible involvement of versican/PG-M. Am. J. Pathol. 2007, 170, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A. Hyaluronan regulation of endothelial barrier function in cancer. Adv. Cancer Res. 2014, 123, 191–209. [Google Scholar] [PubMed]
- Doliana, R.; Canton, A.; Bucciotti, F.; Mongiat, M.; Bonaldo, P.; Colombatti, A. Structure, chromosomal localization, and promoter analysis of the human elastin microfibril interfase located proteIN (EMILIN) gene. J. Biol. Chem. 2000, 275, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Mungiguerra, G.; Bot, S.; Mucignat, M.T.; Giacomello, E.; Doliana, R.; Colombatti, A. Self-assembly and supramolecular organization of EMILIN. J. Biol. Chem. 2000, 275, 25471–25480. [Google Scholar] [CrossRef] [PubMed]
- Colombatti, A.; Spessotto, P.; Doliana, R.; Mongiat, M.; Bressan, G.M.; Esposito, G. The EMILIN/Multimerin family. Front. Immunol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Bot, S.; Andreuzzi, E.; Capuano, A.; Schiavinato, A.; Colombatti, A.; Doliana, R. Multiple-interactions among EMILIN1 and EMILI. Matrix Biol. 2015, 41, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jeimy, S.B.; Tasneem, S.; Cramer, E.M.; Hayward, C.P. Multimerin 1. Platelets 2008, 19, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.; Ahorn, H.; Novatchkova, M.; Garin-Chesa, P.; Park, J.E.; Weber, G.; Eisenhaber, F.; Rettig, W.J.; Lenter, M.C. Molecular cloning and characterization of EndoGlyx-1, an EMILIN-like multisubunit glycoprotein of vascular endothelium. J. Biol. Chem. 2001, 276, 48588–48595. [Google Scholar] [PubMed]
- Doliana, R.; Mongiat, M.; Bucciotti, F.; Giacomello, E.; Deutzmann, R.; Volpin, D.; Bressan, G.M.; Colombatti, A. EMILIN, a component of the elastic fiber and a new member of the C1q/tumor necrosis factor superfamily of proteins. J. Biol. Chem. 1999, 274, 16773–16781. [Google Scholar] [CrossRef] [PubMed]
- Doliana, R.; Bot, S.; Mungiguerra, G.; Canton, A.; Cilli, S.P.; Colombatti, A. Isolation and characterization of EMILIN-2, a new component of the growing EMILINs family and a member of the EMI domain-containing superfamily. J. Biol. Chem. 2001, 276, 12003–12011. [Google Scholar] [CrossRef] [PubMed]
- Corallo, D.; Schiavinato, A.; Trapani, V.; Moro, E.; Argenton, F.; Bonaldo, P. Emilin3 is required for notochord sheath integrity and interacts with Scube2 to regulate notochord-derived Hedgehog signals. Development 2013, 140, 4594–4601. [Google Scholar] [CrossRef] [PubMed]
- Leimeister, C.; Steidl, C.; Schumacher, N.; Erhard, S.; Gessler, M. Developmental Expression and Biochemical Characterization of Emu Family Members. Dev. Biol. 2002, 249, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moncasi, M.P.; Garin-Chesa, P.; Stockert, E.; Jaffe, E.A.; Old, L.J.; Rettig, W.J. Identification of a high molecular weight endothelial cell surface glycoprotein, endoGlyx-1, in normal and tumor blood vessels. Lab. Investig. 1994, 71, 366–373. [Google Scholar] [PubMed]
- Lorenzon, E.; Colladel, R.; Andreuzzi, E.; Marastoni, S.; Todaro, F.; Schiappacassi, M.; Ligresti, G.; Colombatti, A.; Mongiat, M. MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF-A/VEGFR2 pathway. Oncogene 2012, 31, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Colladel, R.; Pellicani, R.; Andreuzzi, E.; Paulitti, A.; Tarticchio, G.; Todaro, F.; Colombatti, A.; Mongiat, M. MULTIMERIN2 binds VEGF-A primarily via the carbohydrate chains exerting an angiostatic function and impairing tumor growth. Oncotarget 2016, 7, 2022–2037. [Google Scholar] [PubMed]
- Noy, P.J.; Swain, R.K.; Khan, K.; Lodhia, P.; Bicknell, R. Sprouting angiogenesis is regulated by shedding of the C-type lectin family 14, member A (CLEC14A) ectodomain, catalyzed by rhomboid-like 2 protein (RHBDL2). FASEB J. 2016, 30, 2311–2323. [Google Scholar] [CrossRef] [PubMed]
- Noy, P.J.; Lodhia, P.; Khan, K.; Zhuang, X.; Ward, D.G.; Verissimo, A.R.; Bacon, A.; Bicknell, R. Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 2015, 34, 5821–5831. [Google Scholar] [CrossRef] [PubMed]
- Braghetta, P.; Ferrari, A.; de Gemmis, P.; Zanetti, M.; Volpin, D.; Bonaldo, P.; Bressan, G.M. Overlapping, complementary and site-specific expression pattern of genes of the EMILIN/Multimerin family. Matrix Biol. 2004, 22, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.K.; Hesson, L.B.; Dansranjavin, T.; Dallol, A.; Bieche, I.; Vacher, S.; Tommasi, S.; Dobbins, T.; Gentle, D.; Euhus, D.; et al. Identification of 5 novel genes methylated in breast and other epithelial cancers. Mol. Cancer 2010, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Marastoni, S.; Ligresti, G.; Lorenzon, E.; Schiappacassi, M.; Perris, R.; Frustaci, S.; Colombatti, A. The extracellular matrix glycoprotein elastin microfibril interface located protein 2: A dual role in the tumor microenvironment. Neoplasia 2010, 12, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 2012, 14, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Klenotic, P.A.; Zhang, C.; Lin, Z. Emerging roles of CCN proteins in vascular development and pathology. J. Cell Commun. Signal. 2016, 10, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.E.; Muntean, A.G.; Chen, C.C.; Stolz, D.B.; Watkins, S.C.; Lau, L.F. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell. Biol. 2002, 22, 8709–8720. [Google Scholar] [CrossRef] [PubMed]
- Babic, A.M.; Kireeva, M.L.; Kolesnikova, T.V.; Lau, L.F. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1998, 95, 6355–6360. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, Y.; Qin, J.; Kuang, C.Y.; Song, M.B.; Yu, S.Y.; Cui, B.; Chen, J.F.; Huang, L. CCN1 promotes the differentiation of endothelial progenitor cells and reendothelialization in the early phase after vascular injury. Basic Res. Cardiol. 2010, 105, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Chintala, H.; Krupska, I.; Yan, L.; Lau, L.; Grant, M.; Chaqour, B. The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development 2015, 142, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Hall-Glenn, F.; de Young, R.A.; Huang, B.L.; van Handel, B.; Hofmann, J.J.; Chen, T.T.; Choi, A.; Ong, J.R.; Benya, P.D.; Mikkola, H.; et al. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS ONE 2012, 7, e30562. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Chuang, S.M.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis. 2014, 5, e1485. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Natesan, V.; Shi, H.; Hamik, A.; Kawanami, D.; Hao, C.; Mahabaleshwar, G.H.; Wang, W.; Jin, Z.G.; Atkins, G.B.; et al. A novel role of CCN3 in regulating endothelial inflammation. J. Cell Commun. Signal. 2010, 4, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.G.; Leu, S.J.; Chen, N.; Tebeau, C.M.; Lin, S.X.; Yeung, C.Y.; Lau, L.F. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J. Biol. Chem. 2003, 278, 24200–24208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; van der Voort, D.; Shi, H.; Zhang, R.; Qing, Y.; Hiraoka, S.; Takemoto, M.; Yokote, K.; Moxon, J.V.; Norman, P.; et al. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J. Clin. Investig. 2016, 126, 1282–1299. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Cheng, H.C.; Wang, J.; Wang, S.W.; Tai, H.C.; Lin, C.W.; Tang, C.H. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget 2014, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, W.; Lin, Z.; Lu, J.; Wan, H.; Zhou, Z.; Liu, Z. CCN4 regulates vascular smooth muscle cell migration and proliferation. Mol. Cells 2013, 36, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Chen, P.C.; Tsao, C.W.; Chang, A.C.; Lein, M.Y.; Lin, C.C.; Wang, S.W.; Lin, C.W.; Tang, C.H. WISP-1 a novel angiogenic regulator of the CCN family promotes oral squamous cell carcinoma angiogenesis through VEGF-A expression. Oncotarget 2015, 6, 4239–4252. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.B.; Rwayitare, K.; Richey, L.; Lem, J.; Castellot, J.J., Jr. CCN5 Expression in mammals. III. Early embryonic mouse development. J. Cell Commun. Signal. 2012, 6, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Connor, A.R.; Sounni, N.E.; Eckhard, U.; Morrison, C.J.; Noel, A.; Overall, C.M. Degradomic and yeast 2-hybrid inactive catalytic domain substrate trapping identifies new membrane-type 1 matrix metalloproteinase (MMP14) substrates: CCN3 (Nov) and CCN5 (WISP2). Matrix Biol. 2016. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111822
Mongiat M, Andreuzzi E, Tarticchio G, Paulitti A. Extracellular Matrix, a Hard Player in Angiogenesis. International Journal of Molecular Sciences. 2016; 17(11):1822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111822
Chicago/Turabian StyleMongiat, Maurizio, Eva Andreuzzi, Giulia Tarticchio, and Alice Paulitti. 2016. "Extracellular Matrix, a Hard Player in Angiogenesis" International Journal of Molecular Sciences 17, no. 11: 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111822